
Floridoside Phosphotriester Derivatives: Synthesis and Inhibition of Human Neutrophils’ Oxidative Burst

 , ,
, ,  ,
,  and
and
Abstract
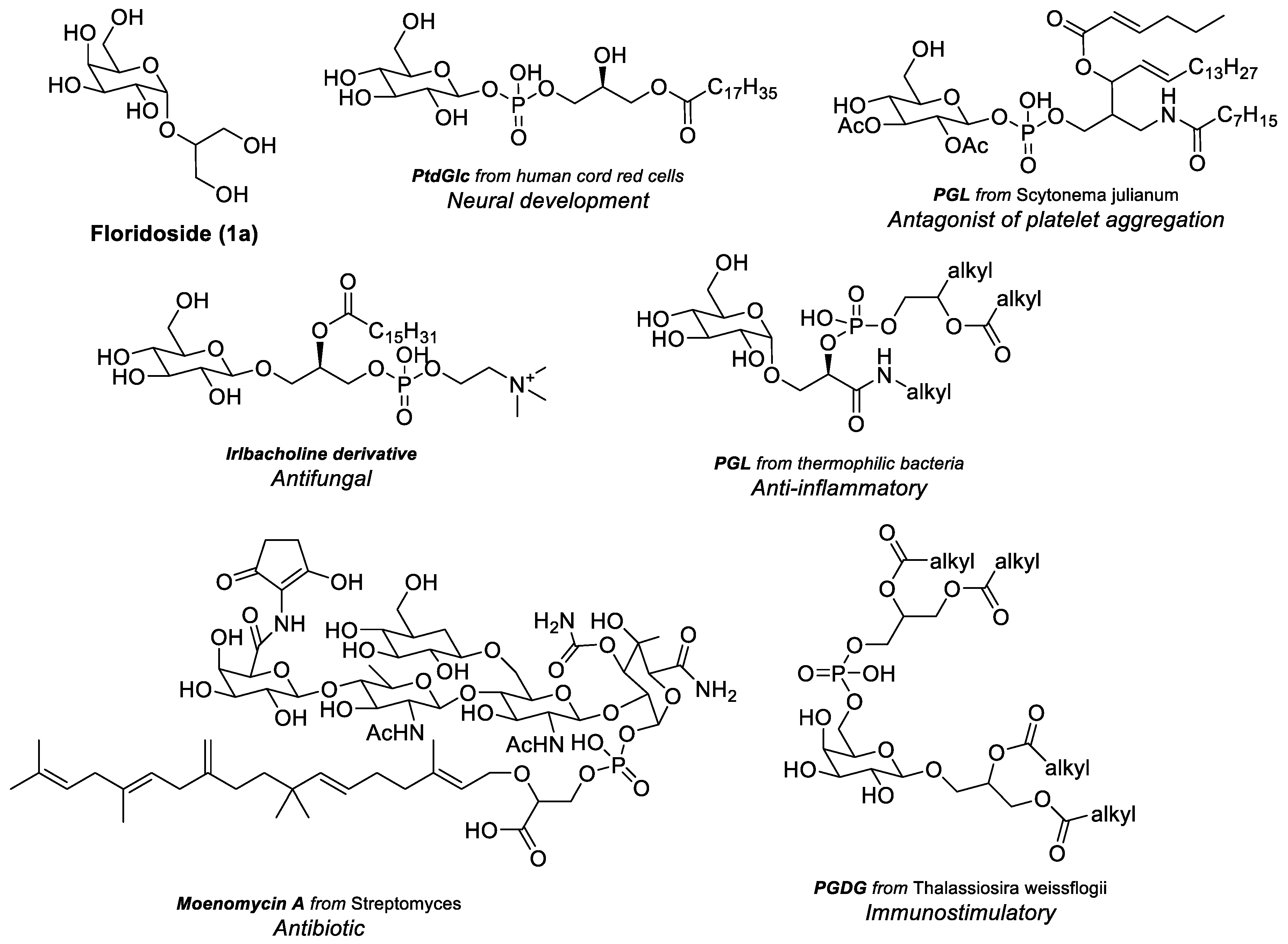
1. Introduction
2. Results and Discussion
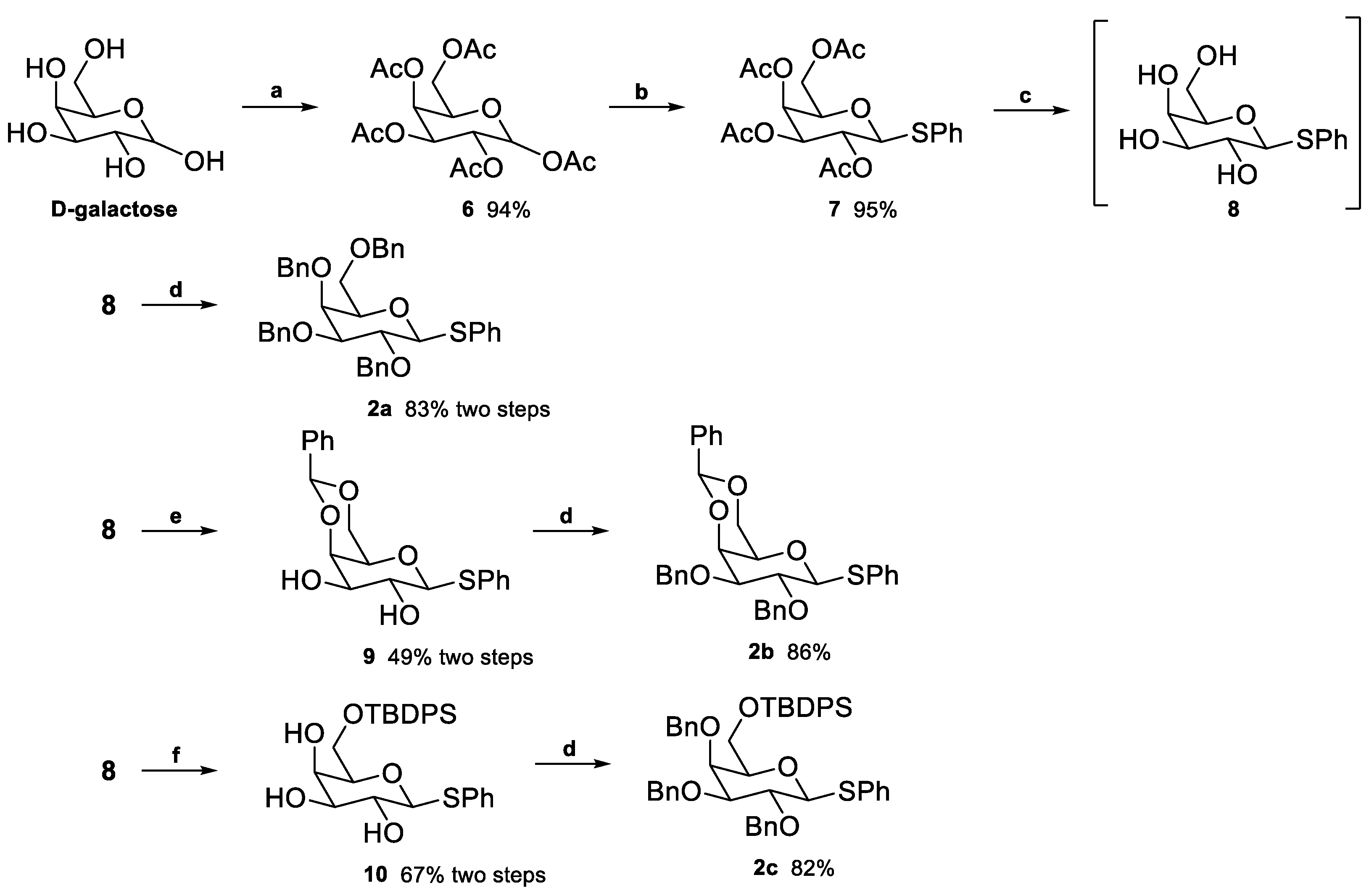
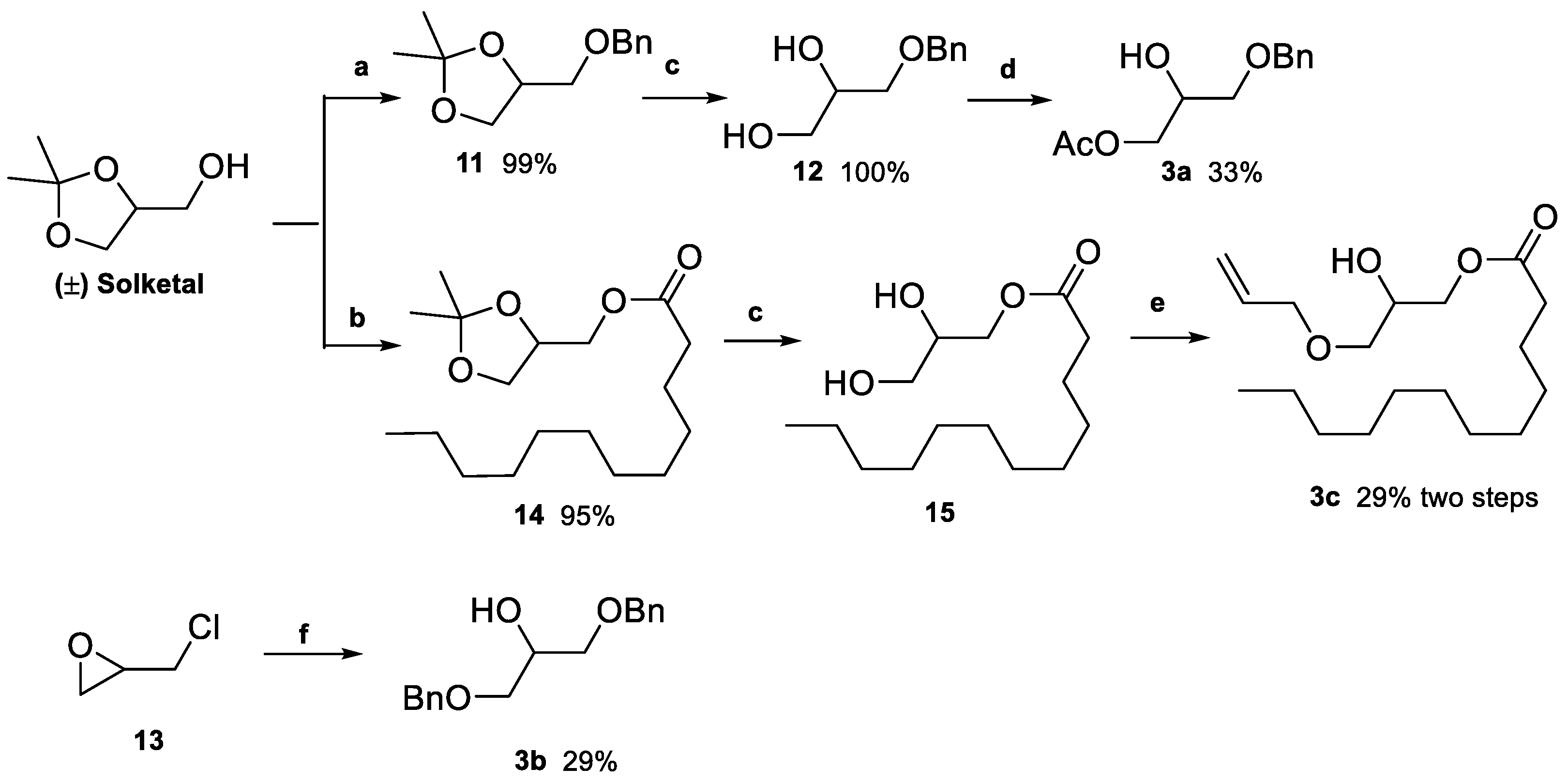
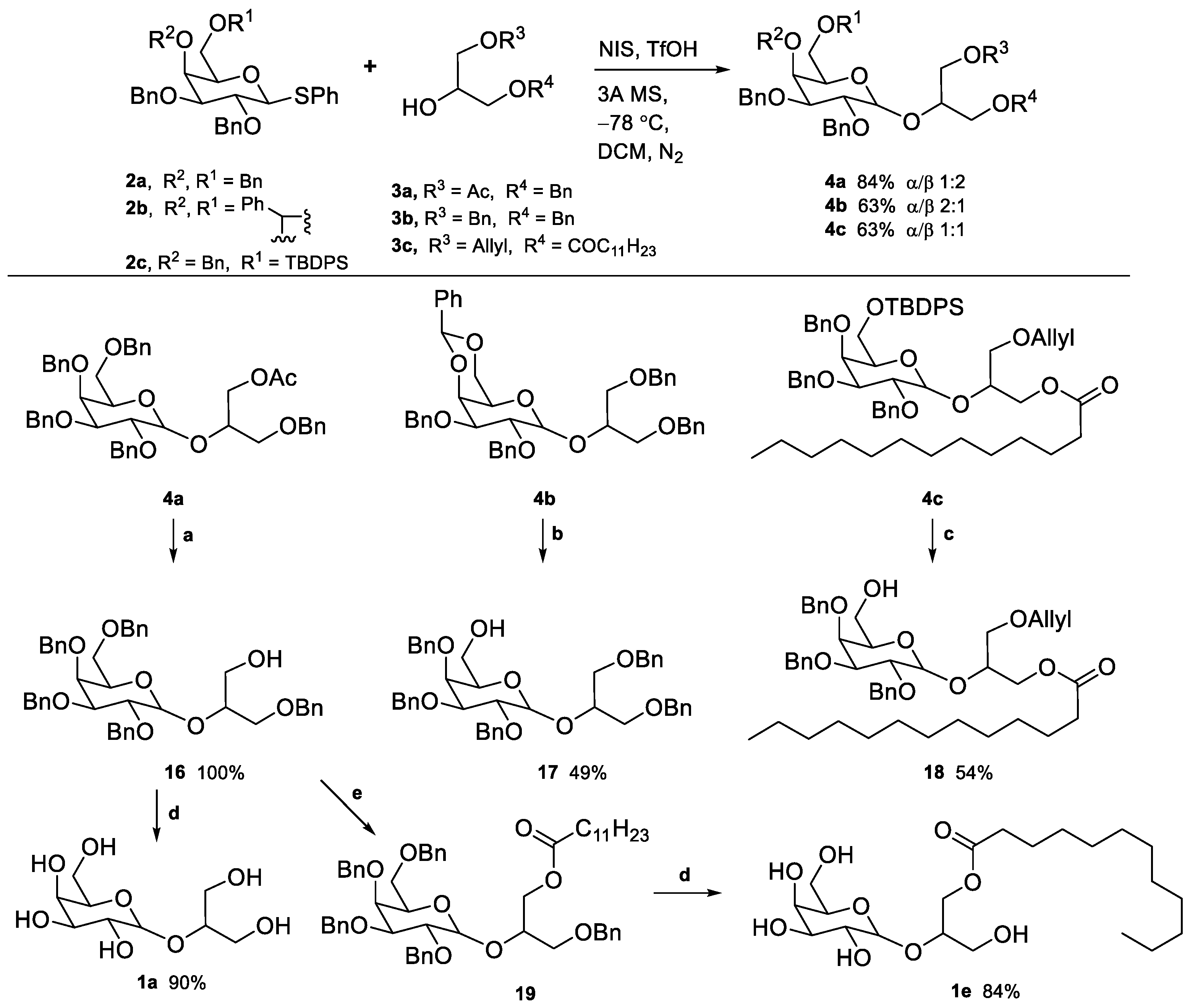
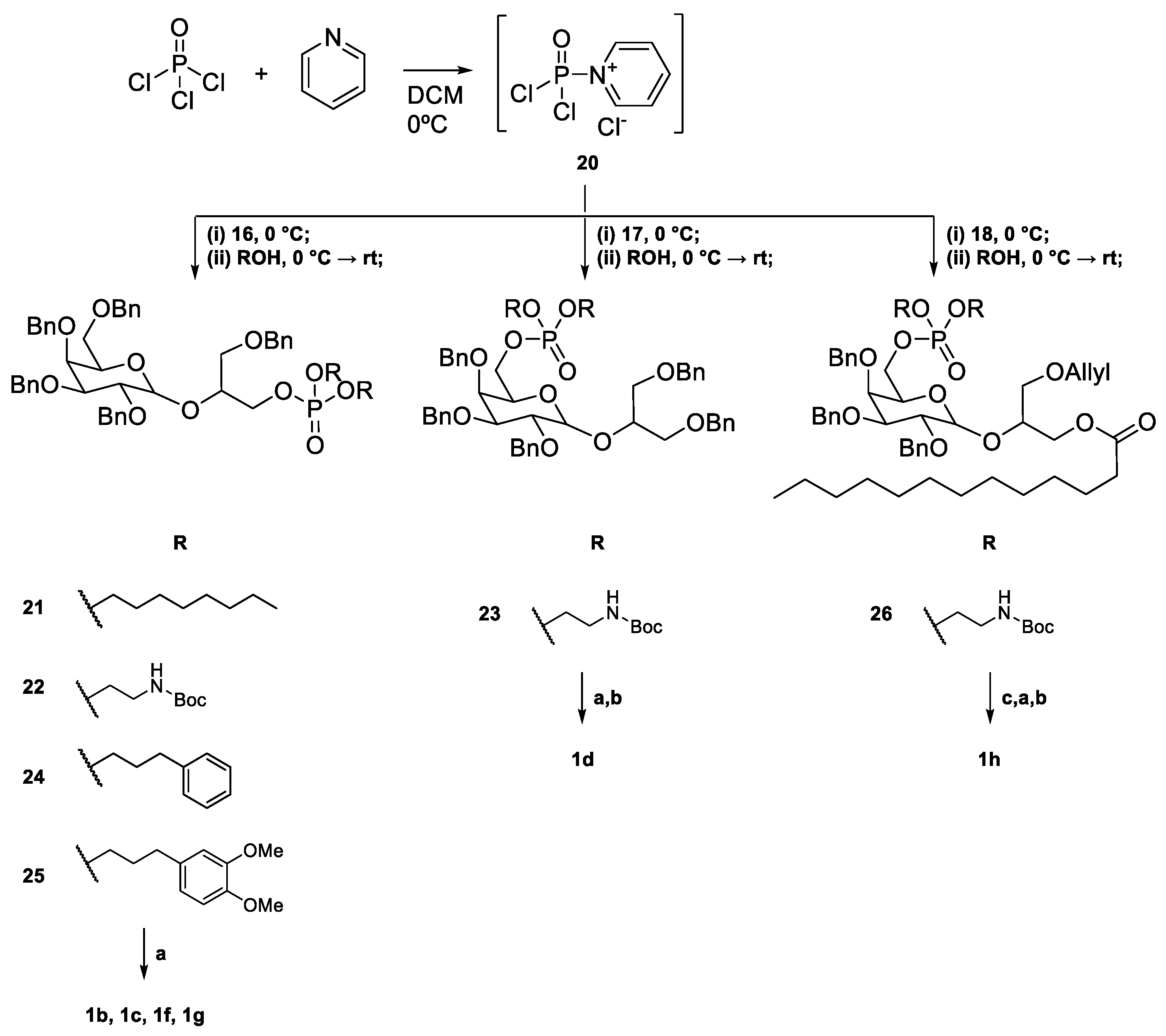
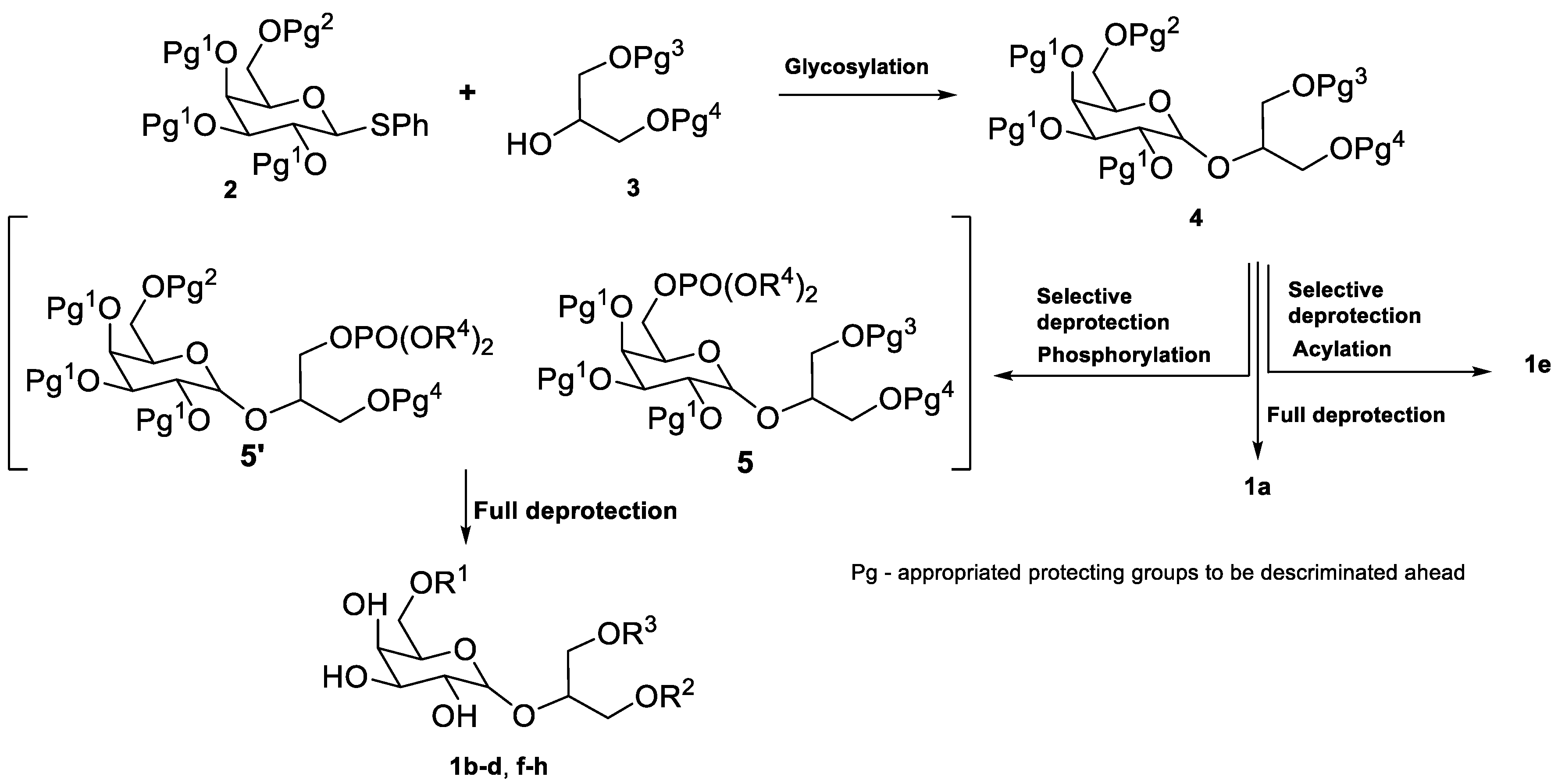
2.1. Synthesis of Floridoside Phosphotriesters 1a–h
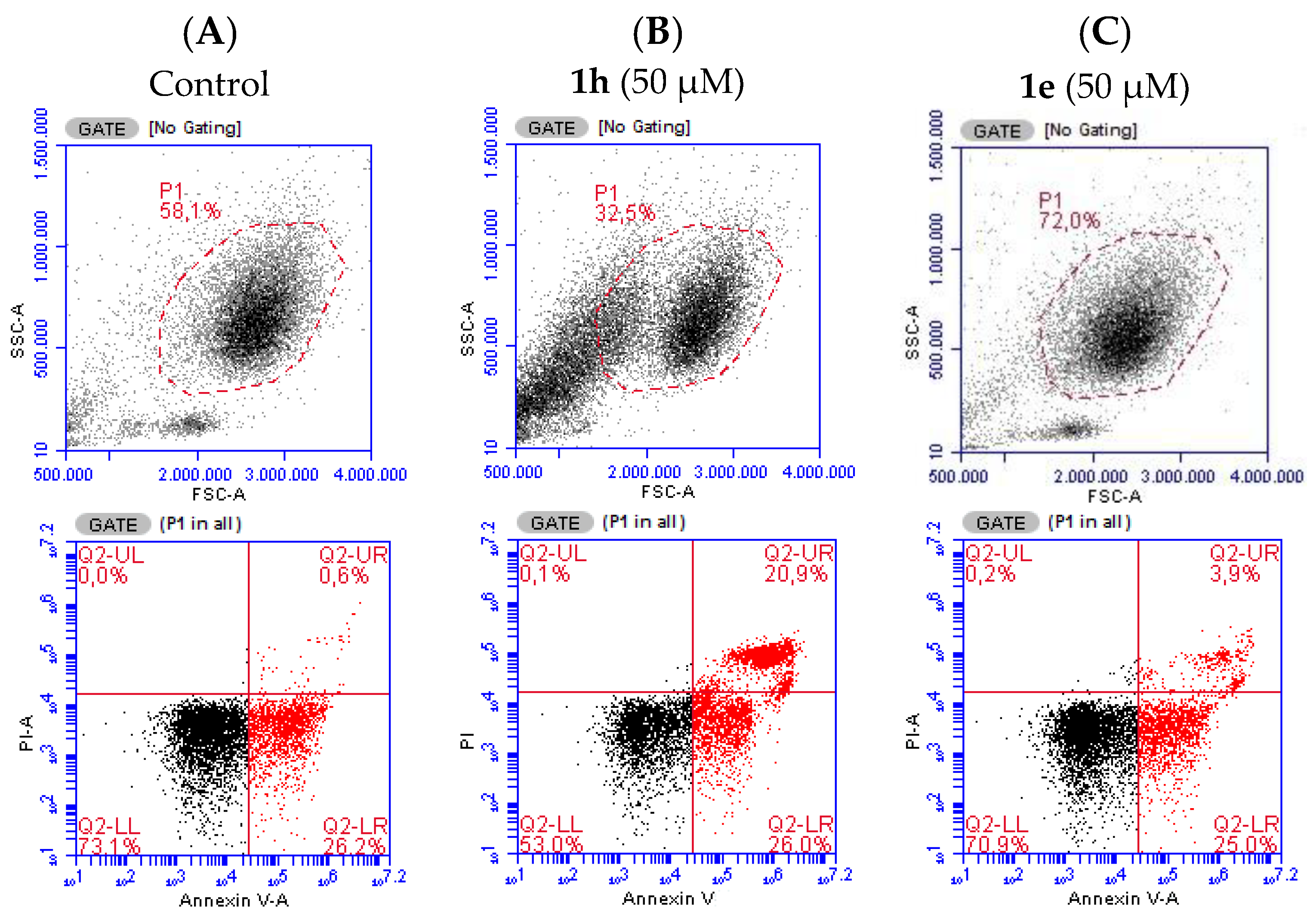
2.2. Evaluation of Cell Death in Neutrophils
2.3. Modulation of Neutrophils’ Oxidative Burst
3. Materials and Methods
3.1. General Information for Synthetic Procedures
3.2. Synthetic Procedures
- 1-O-Acetyl-3-O-benzyl-2-O-(2′,3′,4′,6′-tetra-O-benzyl-β-D-galactopyranosyl)-sn-glycerol (4a) Eluted with PE/EtOAc 7:3. Obtained as a yellow-orange viscous oil in 84% yield (α/β 1:2). α-anomer (2 diastereomers): 1H NMR (400 MHz, CDCl3) δ 7.42–7.21 (m, 25H), 5.16–5.12 (m, 1H, H-1), 4.97–4.91 (m, 1H, OCH2Ar), 4.86–4.64 (m, 4H, OCH2Ar), 4.59–4.54 (m, 1H, OCH2Ar), 4.53–4.51 (m, 1H, OCH2Ar), 4.47–4.45 (m, 1H, OCH2Ar), 4.44–4.35 (m, 2H, OCH2Ar), 4.34–4.27 (m, 1H, H-7a), 4.21–4.00 (m, 4H, H-2, H-4, H-5, H-7b), 3.98–3.93 (m, 2H, H-3, H-8), 3.66–3.46 (m, 4H, H-6, H-9), 1.99–1.92 (m, 3H, OAc). 13C NMR (101 MHz, CDCl3) δ 171.04, 170.98, 139.09, 138.91, 138.84, 138.78, 138.28, 138.11, 128.62, 128.59, 128.56, 128.53, 128.51, 128.48, 128.44, 128.41, 128.08, 128.06, 127.96, 127.94, 127.91, 127.87, 127.83, 127.79, 127.75, 127.66, 127.64, 127.60, 97.37 (C-1), 97.27 (C-1), 79.13, 77.56, 77.44, 77.24, 76.92, 76.61, 76.38, 75.27, 75.10, 75.01, 74.97, 74.19, 74.03, 73.74, 73.65, 73.62, 73.39, 73.32, 73.26, 73.19, 70.02, 69.83, 69.71, 69.66, 69.24, 68.90, 64.84, 64.32, 60.61, 21.02, 20.92. β-anomer (2 diastereomers): 1H NMR (400 MHz, CDCl3) δ 7.38–7.22 (m, 25H, ArH), 4.96–4.90 (m, 2H, OCH2Ar), 4.78–4.68 (m, 3H, OCH2Ar), 4.61 (d, J = 11.6 Hz, 1H, OCH2Ar), 4.51 (s, 2H, OCH2Ar), 4.49 (d, J = 7.8 Hz, 1H, H-1), 4.44–4.39 (m, 2H, OCH2Ar), 4.35 (dd, J = 11.7, 3.4 Hz, 1H, H-7a), 4.22 (dd, J = 11.8, 6.3 Hz, 1H, H-7b), 4.10–4.04 (m, 1H, H-8), 3.90–3.87 (m, 1H, H-4), 3.80 (dd, J = 9.6, 7.8 Hz, 1H, H-2), 3.74 (dd, J = 9.9, 4.4 Hz, 1H, H-9a), 3.60 (dd, J = 7.6, 2.4 Hz, 1H, H-9b), 3.58–3.48 (m, 4H, H-6, H-5, H-3, 1.88 (s, 3H, OAc). 13C NMR (101 MHz, CDCl3) δ 138.85, 138.62, 138.49, 138.14, 137.91, 128.45, 128.41, 128.37, 128.34, 128.27, 128.24, 128.21, 128.07, 127.97, 127.91, 127.85, 127.82, 127.70, 127.67, 127.60, 127.56, 127.46, 103.99, 82.19, 79.38, 77.37, 77.25, 77.05, 76.73, 76.46, 75.00, 74.57, 73.54, 73.45, 73.33, 73.14, 73.09, 69.68, 68.88, 64.39, 50.84. HRMS-ESI: Calculated for [C46H50O9+Na+] 769.3347; found 769.3340.
- 1,3-di-O-Benzyl-2-O-(2′,3′-di-O-benzyl-4′,6′-di-O-benzylidene-α-D-galactopyranosyl)-sn-glycerol (4b) Eluted with PE/EtOAc 7:3. Both anomers were obtained as yellowish oils in 63% combined yield (α/β 2:1) α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.52–7.48 (m, 2H, ArH), 7.43–7.39 (m, 2H, ArH), 7.39–7.22 (m, 21H, ArH), 5.40 (s, 1H, H-7), 5.25 (d, J = 3.4 Hz, 1H, H-1), 4.84–4.63 (m, 4H, CH2Ar), 4.55–4.41 (m, 4H, CH2Ar), 4.15–4.09 (m, 2H, H-3, H-5), 4.06–3.98 (m, 3H, H-2, H-4, H-8/10), 3.92–3.89 (m, 1H, H-9), 3.68 (dd, J = 12.5, 1.9 Hz, 1H, H-8/10), 3.65–3.62 (m, 2H, H-6), 3.59 (m, 2H, H-8/10). 13C NMR (101 MHz, CDCl3) δ 139.14, 138.88, 138.31, 138.23, 138.09, 128.91, 128.53, 128.51, 128.42, 128.40, 128.36, 128.25, 128.20, 127.93, 127.83, 127.76, 127.70, 127.65, 127.63, 127.60, 126.48, 101.13 (C-7), 97.27 (C-1), 76.21 (C-4), 75.59 (C-2), 74.90 (C-3), 74.79 (C-5), 73.54, 73.29, 73.26, 72.22, 71.01 (C-6), 70.08 (C-9), 69.52 (C-8), 62.60 (C-10). HRMS-ESI: Calculated for [C44H46O8+Na+]: 727.3085; found: 727.3094.
- 3-(Allyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-(((tert-butyldiphenylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propyl dodecanoate (4c) Eluted with a mixture of PE/EtOAc 9.5:0.5. The product was obtained as a colourless oil in 63% yield (α/β 1:1). Since the two anomers were unseparated, the 1H NMR nuclide count is doubled. 1H NMR (400 MHz, CDCl3) δ 7.73–7.56 (m, 8H, ArH), 7.50–7.18 (m, 42H, ArH), 5.93–5.73 (m, 2H, H-15), 5.30–5.06 (m, 5H, H-1α, H-16), 5.02–4.56 (m, 12H, CH2Ar), 4.53–4.43 (m, 1H, H-1β), 4.38–4.32 (m, 1H, H-7), 4.28–4.12 (m, 3H, H-7), 4.10–3.87 (m, 10H, H-2α, H-4, H-5α, H-8, H-14), 3.84–3.63 (m, 6H, H-2β, H-6, H-91H), 3.61–3.47 (m, 5H, H-3, H-93H), 3.43–3.37 (m, 1H, H-5β), 2.28–2.14 (m, 4H, H-10), 1.75–1.51 (m, 4H, H-11), 1.41–1.19 (m, 32H, H-12), 1.13–1.01 (m, 18H, C(CH3)3), 0.95–0.85 (m, 6H, H-13). 13C NMR (101 MHz, CDCl3) δ 173.75, 173.58, 173.49, 173.43, 139.01, 138.95, 138.82, 138.78, 138.74, 138.71, 138.69, 138.64, 135.64, 135.60, 135.57, 135.55, 135.33, 134.82, 134.58, 134.53, 133.37, 133.32, 133.27, 129.82, 129.78, 129.76, 129.74, 128.37, 128.35, 128.32, 128.28, 128.21, 128.16, 128.14, 128.11, 128.08, 128.01, 127.96, 127.94, 127.86, 127.78, 127.74, 127.68, 127.60, 127.57, 127.54, 127.47, 127.44, 127.40, 127.35, 117.04, 116.99, 116.91, 109.22, 103.98, 103.43, 96.87, 96.77, 82.19, 82.14, 79.50, 79.46, 78.91, 77.37, 77.25, 77.05, 76.73, 76.47, 76.38, 76.32, 76.10, 75.72, 75.23, 74.99, 74.93, 74.88, 74.83, 74.66, 73.89, 73.85, 73.53, 73.27, 73.24, 73.20, 73.13, 72.92, 72.81, 72.32, 72.24, 72.21, 71.22, 71.09, 69.75, 69.63, 69.54, 66.85, 64.59, 64.05, 63.91, 62.59, 62.41, 62.32, 34.19, 34.12, 31.93, 29.73, 29.64, 29.52, 29.49, 29.36, 29.29, 29.17. HRMS-ESI: Calculated for [C61H80O9Si+Na+] 1007.5459; found 1007.5446.
- 3-O-benzyl-2-O-(2′,3′,4′,6′-tetra-O-benzyl-β-D-galactopyranosyl)-sn-glycerol (16) The starting material 4a was dissolved in MeOH, and 0.5 eq of NaOMe was added. The reaction proceeded at rt. After full consumption of the starting material, a few drops of HCl 1 M were added to neutralize the reaction. The solvent was evaporated, and the crude dried in a vacuum. The mixture was purified by “flash” chromatography and eluted with petroleum ether/EtOAc 6:4. α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.41–7.24 (m, 25H, ArH), 5.01 (d, J = 3.7 Hz, 1H, H-1), 4.93 (d, J = 11.4 Hz, 1H, CH2Ar), 4.87 (d, J = 11.5 Hz, 1H, CH2Ar), 4.79–4.75 (m, 2H, CH2Ar), 4.70 (d, J = 11.5 Hz, 1H, CH2Ar), 4.57 (d, J = 11.5 Hz, 1H, CH2Ar), 4.50–4.42 (m, 2H, CH2Ar), 4.42–4.32 (m, 2H, CH2Ar), 4.15–4.10 (m, 1H, H-5), 4.10–4.04 (m, 1H, H-2), 4.01–3.95 (m, 2H, H-3, H-4), 3.89–3.81 (m, 1H, H-8), 3.75–3.66 (m, 1H, H-6a), 3.65–3.42 (m, 5H, H-6b, H-7, H-9). NMR data are consistent with the literature [80]. β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.45–7.20 (m, 25H, ArH), 4.94 (d, J = 11.4 Hz, 1H, CH2Ar), 4.90–4.81 (m, 1H, CH2Ar), 4.79–4.67 (m, 3H, CH2Ar), 4.64–4.37 (m, 6H, CH2Ar, H-1), 3.97–3.81 (m, 4H), 3.81–3.42 (m, 7H).
- ((2R,3S,4S,5R)-3,4,5-Tris(benzyloxy)-6-((1,3-bis(benzyloxy)propan-2-yl)oxy)tetrahydro-2H-pyran-2-yl)methanol (17) In a dried round-bottom flask under N2 atmosphere, 4b in THF was added, followed by a catalytic amount (0.15 eq) of Cu(OTf)2. Next, 3 eq of BH3.SMe2 (2M) or BH3·THF (1M) were quickly added. After completion, water was added dropwise to quench. After the gas evolution stopped, EtOAc was added, and the organic phase was washed with sat. NaHCO3 and brine and dried over Na2SO4. The mixture was purified by “flash” chromatography and eluted with a mixture of PE/EtOAc 75:25. The product was obtained in 49% yield as a colourless oil (α/β 1:1). α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.43–7.22 (m, 25H, ArH), 5.24 (d, J = 3.8 Hz, 1H, H-1), 4.95 (d, J = 11.6 Hz, 1H, CH2Ar), 4.89 (d, J = 11.6 Hz, 1H, CH2Ar), 4.74 (d, J = 11.6 Hz, 1H, CH2Ar), 4.72–4.69 (m, 2H, CH2Ar), 4.62 (d, J = 11.6 Hz, 1H, CH2Ar), 4.54–4.44 (m, 4H, CH2Ar), 4.17–4.10 (m, 1H, H-5), 4.08–3.99 (m, 2H, H-2, H-8), 3.93 (dd, J = 10.1, 2.9 Hz, 1H, H-3), 3.86–3.83 (m, 1H, H-4), 3.63–3.55 (m, 5H, H-6, H-7, H-9a), 3.43–3.31 (m, 1H, H-9b). 13C NMR (101 MHz, CDCl3) δ 138.91, 138.55, 138.30, 138.08, 138.04, 128.50, 128.45, 128.42, 128.40, 128.27, 127.87, 127.80, 127.65, 127.55, 127.49, 96.69 (C-1), 79.07 (C-3), 77.25 (C-2), 76.33 (C-5), 75.38 (C-4), 74.64, 74.46, 73.45, 73.42, 73.28, 72.77, 70.73 (C-6), 70.37, 70.30, 62.64, 62.48. β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.39–7.23 (m, 25H, ArH), 5.03–4.91 (m, 2H, CH2Ar), 4.81 (d, J = 11.8 Hz, 1H, CH2Ar), 4.77–4.71 (m, 2H, CH2Ar), 4.66 (d, J = 11.9 Hz, 1H, CH2Ar), 4.58 (d, J = 7.7 Hz, 1H, H-1), 4.54 (d, J = 2.6 Hz, 2H, CH2Ar), 4.51–4.46 (m, 2H, CH2Ar), 4.07 (p, J = 5.2 Hz, 1H, H-8), 3.84 (dd, J = 9.7, 7.6 Hz, 1H, H-3), 3.76–3.68 (m, 4H, H-4, H-5, H-7a, H-9a), 3.67–3.60 (m, 2H, H-7b, H-9b), 3.51 (dd, J = 9.7, 3.0 Hz, 1H, H-2), 3.47–3.38 (m, 1H, H-6a), 3.35–3.30 (m, 1H, H-6b). 13C NMR (101 MHz, CDCl3) δ 138.92, 138.49, 138.30, 138.24, 128.66, 128.45, 128.44, 128.33, 128.21, 128.07, 127.96, 127.67, 127.63, 127.57, 127.53, 127.41, 103.72 (C-1), 82.32, 79.68, 77.85, 77.23, 75.03, 74.66, 74.13, 73.50, 73.37, 73.31, 73.01, 70.45. HRMS-ESI: Calculated for [C44H48O8+Na+]: 727.3241; found: 727.3232. NMR data are consistent with the literature [81].
- 3-(Allyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)propyl dodecanoate (18) To starting material 4c (500 mg, 0.51 mmol) were added 3 equivalents of tetrabutylammonium fluoride (1 M in THF). After 2h and completion of the reaction, the volatiles were evaporated, and the crude mixture was purified by “flash” chromatography and eluted with PE/EtOAc 7:3. The product was obtained as colourless oil in 54% yield (α/β 1:1). α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.43–7.24 (m, 15H, ArH), 5.85 (ddt, J = 17.3, 10.3, 5.6 Hz, 1H, H-15), 5.24 (dq, J = 17.3, 1.7 Hz, 1H, H-16a), 5.19–5.13 (m, 2H, H-1, H-16b), 4.97 (d, J = 11.6 Hz, 1H, CH2Ar), 4.88 (d, J = 11.7 Hz, 1H, CH2Ar), 4.80–4.75 (m, 2H, CH2Ar), 4.72 (d, J = 11.8 Hz, 1H, CH2Ar), 4.64 (d, J = 11.7 Hz, 1H, CH2Ar), 4.25 (dd, J = 11.7, 4.8 Hz, 1H, H-7a), 4.15 (dd, J = 11.7, 6.0 Hz, 1H, H-7b), 4.10–3.90 (m, 7H, H-2, H-3, H-4, H-5, H-8, H-14), 3.70 (dd, J = 11.3, 6.5 Hz, 1H, H-6a), 3.60–3.52 (m, 2H, H-9), 3.52–3.46 (m, 1H, H-6b), 2.24 (dd, J = 8.1, 7.1 Hz, 2H, H-10), 1.60–1.54 (m, 2H, H-11), 1.25 (d, J = 5.3 Hz, 16H, H-12), 0.88 (t, J = 6.8 Hz, 3H, H-13). 13C NMR (101 MHz, CDCl3) δ 173.64, 138.89, 138.58, 138.34, 134.53 (C-15), 128.69, 128.61, 128.55, 128.51, 128.10, 127.93, 127.81, 127.71, 127.63, 117.50 (C-16), 96.93 (C-1), 79.19, 77.36, 76.92, 76.54, 75.23, 74.55, 74.06, 73.61, 73.26, 72.38 (C-14), 70.73, 70.32, 63.85, 62.79, 34.29, 32.04, 29.74, 29.60, 29.47, 29.41, 29.39, 29.29, 24.99, 22.82, 14.25. β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.40–7.27 (m, 15H), 5.93–5.77 (m, 1H), 5.29–5.12 (m, 2H), 4.98–4.89 (m, 2H), 4.83–4.69 (m, 3H), 4.65 (dd, J = 11.8, 7.9 Hz, 1H), 4.51 (dd, J = 7.7, 2.6 Hz, 1H), 4.34 (ddd, J = 26.5, 11.6, 4.1 Hz, 1H), 4.22 (dt, J = 12.0, 6.1 Hz, 1H), 4.09–3.95 (m, 3H), 3.85–3.67 (m, 4H), 3.60–3.43 (m, 3H), 3.42–3.34 (m, 1H), 2.34–2.13 (m, 2H), 1.65–1.47 (m, 2H), 1.34–1.18 (m, 16H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 174.06, 173.74, 138.84, 138.77, 138.44, 138.39, 138.24, 138.21, 134.52, 128.69, 128.56, 128.48, 128.44, 128.41, 128.26, 128.09, 128.02, 127.92, 127.71, 127.65, 127.52, 127.50, 117.25, 117.15, 104.19, 103.90, 82.30, 82.26, 79.54, 79.47, 77.36, 77.24, 77.04, 76.78, 76.72, 75.49, 75.07, 75.03, 74.78, 74.21, 74.18, 73.52, 73.47, 73.31, 72.90, 72.40, 72.37, 70.02, 69.98, 64.10, 63.79, 62.48, 61.99, 34.32, 34.10, 31.92, 29.63, 29.48, 29.35, 29.28, 29.15, 29.13, 24.88, 24.84, 22.70, 14.14. HRMS-ESI: Calculated for [C45H62O9+Na+] 769.4292; found 769.4270.
- 3-(benzyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propyl dodecanoate (19) The starting galactoglycerol 16 (62 mg, 0.088 mmol) was dissolved in dry DCM (0.25 M) under an N2 atmosphere. Dicyclohexyl carbodiimide (20 mg, 0.097 mmol), DMAP (2.2 mg, 0.018 mmol), and dodecanoic acid (19.4 mg, 0.097 mmol) were sequentially added. The reaction proceeded at room temperature. After completion of the reaction, DCM was added, and the precipitate was filtered. The filtrate was washed with water (3 × 10 mL) and brine (10 mL). The organic phase was dried over Na2SO4. The mixture was purified by “flash” chromatography and eluted with PE/EtOAc 7:3. The product was obtained as a white waxy solid in 85% yield (α/β 1:2). α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.41–7.21 (m, 25H, ArH), 5.18–5.13 (m, 1H, H-1), 4.97–4.91 (m, 1H, CH2Ar), 4.87–4.81 (m, 1H, CH2Ar), 4.77–4.66 (m, 3H, CH2Ar), 4.65–4.44 (m, 3H, CH2Ar), 4.44–4.20 (m, 3H, CH2Ar, H-7a), 4.18–4.00 (m, 3H, H2, H-5, H-7b), 3.98–3.92 (m, 1H, H-3), 3.90–3.86 (m, 1H, H-8), 3.65–3.45 (m, 5H, H-4, H-6, H-9), 2.39–2.10 (m, 2H, H-10), 1.65–1.44 (m, 2H, H-11), 1.35–1-17 (m, 16H, H-12), 0.88 (t, J = 6.8 Hz, 3H, H-13). β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.41–7.21 (m, 25H, ArH), 4.97–4.91 (m, 1H, CH2Ar), 4.77–4.66 (m, 4H, CH2Ar), 4.65–4.44 (m, 4H, CH2Ar, H-1), 4.44–4.20 (m, 4H, CH2Ar, H-7a), 4.18–4.00 (m, 1H, H-7b, H-8), 3.98–3.92 (m, 1H, H-4), 3.90–3.86 (m, 1H, H-3), 3.84–3.71 8m, 2H, H-2, H-9a) 3.65–3.45 (m, 4H, H-5, H-6, H-9b), 2.39–2.10 (m, 2H, H-10), 1.65–1.44 (m, 2H, H-11), 1.35–1-17 (m, 16H, H-12), 0.88 (t, J = 6.8 Hz, 3H, H-13). αβ-anomers: 13C NMR (101 MHz, CDCl3) δ 173.86, 173.75, 139.03, 138.85, 138.72, 138.30, 138.22, 138.13, 138.05, 128.57, 128.54, 128.48, 128.45, 128.43, 128.39, 128.33, 128.20, 128.06, 128.00, 127.97, 127.93, 127.91, 127.88, 127.86, 127.84, 127.80, 127.76, 127.74, 127.70, 127.67, 127.65, 127.56, 127.51, 104.12, 97.21, 97.15, 82.33, 79.51, 79.07, 76.32, 76.16, 75.20, 75.09, 74.95, 74.68, 74.09, 73.88, 73.66, 73.63, 73.58, 73.53, 73.28, 73.05, 70.02, 69.85, 69.55, 69.13, 68.99, 68.86, 64.47, 60.54, 34.28, 34.21, 34.16, 32.05, 29.76, 29.64, 29.61, 29.50, 29.48, 29.44, 29.41, 29.30, 29.26, 25.05, 24.95, 22.83, 14.34, 14.26. HRMS-ESI: Calculated for [C56H70O9+Na+]: 909.4912; found 909.4899.
- 3-O-Benzyl-2-O-(2′,3′,4′,6′-tetra-O-benzyl-D-galactopyranosyl)propyl dioctyl phosphate (21): Starting with 16, the alcohol used was n-octanol. The crude mixture was eluted with PE/EtOAc 8:2. The product was obtained as colourless oil in 52% yield (α/β 1:1). α-anomer: 1H NMR (400 MHz, CDCl3) δ 7.39–7.19 (m, 25H, ArH), 5.17 (d, J = 3.8 Hz, 1H, H-1), 4.92 (d, J = 11.4 Hz, 1H, CH2Ar), 4.82 (d, J = 11.7 Hz, 1H, CH2Ar), 4.73–4.63 (m, 3H, OCH2Ar), 4.57–4.48 (m, 4H, CH2Ar), 4.41 (d, J = 11.7 Hz, 1H, CH2Ar), 4.23–4.11 (m, 3H, H-6, H-8), 4.11–3.95 (m, 8H, H-2, H-3, H-4, H-5, H-10), 3.66–3.60 (m, 2H, H-9), 3.55 (dd, J = 6.7, 1.6 Hz, 2H, H-7), 1.63–1.55 (m, 4H, H-11), 1.30–1.19 (m, 20H, H-12), 0.87 (m, 6H, H-13). 13C NMR (101 MHz, CDCl3) δ 138.73–137.95, 128.39–127.36, 97.08 (C-1), 78.91 (C-2), 76.24 (C-3), 75.03 (C-4), 74.81, 74.47 (C-5), 73.47, 73.40 (CH2, CH2Ar), 73.11, 72.86, 69.42 (C-8), 69.22 (C-9), 68.74 (C-7), 67.91 (C-10), 67.10 (C-6), 31.79 (C-10), 30.35 (C-11), 30.28 (C-11), 29.20–22.64 (C-12), 14.09 (C-13). LCMS: 1009.5 [M+H]+; 1026.5 [M+NH4]+.
- Di-tert-butyl (2-(((3-(benzyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propoxy)(2-(methylamino)ethoxy)phosphoryl)bis(oxy)bis(ethane-2,1-diyl)dicarbamate (22) Starting with 16, the alcohol used was N-Boc ethanolamine. The crude mixture was eluted with PE/EtOAc 6:4. The product was confirmed to be present by 1H, albeit heavily contaminated with tris(N-Boc ethanolamine) phosphate, which co-elutes with the intended product. The mixture was used in the next reaction without further purification. LCMS: 1088.5 [M+NH4]+; 1093.4 [M+Na]+.
- Di-tert-butyl (2-(((2R,3S,4S,5R)-3,4,5-tris(benzyloxy)-6-((1,3-bis(benzyloxy)propan-2-yl)oxy)tetrahydro-2H-pyran-2-yl)methoxy)phosphoryl) bis(oxy)bis(ethane-2,1-diyl)dicarbamate (23) Starting with 17, the alcohol used was N-Boc ethanolamine. The mixture was purified by flash chromatography and eluted with PE/EtOAc 6:4. The product was confirmed to be present by 1H, albeit heavily contaminated with N-Boc ethanolamine, which co-elutes with the intended product. The mixture was used in the next reaction without further purification.
- 3-(Benzyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propyl bis(3-phenylpropyl) phosphate (24) Starting with 16, the alcohol used was 3-phenylpropan-1-ol. The crude mixture was eluted with PE/EtOAc 7:3. The product was obtained as a colourless viscous oil (α/β 1:10). β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.40–7.05 (m, 35H, ArH), 5.15 (d, J = 3.7 Hz, 1H, H-1α), 5.00–4.89 (m, 2H, CH2Ar), 4.76–4.65 (m, 3H, CH2Ar), 4.64–4.44 (m, 4H, CH2Ar, H-1β), 4.43–4.31 (m, 2H, H-7), 4.23–3.93 (m, 7H, H-8, H-9, H-10), 3.92–3.85 (m, 1H, H-4), 3.84–3.72 (m, 2H, H-2, H-6a), 3.71–3.42 (m, 5H, H-3, H-5, H-6b, H-9), 2.77–2.54 (m, 4H, H-12), 2.08–1.81 (m, 4H, H-11). 13C NMR (101 MHz, CDCl3) δ 141.04, 140.99, 140.97, 140.91, 140.71, 138.87, 138.82, 138.71, 138.65, 138.59, 138.53, 138.49, 138.16, 138.08, 137.99, 137.93, 137.90, 137.88, 128.54, 128.49, 128.46, 128.38, 128.35, 128.29, 128.22, 128.17, 128.07, 127.91, 127.88, 127.85, 127.82, 127.78, 127.70, 127.65, 127.63, 127.60, 127.56, 127.45, 127.40, 126.17, 126.12, 126.08, 126.04, 125.89, 103.94, 103.54, 97.55 (C-1α), 82.17, 82.13, 79.46, 79.42, 78.87, 76.51, 76.43, 76.37, 75.11, 75.06, 74.80, 74.68, 74.60, 73.57, 73.52, 73.48, 73.43, 73.38, 73.33, 73.23, 73.16, 73.10, 72.89, 69.58, 69.37, 69.05, 68.81, 68.59, 68.46, 68.40, 67.13, 67.08, 67.03, 66.90, 66.84, 34.26, 32.11, 31.88, 31.81, 31.73, 31.68, 31.64, 31.62, 31.51, 29.73. HRMS-ESI: Calculated for [C62H69O11P+Na+] 1043.4470; found 1043.4459.
- 3-(Benzyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propyl bis(3-(3,4-dimethoxyphenyl)propyl) phosphate (25) The alcohol used was 3-(3,4-dimethoxyphenyl)propan-1-ol. The crude mixture was eluted with PE/EtOAc 7:3. The product was obtained as colourless viscous oil (α/β 1:1), contaminated with the tri-substituted phosphate of the alcohol used. 1H NMR (400 MHz, CDCl3) δ 7.42–7.17 (m, 50H, ArH), 6.84–6.62 (m, 12H, H-13, H-14, H-15), 5.20–5.12 (m, 1H, H-1α), 4.97–4.87 (m, 3H, CH2Ar), 4.82–4.64 (m, 7H), 4.61–4.29 (m, 11H, H-1β), 4.25–3.99 (m, 24H, H-2α, H-10), 3.96–3.72 (m, 40H, H-2β, -OMe), 3.71–3.42 (m, 12H), 2.79–2.54 (m, 8H, H-12), 2.10–1.85 (m, 8H, H-11). The nuclide count is doubled to account for both anomers. 13C NMR (101 MHz, CDCl3) δ 170.95, 148.73, 148.69, 147.20, 147.15, 140.70, 138.63, 138.45, 138.38, 138.25, 137.94, 137.81, 137.68, 133.30, 133.28, 133.06, 128.29, 128.23, 128.21, 128.15, 128.13, 128.10, 128.05, 128.03, 128.00, 127.96, 127.83, 127.66, 127.62, 127.60, 127.54, 127.48, 127.45, 127.41, 127.38, 127.35, 127.32, 127.24, 127.16, 127.13, 125.91, 120.07, 120.04, 111.58, 111.54, 111.51, 111.10, 111.08, 97.38, 96.94, 81.98, 79.20, 78.67, 77.55, 76.10, 74.86, 74.77, 74.58, 74.37, 73.28, 73.21, 73.01, 72.92, 72.83, 72.65, 69.38, 68.82, 68.22, 66.86, 66.75, 66.69, 60.19, 55.72, 55.70, 55.64, 55.62, 31.95, 31.88, 31.75, 31.69, 31.47, 31.05, 31.02, 30.90, 20.85, 14.00. HRMS-ESI: Calculated for [C66H77O15P+Na+] 1163.4892; found 1163.4875.
- 3-(Allyloxy)-2-(((3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-(((bis(2-((tert-butoxycarbonyl)amino)ethoxy)phosphoryl)oxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)propyl dodecanoate (26) Starting with 18, the alcohol used was N-Boc ethanolamine. The product was eluted with PE/EtOAc 8:2. The product was obtained contaminated with N-Boc ethanolamine, and the mixture appeared as colourless oil. The product was used without further purification (α/β 1:1). β-anomer: 1H NMR (400 MHz, CDCl3) δ 7.40–7.25 (m, 15H, ArH), 5.93–5.76 (m, 1H, H-15), 5.28–5.10 (m, 3H, H-16, NH), 5.02–4.95 (m, 1H, CH2Ar), 4.93 (d, J = 10.7 Hz, 1H, CH2Ar), 4.80 (d, J = 11.8 Hz, 1H, CH2Ar), 4.77–4.69 (m, 2H, CH2Ar), 4.61 (d, J = 11.5 Hz, 1H, CH2Ar), 4.56–4.49 (m, 1H, H-1), 4.35–4.28 (m, 1H, H-9a), 4.21 (dd, J = 12.0, 6.0 Hz, 1H, H-9b), 4.18–3.92 (m, 9H, H-6, H-8, H-14, H-17), 3.84–3.77 (m, 2H, H-2, H-4), 3.72–3.67 (m, 1H, H-7a), 3.62–3.49 (m, 3H, H-3, H-5, H-7b), 3.42–3.27 (m, 4H, H-18), 2.30 (t, J = 7.6 Hz, 1H, H-10a), 2.19–2.12 (m, 1H, H-10b), 1.63–1.48 (m, 2H, H-11), 1.44 (m, 18H, tert-butyl), 1.35–1.18 (m, 16H, H-12), 0.88 (t, J = 6.7 Hz, 3H, H-13). 13C NMR (101 MHz, CDCl3) δ 173.74, 173.65, 155.84, 138.80, 138.73, 138.36, 138.32, 138.26, 134.55, 134.51, 128.43, 128.34, 128.24, 128.07, 127.89, 127.77, 127.70, 127.62, 127.51, 127.49, 117.17, 117.10, 103.91, 103.65, 81.88, 79.72, 79.19, 79.14, 77.24, 76.56, 76.34, 75.02, 74.98, 74.48, 73.53, 73.49, 73.24, 72.97, 72.35, 69.66, 69.61, 67.24, 67.18, 66.36, 66.31, 64.02, 63.82, 59.54, 40.90, 38.16, 34.23, 34.08, 31.94, 31.92, 31.26, 29.71, 29.67, 29.63, 29.51, 29.49, 29.38, 29.35, 29.31, 29.30, 29.19, 29.14, 28.40, 24.92, 24.83, 22.70. HRMS-ESI: Calculated for [C59H89N2O16P+Na+]: 1135.5842; found 1135.5824.
- 2-O-D-(galactopyranosyl)glycerol (1a) Galactoglycerol 16 was subjected to hydrogenation conditions. The product 1a was obtained as a yellowish, highly hygroscopic oil (α/β 3:1). 1H NMR (400 MHz, MeOD) δ 5.03 (d, J = 3.2 Hz, 1H, H-1α), 4.38 (d, J = 7.6 Hz, 1H, H-1β), 4.04–3.99 (m, 1H), 3.90–3.88 (m, 1H), 3.85–3.64 (m, 16H), 3.61–3.47 (m, 4H). HRMS-ESI: Calculated for [C9H18O8+Na+] 277.0894; found 277.0891.
- 3-Hydroxy-2-galactopyranosylpropyl dioctyl phosphate (1b) Starting material 21 was subjected to hydrogenation conditions. Product 1b was obtained as a colourless oil (α/β 1:1). 1H NMR (400 MHz, MeOD) δ 5.07–5.00 (m, 1H, H-1α), 4.38 (d, J = 7.5 Hz, 1H, H-1β), 4.25–4.14 (m, 4H, H-6), 4.13–4.04 (m, 9H, H-4, H-10), 4.04–3.94 (m, 3H, H-4, H-8), 3.93–3.86 (m, 2H, H-5), 3.84–3.63 (m, 9H, H-2α, H-7, H-9), 3.57–3.50 (m, 1H, H-2β), 3.50–3.43 (m, 2H, H-3α), 1.75–1.65 (m, 8H, H-11), 1.38–1.20 (m, 40H, H-12), 0.93–0.86 (m, 12H, H-13). 13C NMR (101 MHz, MeOD) δ 104.99, 100.60, 76.88, 74.81, 72.48, 71.49, 71.08, 70.23, 69.57, 62.82, 62.53, 40.43, 32.97, 31.37, 31.30, 30.75, 30.35, 30.23, 26.61, 23.71, 14.43. HRMS-ESI: Calculated for [C25H51O11P+Na+] 581.3061; found 581.3055.
- 3-Hydroxy-2-galactopyranosylpropyl (bis(2-aminoethyl)) phosphate (1c) Starting material 22 was subjected to hydrogenation conditions and then Boc removal conditions. The product 1c was obtained as an orange viscous oil (α/β 1:1). 1H NMR (400 MHz, D2O) δ 5.21–5.17 (m, 1H, H-1), 4.49–4.42 (m, 5H, H-6a, H-10), 4.38–4.31 (m, 1H, H-6b), 4.10–4.04 (m, 2H, H-4, H-8), 4.03–3.99 (m, 1H, H-5), 3.93–3.84 (m, 2H, H-2, H-3), 3.84–3.73 (m, 4H, H-7, H-9), 3.43–3.38 (m, 4H, H-11). 13C NMR (101 MHz, D2O) δ 98.83, 76.54, 76.47, 71.94, 69.95, 69.84, 68.88, 68.00, 67.94, 65.52, 65.47, 61.81, 61.36, 40.12, 40.04. LCMS: 421 [M+H]+, 443 [M+Na]+.
- Bis(2-Aminoethyl) (galactopyranosyl)glycerol)-6-phosphate (1d) Starting material 23 was subjected to hydrogenation conditions and then Boc removal conditions. Product 1d was obtained as an orange viscous oil. Only the α-anomer was recovered. α-anomer: 1H NMR (400 MHz, MeOD) δ 5.08 (d, J = 3.5 Hz, 1H, H-1), 4.25–4.10 (m, 5H, H-5, H-10), 4.07–4.00 (m, 2H, H-6), 3.99–3.95 (m, 1H, H-4), 3.87–3.62 (m, 7H, H-2, H-7, H-8, H-9), 3.28–3.17 (m, 4H, H-11). 13C NMR (101 MHz, MeOD) δ 97.91, 78.83, 69.00, 68.92, 64.20, 61.36, 61.07, 60.63, 59.87, 39.28, 39.22. HRMS: Calculated for [C11H24O11NP–C2H6N]−: 376.1014; found 376.1009. Calculated for [C11H24O11NP + Na+–C2H5N]: 400.0985; found 400.0974.
- 3-Hydroxy-2-(galactopyranosyl)propyl dodecanoate (1e) Starting material 19 was subjected to hydrogenation conditions. The product 1e was obtained as a colourless viscous oil (α/β 1:1). The 1H NMR signals of the diastereomers were assigned according to the anomers α and β. 1H NMR (400 MHz, MeOD) δ 5.09–5.02 (m, 2H, H-1α), 4.43–4.38 (m, 2H, H-1β), 4.34–4.17 (m, 4H, H-7), 4.05–3.89 (m, 4H, H-4, H-8), 3.87–3.62 (m, 10H, H-2α, H-3α, H-6, H-9), 3.60–3.46 (m, 4H, H-2β, H-3β, H-5), 2.43–2.32 (m, 4H, H-10), 1.70–1.57 (m, 4H, H-11), 1.41–1.18 (m, 32H, H-12), 0.96–0.87 (m, 6H, H-13). 13C NMR (101 MHz, MeOD) δ 174.04, 174.01, 103.72, 103.34, 98.96, 98.75, 78.12, 77.74, 76.79, 76.11, 75.43, 75.36, 73.46, 71.34, 71.15, 71.08, 70.03, 69.71, 69.58, 68.88, 68.77, 63.56, 63.34, 63.20, 62.95, 61.93, 61.59, 61.42, 61.08, 60.59, 33.56, 31.67, 29.34, 29.22, 29.07, 29.03, 28.83, 24.62, 24.57, 22.33, 13.04. HRMS-ESI: Calculated for [C21H40O9P+Na+] 459.2564; found 459.2558.
- 3-Hydroxy-2-(galactopyranosyl)propyl bis(3-phenylpropyl) phosphate (1f) Starting material 24 was subjected to hydrogenation conditions. The product 1f was obtained as a colourless viscous oil (α/β 1:10). β-anomer: 1H NMR (400 MHz, MeOD) δ 7.32–7.13 (m, 10H, ArH), 4.42–4.35 (m, 1H, H-1), 4.27–4.18 (m, 2H, H-6), 4.14–4.05 (m, 4H, H-10), 4.01–3.93 (m, 1H, H-5), 3.84–3.80 (m, 1H, H-8), 3.78–3.66 (m, 4H, H-3, H-4, H-7), 3.59–3.43 (m, 3H, H-2, H-9), 2.73 (t, J = 7.6 Hz, 4H, H-12), 2.05–1.96 (m, 4H, H-11). 13C NMR (101 MHz, MeOD) δ 140.93, 129.17, 128.14, 128.12, 125.71, 103.74, 77.88, 71.08, 67.29, 31.68, 31.61, 31.20, 29.36. HRMS-ESI: Calculated for [C27H39O11P+Cl−]: 605.1924; found 605.1925.
- 3-Hydroxy-2-(galactopyranosyl)propyl (bis(3-(3,4-dimethoxyphenyl)propyl) phosphate (1g) Starting material 25 was subjected to hydrogenation conditions. The product was obtained as a colourless viscous oil and a mixture of 4 diastereomers (α/β 1:0.75). 1H NMR (400 MHz, MeOD) δ 6.86–6.70 (m, 12H, ArH), 5.05–5.01 (m, 1H, H-1α), 4.39–4.34 (m, 1H, H-1β), 4.28–4.19 (m, 4H, H-6), 4.13–4.03 (m, 8H, H-10), 4.00–3.93 (m, 2H), 3.92–3.85 (m, 4H), 3.85–3.63 (m, 33H, -OCH3, H-2α, H-7, H-9), 3.58–3.42 (m, 4H, H-2β), 2.66 (d, J = 3.8 Hz, 8H, H-11), 2.06–1.94 (m, 8H, H-12). 13C NMR (101 MHz, MeOD) δ 135.34, 135.32, 129.60, 121.88, 113.69, 113.32, 105.08, 100.69, 76.93, 74.90, 72.94, 72.56, 71.58, 71.16, 70.43, 68.82, 62.92, 62.75, 62.35, 56.63, 56.55, 33.20, 33.13, 32.20. HRMS-ESI: Calculated for [C31H47O15P+Na+]: 713.2545; found 713.2531.
- 2-(6-(bis(2-Aminoethoxy))phosphoryl)galactopyranosyl-3-hydroxypropyl dodecanoate (1h) The starting material 26 (119 mg) was dissolved in 2 mL of DCM/MeOH 1:1. A catalytic amount of PdCl2 (0.2 mol eq) was added, and the reaction proceeded at rt. After completion (followed by TLC), the reaction was quenched with a few drops of Et3N, and the black precipitate that formed was filtered. The orange solution obtained was diluted with DCM and washed with HCl 0.1M (2 × 10 mL), saturated NaHCO3 (10 mL), and brine (10 mL). The organic phase was dried over Na2SO4, and the solvent was evaporated. The resulting crude was subjected to hydrogenation and then Boc removal conditions. Product 1h (23.2 mg, 36% yield) was obtained as an orange viscous oil (α/β 1:2). α-anomer: 1H NMR (400 MHz, MeOD) δ 5.13–5.07 (m, 1H, H-1), 4.49–4.15 (m, 10H, H-4, H-5, H-6, H-9, H-14), 4.04–3.83 (m, 2H, H-3, H-8), 3.83–3.64 (m, 3H, H-2, H-7), 3.39–3.27 (m, 4H, H-15), 2.41–2.33 (m, 2H, H-10), 1.69–1.58 (m, 2H, H-11), 1.41–1.25 (m, 16H, H-12), 0.92 (t, J = 6.7 Hz, 3H, H-13). β-anomer: 1H NMR (400 MHz, MeOD) δ 4.49–4.15 (m, 10H, H-1, H-5, H-6, H-9, H-14), 4.04–3.83 (m, 2H, H-4, H-8), 3.83–3.64 (m, 2H, H-7), 3.60–3.52 (m, 2H, H-2, H-3), 3.39–3.27 (m, 4H, H-15), 2.41–2.33 (m, 2H, H-10), 1.69–1.58 (m, 2H, H-11), 1.41–1.25 (m, 16H, H-12), 0.92 (t, J = 6.7 Hz, 3H, H-13). αβ-anomers: 13C NMR (101 MHz, MeOD) δ 175.48, 105.20, 104.90, 101.05, 100.22, 80.07, 79.85, 78.92, 77.76, 74.66, 74.58, 74.52, 74.44, 74.35, 72.44, 72.18, 70.86, 70.67, 70.07, 69.91, 69.33, 68.82, 65.87, 65.81, 64.66, 64.58, 64.44, 64.36, 63.07, 62.96, 62.42, 62.14, 40.86, 40.78, 35.06, 34.99, 34.95, 34.91, 33.04, 30.75, 30.72, 30.63, 30.60, 30.45, 30.41, 30.25, 30.23, 27.57, 27.56, 26.07, 26.00, 25.93, 23.71, 14.42. HRMS-ESI: Calculated for [C23H46NO12P+Na+]: 582.2650; found 582.2644.
3.3. General Information for Biological Assays
3.4. Isolation of Human Neutrophils
3.5. Assessment of Neutrophils’ Apoptosis Versus Necrosis
3.6. Evaluation of Neutrophils’ Oxidative Burst
3.6.1. Oxidation of Luminol
3.6.2. Oxidation of APF
3.6.3. Oxidation of Amplex Red
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac | Acetyl |
| Ac2O | Acetic Anhydride |
| AcOH | Acetic Acid |
| APF | Aminophenyl fluorescein |
| AR | Amplex Red |
| AV | Annexin V |
| Bn | Benzyl |
| Boc | N-Tert-Butyloxycarbonyl |
| DBTO | Dibutyl Tin Oxide |
| DCC | N,N’-Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DGDG | Digalactosyldiacylglycerol |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethyl Formamide |
| DNA | Deoxyribonucleic Acid |
| DPI | Diphenyleneiodonium Chloride |
| Et3N | Triethylamine |
| EtOH | Ethanol |
| FSC | Forward Scatter |
| GPL | Glycophospholipids |
| GSH-Px | Glutathione Peroxidase |
| HO-1 | Hemoxygenase-1 |
| HRMS | High-Resolution Mass Spectra |
| iNOS | Inducible Nitric Oxide Synthase |
| LPS | Lipopolysaccharide |
| MeOH | Methanol |
| MGDG | Monogalactosyldiacylglycerol |
| MMP-2 | Matrix Metalloproteinase-2 |
| MPO | Myeloperoxidase |
| MS | Molecular Sieves |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NIS | N-iodosuccinimide |
| NMR | Nuclear Magnetic Resonance |
| OAc | Acetate |
| OMe | Methoxide |
| PGL | Phosphoglycolipids |
| PhSH | Thiophenol |
| PI | Propidium Iodide |
| PMA | Phorbol 12-Myristate 13-Acetate |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| rt | Room Temperature |
| SEM | Standard Error of Mean |
| SOD | Superoxide Dismutase |
| SSC | Side Scatter |
| TBAF | Tetrabutylammonium Fluoride |
| TBDPS | Tert-Butyldiphenylsilyl |
| TFA | Trifluoroacetic Acid |
| TfOH | Trifluoromethanesulphonic Acid |
| THF | Tetrahydrofuran |
| TsOH | P-Toluenesulfonic Acid |
References
- Fischer, W. Bacterial Phosphoglycolipids and Lipoteichoic Acids. In Glycolipids, Phosphoglycolipids, and Sulfoglycolipids; Springer: Boston, MA, USA, 1990; pp. 123–234. [Google Scholar] [CrossRef]
- Pinheiro, L.; Freitas, M.; Branco, P.S. Phosphate-Containing Glycolipids: A Review on Synthesis and Bioactivity. ChemMedChem 2024, 19, e202400315. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuka, Y.; Kasama, T.; Ohashi, Y.; Uzawa, J.; Ono, Y.; Shimizu, K.; Hirabayashi, Y. A new phosphoglycerolipid; phosphatidylglucose’, found in human cord red cells by multi-reactive monoclonal anti-i cold agglutinin, mAb GL-1/GL-2. FEBS Lett. 2001, 497, 141–147. [Google Scholar] [CrossRef]
- Guy, A.T.; Nagatsuka, Y.; Ooashi, N.; Inoue, M.; Nakata, A.; Greimel, P.; Inoue, A.; Nabetani, T.; Murayama, A.; Ohta, K.; et al. Glycerophospholipid regulation of modality-specific sensory axon guidance in the spinal cord. Science 2015, 349, 974–977. [Google Scholar] [CrossRef]
- Li, X.; Hanafusa, K.; Kage, M.; Yokoyama, N.; Nakayama, H.; Hotta, T.; Oshima, E.; Kano, K.; Matsuo, I.; Nagatsuka, Y.; et al. Iwabuchi, Lysophosphatidylglucoside is a GPR55-mediated chemotactic molecule for human monocytes and macrophages. Biochem. Biophys. Res. Commun. 2021, 569, 86–92. [Google Scholar] [CrossRef]
- Antonopoulou, S.; Nomikos, T.; Oikonomou, A.; Kyriacou, A.; Andriotis, M.; Fragopoulou, E. Pantazidou, Characterization of bioactive glycolipids from Scytonema julianum (cyanobacteria). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005, 140, 219–231. [Google Scholar] [CrossRef]
- Tempesta, M.S.; Jolad, S.D.; King, S.; Mao, G.; Bruening, R.C.; Kuo, J.E.; Truong, T.V.; Bierer, D.E.; Dener, J.M. Novel Class of Phosphocholine Derivatives Having Antifungal Activity. WO1994008563A1, 8 October 1993. [Google Scholar]
- Yang, F.L.; Hua, K.F.; Yang, Y.L.; Zou, W.; Chen, Y.P.; Liang, S.M.; Hsu, H.Y.; Wu, S.H. TLR-independent induction of human monocyte IL-1 by phosphoglycolipids from thermophilic bacteria. Glycoconj. J. 2008, 25, 427–439. [Google Scholar] [CrossRef]
- Ostash, B.; Makitrynskyy, R.; Yushchuk, O.; Fedorenko, V. Structural diversity, bioactivity, and biosynthesis of phosphoglycolipid family antibiotics: Recent advances. BBA Adv. 2022, 2, 100065. [Google Scholar] [CrossRef]
- Manzo, E.; Gallo, C.; Sartorius, R.; Nuzzo, G.; Sardo, A.; De Berardinis, P.; Fontana, A. Cutignano, Immunostimulatory Phosphatidylmonogalactosyldiacylglycerols (PGDG) from the Marine Diatom Thalassiosira weissflogii: Inspiration for a Novel Synthetic Toll-Like Receptor 4 Agonist. Mar. Drugs 2019, 17, 103. [Google Scholar] [CrossRef]
- Osawa, T.; Fujikawa, K.; Shimamoto, K. Structures, functions, and syntheses of glycero-glycophospholipids. Front. Chem. 2024, 12. [Google Scholar] [CrossRef]
- Simon-Colin, C.; Kervarec, N.; Pichon, R. Deslandes, Complete 1H and 13C spectral assignment of floridoside. Carbohydr. Res. 2002, 337, 279–280. [Google Scholar] [CrossRef]
- Bondu, S.; Cerantola, S.; Kervarec, N.; Deslandes, E. Impact of the salt stress on the photosynthetic carbon flux and 13C-label distribution within floridoside and digeneaside in Solieria chordalis. Phytochemistry 2009, 70, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Courtois, A. Floridoside Extracted from the Red Alga Mastocarpus stellatus Is a Potent Activator of the Classical Complement Pathway. Mar. Drugs 2008, 6, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, M.; Pade, N. Heterosides–compatible solutes occurring in prokaryotic and eukaryotic phototrophs. Plant Biol. 2015, 17, 927–934. [Google Scholar] [CrossRef]
- Bisson, M.A.; Kirst, G.O. Osmotic Adaption in the Marine Alga Griffithsia monilis (Rhodophyceae): The Role of Ions and Organic Compounds. Funct. Plant Biol. 1979, 6, 523. [Google Scholar] [CrossRef]
- Simon-Colin, C.; Kervarec, N.; Pichon, R.; Deslandes, E. NMR 13C-isotopic enrichment experiments to study carbon-partitioning into organic solutes in the red alga Grateloupia doryphora. Plant Physiol. Biochem. 2004, 42, 21–26. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; van der Maarel, M.J.E.C. Floridoside production by the red microalga Galdieria sulphuraria under different conditions of growth and osmotic stress. AMB Express 2016, 6, 71. [Google Scholar] [CrossRef]
- Luo, Q.; Zhu, Z.; Zhu, Z.; Yang, R.; Qian, F.; Chen, H.; Yan, X. Different Responses to Heat Shock Stress Revealed Heteromorphic Adaptation Strategy of Pyropia haitanensis (Bangiales, Rhodophyta). PLoS ONE 2014, 9, e94354. [Google Scholar] [CrossRef]
- Kim, M.J.; Li, Y.X.; Dewapriya, P.; Ryu, B.; Kim, S.K. Floridoside suppresses pro-inflammatory responses by blocking MAPK signaling in activated microglia. BMB Rep. 2013, 46, 398–403. [Google Scholar] [CrossRef]
- Ochsenkühn, M.A.; Röthig, T.; D’Angelo, C.; Wiedenmann, J.; Voolstra, C.R. The role of floridoside in osmoadaptation of coral-associated algal endosymbionts to high-salinity conditions. Sci. Adv. 2017, 3, e1602047. [Google Scholar] [CrossRef]
- Li, Y.X.; Li, Y.; Lee, S.H.; Qian, Z.J.I.; Kim, S.E.K. Inhibitors of oxidation and matrix metalloproteinases, floridoside, and d-lsofloridoslde from marine red alga Laurencia undulata. J. Agric. Food Chem. 2010, 58, 578–586. [Google Scholar] [CrossRef]
- Pullar, J.M.; Vissers, M.C.M.; Winterbourn, C.C. Living with a Killer: The Effects of Hypochlorous Acid on Mammalian Cells. IUBMB Life 2000, 50, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Myeloperoxidaseederived oxidation: Mechanisms of biological damage and its prevention Overview of the Action of Myeloperoxidase and Other Heme Peroxidases. J. Clin. Biochem. Nutr. 2011, 48, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.J.; Morrow, J.D.; Ning, M.; Koroshetz, W.; Lo, E.H.; Terry, E.; Milne, G.L.; Hubbard, J.; Lee, H.; Stevenson, E.; et al. Oxidative Stress and Matrix Metalloproteinase-9 in Acute Ischemic Stroke. Stroke 2008, 39, 100–104. [Google Scholar] [CrossRef]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the Neutrophil Phagosome: Oxidants, Myeloperoxidase, and Bacterial Killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukoc. Biol. 2004, 76, 760–781. [Google Scholar] [CrossRef]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef]
- Halliwell, B. Antioxidants in Human Health and Disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef]
- Mydock, L.K.; Demchenko, A.V. Mechanism of chemical O-glycosylation: From early studies to recent discoveries. Org. Biomol. Chem. 2010, 8, 497–510. [Google Scholar] [CrossRef]
- Escopy, S.; Demchenko, A.V. Transition-Metal-Mediated Glycosylation with Thioglycosides. Chem. A Eur. J. 2022, 28, e202103747. [Google Scholar] [CrossRef]
- Singh, Y.; Geringer, S.A.; Demchenko, A.V. Synthesis and Glycosidation of Anomeric Halides: Evolution from Early Studies to Modern Methods of the 21st Century. Chem. Rev. 2022, 122, 11701–11758. [Google Scholar] [CrossRef] [PubMed]
- Tokatly, A.I.; Vinnitskiy, D.Z.; Ustuzhanina, N.E.; Nifantiev, N.E. Protecting Groups as a Factor of Stereocontrol in Glycosylation Reactions. Russ. J. Bioorganic Chem. 2021, 47, 53–70. [Google Scholar] [CrossRef]
- Mukherjee, M.M.; Ghosh, R.; Hanover, J.A. Recent Advances in Stereoselective Chemical O-Glycosylation Reactions. Front. Mol. Biosci. 2022, 9, 1–30. [Google Scholar] [CrossRef]
- Chen, A.; Yang, B.; Zhou, Z.; Zhu, F. Recent advances in transition-metal-catalyzed glycosyl cross-coupling reactions. Chem Catal. 2022, 2, 3430–3470. [Google Scholar] [CrossRef]
- Andreu, A.; Ćorović, M.; Garcia-Sanz, C.; Santos, A.S.; Milivojević, A.; Ortega-Nieto, C.; Mateo, C.; Bezbradica, D.; Palomo, J.M. Enzymatic Glycosylation Strategies in the Production of Bioactive Compounds. Catalysts 2023, 13, 1359. [Google Scholar] [CrossRef]
- Schmidt, R.R. New Methods for the Synthesis of Glycosides and Oligosaccharides—Are There Alternatives to the Koenigs-Knorr Method? [New Synthetic Methods (56)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 212–235. [Google Scholar] [CrossRef]
- Zhu, Z.-Y.; Cui, D.; Gao, H.; Dong, F.-Y.; Liu, X.; Liu, F.; Chen, L.; Zhang, Y. Efficient synthesis and activity of beneficial intestinal flora of two lactulose-derived oligosaccharides. Eur. J. Med. Chem. 2016, 114, 8–13. [Google Scholar] [CrossRef]
- Jacobson, J.M.; Kitov, P.I.; Bundle, D.R. The synthesis of a multivalent heterobifunctional ligand for specific interaction with Shiga toxin 2 produced by E. coli O157:H7. Carbohydr. Res. 2013, 378, 4–14. [Google Scholar] [CrossRef]
- Trinderup, H.H.; Andersen, S.M.; Heuckendorff, M.; Jensen, H.H. How do Various Reaction Parameters Influence Anomeric Selectivity in Chemical Glycosylation with Thioglycosides and NIS/TfOH Activation? Eur. J. Org. Chem. 2021, 2021, 3251–3259. [Google Scholar] [CrossRef]
- Dinkelaar, J.; De Jong, A.R.; Van Meer, R.; Somers, M.; Lodder, G.; Overkleeft, H.S.; Codée, J.D.C.; Van Der Marel, G.A. Stereodirecting effect of the pyranosyl C-5 substituent in glycosylation reactions. J. Org. Chem. 2009, 74, 4982–4991. [Google Scholar] [CrossRef]
- Crich, D.; Cai, W. Chemistry of 4,6-O-benzylidene-D-glycopyranosyl triflates: Contrasting behavior between the gluco and manno series. J. Org. Chem. 1999, 64, 4926–4930. [Google Scholar] [CrossRef]
- Tam, P.H.; Lowary, T.L. Epimeric and amino disaccharide analogs as probes of an α-(1→6)-mannosyltransferase involved in mycobacterial lipoarabinomannanbiosynthesis. Org. Biomol. Chem. 2010, 8, 181–192. [Google Scholar] [CrossRef]
- Aragozzini, F.; Maconi, E.; Potenza, D.; Scolastico, C. Enantioselective Microbial Reduction of Monoesters of 1,3-Dihydroxypropanone: Synthesis of (S)- and (R)-1,2-O-Isopropylideneglycerol. Synthesis 1989, 1989, 225–227. [Google Scholar] [CrossRef]
- Yoshida, M.; Saito, K.; Kato, H.; Tsukamoto, S.; Doi, T. Total Synthesis and Biological Evaluation of Siladenoserinol A and its Analogues. Angew. Chem.-Int. Ed. 2018, 57, 5147–5150. [Google Scholar] [CrossRef] [PubMed]
- Köhler, P.; Grosch, W. Study of the Effect of DATEM. 1. Influence of Fatty Acid Chain Length on Rheology and Baking. J. Agric. Food Chem. 1999, 47, 1863–1869. [Google Scholar] [CrossRef]
- Bondarev, I. Prevention and Treatment of Cancer and Other Diseases. WO 2007/106561A2, 14 March 2007. [Google Scholar]
- Veeneman, G.H.; van Leeuwen, S.H.; van Boom, J.H. Iodonium ion promoted reactions at the anomeric centre. II An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters. Tetrahedron Lett. 1990, 31, 1331–1334. [Google Scholar] [CrossRef]
- Hada, N.; Morita, T.; Ueda, T.; Masuda, K.; Nakane, H.; Ogane, M.; Yamano, K.; Schweizer, F.; Kiuchi, F. Synthesis of the Carbohydrate Moiety of Glycoproteins from the Parasite Echinococcus granulosus and Their Antigenicity against Human Sera. Molecules 2021, 26, 5652. [Google Scholar] [CrossRef]
- Lipták, A.; Jodál, I.; Nánási, P. Stereoselective ring-cleavage of 3-O-benzyl- and 2,3-di-O-benzyl-4,6-O-benzylidenehexopyranoside derivatives with the LiAlH4-AlCl3, reagent. Carbohydr. Res. 1975, 44, 1–11. [Google Scholar] [CrossRef]
- Mikami, T.; Asano, H.; Mitsunobu, O. Acetal-bond Cleavage of 4,6-O-Alkylidenehexopyranosides by Diisobutylaluminium Hydride and by Lithium Triethylborohydride-TiCl4. Chem. Lett. 1987, 16, 2033–2036. [Google Scholar] [CrossRef]
- Oikawa, M.; Liu, W.-C.; Nakai, Y.; Koshida, S.; Fukase, K.; Kusumoto, S. Regioselective Reductive Opening of 4,6-O-Benzylidene Acetals of Glucose or Glucosamine Derivatives by BH3⋅Me2NH-BF3⋅OEt2. Synlett 1996, 1996, 1179–1180. [Google Scholar] [CrossRef]
- Wang, C.-C.; Luo, S.-Y.; Shie, C.-R.; Hung, S.-C. Metal Trifluoromethanesulfonate- Catalyzed Regioselective Borane-Reductive Ring Opening of Benzylidene Acetals: A Concise Synthesis of 1,4-Dideoxy-1,4-imino-l-xylitol. Org. Lett. 2002, 4, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Shie, C.; Tzeng, Z.; Kulkarni, S.S.; Uang, B.; Hsu, C.; Hung, S. Cu(OTf) 2 as an Efficient and Dual-Purpose Catalyst in the Regioselective Reductive Ring Opening of Benzylidene Acetals. Angew. Chem. Int. Ed. 2005, 44, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Ahmadipour, S.; Miller, G.J. Recent advances in the chemical synthesis of sugar-nucleotides. Carbohydr. Res. 2017, 451, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M. The synthesis of mono-alkyl phosphates and their derivatives: An overview of their nature, preparation and use, including synthesis under plausible prebiotic conditions. Org. Biomol. Chem. 2018, 16, 3068–3086. [Google Scholar] [CrossRef]
- Gan, C.H.; Wijaya, H.; Li, L.H.; Wei, C.F.; Peng, Y.J.; Wu, S.H.; Hua, K.F.; Lam, Y. H-Phosphonate Synthesis and Biological Evaluation of an Immunomodulatory Phosphoglycolipid from Thermophilic Bacteria. Org. Lett. 2020, 22, 2569–2573. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Mitsunobe, K.; Fujiwara, S.; Mori, M.; Hashimoto, M.; Suda, Y.; Kusumoto, S.; Fukase, K. Synthesis and biological activity of phosphoglycolipids from Thermus thermophilus. Org. Biomol. Chem. 2013, 11, 5034–5041. [Google Scholar] [CrossRef]
- Lin, H.J.; Adak, A.K.; Reddy, L.V.R.; Wu, S.H.; Lin, C.C. Total synthesis of an immunomodulatory phosphoglycolipid from thermophilic bacteria. Chem. A Eur. J. 2013, 19, 7989–7998. [Google Scholar] [CrossRef]
- Martin, S.F.; Josey, J.A.; Wong, Y.L.; Dean, D.W. General Method for the Synthesis of Phospholipid Derivatives of 1,2-O-Diacyl-sn-Glycerols. J. Org. Chem. 1994, 59, 4805–4820. [Google Scholar] [CrossRef]
- Roussev, C.D.; Ivanova, G.D.; Bratovanova, E.K.; Petkov, D.D. Medium-Controlled Intramolecular Catalysis in the Direct Synthesis of 5′-O-Protected Ribonucleoside 2′- and 3′-Dialkylphosphates. Angew. Chem. Int. Ed. 2000, 39, 779–781. [Google Scholar] [CrossRef]
- Huang, H.; Denne, J.; Yang, C.; Wang, H.; Kang, J.Y. Direct Aryloxylation/Alkyloxylation of Dialkyl Phosphonates for the Synthesis of Mixed Phosphonates. Angew. Chem. Int. Ed. 2018, 57, 6624–6628. [Google Scholar] [CrossRef]
- Lira, L.M.; Vasilev, D.; Pilli, R.A.; Wessjohann, L.A. One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett. 2013, 54, 1690–1692. [Google Scholar] [CrossRef]
- Huang, H.; Ash, J.; Kang, J.Y. Tf2O-Promoted Activating Strategy of Phosphate Analogues: Synthesis of Mixed Phosphates and Phosphinate. Org. Lett. 2018, 20, 4938–4941. [Google Scholar] [CrossRef] [PubMed]
- Yashunsky, D.V.; Borodkin, V.S.; Ferguson, M.A.J.; Nikolaev, A.V. The Chemical Synthesis of Bioactive Glycosylphosphatidylinositols from Trypanosoma cruzi Containing an Unsaturated Fatty Acid in the Lipid. Angew. Chem. Int. Ed. 2006, 45, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hansen, T.; Zhang, Q.; Liu, D.; Sun, Y.; Yan, H.; Codée, J.D.C.; Schmidt, R.R.; Sun, J. 1-Picolinyl-5-azido Thiosialosides: Versatile Donors for the Stereoselective Construction of Sialyl Linkages. Angew. Chem. Int. Ed. 2019, 58, 17000–17008. [Google Scholar] [CrossRef]
- Larsen, E.; Kharazmi, A.; Christensen, L.P.; Christensen, S.B. An antiinflammatory galactolipid from rose hip (Rosa canina) that inhibits chemotaxis of human peripheral blood neutrophils in vitro. J. Nat. Prod. 2003, 66, 994–995. [Google Scholar] [CrossRef]
- Colombo, D.; Compostella, F.; Ronchetti, F.; Scala, A.; Toma, L.; Mukainaka, T.; Nagatsu, A.; Konoshima, T.; Tokuda, H.; Nishino, H. Inhibitory effects of monoacylated 2-O-β-galactosylglycerols on Epstein-Barr virus activation: The significant role of the hexanoyl chain. Cancer Lett. 1999, 143, 1–4. [Google Scholar] [CrossRef]
- Colombo, D.; Compostella, F.; Ronchetti, F.; Scala, A.; Toma, L.; Tokuda, H.; Nishino, H. Glycoglycerolipid analogues active as anti-tumor-promoters: The influence of the anomeric configuration. Eur. J. Med. Chem. 2000, 35, 1109–1113. [Google Scholar] [CrossRef]
- Stonik, V.A.; Stonik, I.V. Carbohydrate-Containing Low Molecular Weight Metabolites of Microalgae. Mar. Drugs 2023, 21, 427. [Google Scholar] [CrossRef]
- Hrabák, A.; Vercruysse, V.; Kahán, I.L.; Vray, B. Indomethacin prevents the induction of inducible nitric oxide synthase in murine peritoneal macrophages and decreases their nitric oxide production. Life Sci. 2001, 68, 1923–1930. [Google Scholar] [CrossRef]
- Yang, D.; Liu, Z.; Li, S.; Han, N.; Zhai, J.; Yin, J. Novel glycolipids from Potentilla anserina L. rhizomes: Anti-inflammatory, hepatoprotective activities, and structure-activity relationship studies. Food Biosci. 2025, 64, 105894. [Google Scholar] [CrossRef]
- Banskota, A.H.; Stefanova, R.; Gallant, P.; McGinn, P.J. Mono- and digalactosyldiacylglycerols: Potent nitric oxide inhibitors from the marine microalga Nannochloropsis granulata. J. Appl. Phycol. 2013, 25, 349–357. [Google Scholar] [CrossRef]
- Banskota, A.H.; Stefanova, R.; Sperker, S.; Melanson, R.; Osborne, J.A.; O’Leary, S.J.B. Five new galactolipids from the freshwater microalga Porphyridium aerugineum and their nitric oxide inhibitory activity. J. Appl. Phycol. 2013, 25, 951–960. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Y.; Lu, Z.; Lv, F.; Bie, X.; Zhao, H. Separation, characterization and anti-inflammatory activities of galactoglycerolipids from Perilla frutescens (L.) Britton. Nat. Prod. Res. 2023, 37, 3610–3615. [Google Scholar] [CrossRef]
- Matsufuji, M.; Nagamatsu, Y.; Yoshimoto, A. Protective effects of bacterial glyceroglycolipid M874B against cell death caused by exposure to heat and hydrogen peroxide. J. Biosci. Bioeng. 2000, 89, 345–349. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Lawton, M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Stahl, E.; Ashworth, M.R.F. Thin-Layer Chromatography, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1969. [Google Scholar] [CrossRef]
- Metten, K.-H.; Welzel, P. Synthesis of the repeating unit of the capsular antigen of Neisseria meningitidis serogroup H. Tetrahedron 1990, 46, 5145–5154. [Google Scholar] [CrossRef]
- Compostella, F.; Colombo, D.; Ferraboschi, P.; Scala, A.; Toma, L.; Ronchetti, F. Synthesis of Isosteric Analogues of Acylglycosylglycerols Active as Chemoprevention Agents. Eur. J. Org. Chem. 2002, 2002, 1429–1435. [Google Scholar] [CrossRef]
- Ribeiro, D.; Freitas, M.; Tomé, S.M.; Silva, A.M.S.; Porto, G.; Fernandes, E. Modulation of human neutrophils’ oxidative burst by flavonoids. Eur. J. Med. Chem. 2013, 67, 280–292. [Google Scholar] [CrossRef]
- Allen, R.C.; Loose, L.D. Phagocytic activation of a luminol-dependent chemiluminescence in rabbit alveolar and peritoneal macrophages. Biochem. Biophys. Res. Commun. 1976, 69, 245–252. [Google Scholar] [CrossRef]
- DeChatelet, L.; Shirley, P.; Johnston, R.J. Effect of phorbol myristate acetate on the oxidative metabolism of human polymorphonuclear leukocytes. Blood 1976, 47, 545–554. [Google Scholar] [CrossRef]
- Freitas, M.; Costa, V.M.; Ribeiro, D.; Couto, D.; Porto, G.; Carvalho, F.; Fernandes, E. Acetaminophen prevents oxidative burst and delays apoptosis in human neutrophils. Toxicol. Lett. 2013, 219, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, K.; Shishido, N.; Aizawa, H.; Hasebe, N.; Kikuchi, K.; Nakamura, M. Effects of MCI-186 upon neutrophil-derived active oxygens. Redox Rep. 2007, 12, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of Novel Fluorescence Probes That Can Reliably Detect Reactive Oxygen Species and Distinguish Specific Species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.; Lima, J.L.F.C.; Fernandes, E. Optical probes for detection and quantification of neutrophils’ oxidative burst. A review. Anal. Chim. Acta 2009, 649, 8–23. [Google Scholar] [CrossRef]
- Vidadala, S.R.; Thadke, S.A.; Hotha, S.; Kashyap, S. Synthesis of thioglycosides from propargyl glycosides exploiting alkynophilic gold catalyst. J. Carbohydr. Chem. 2012, 31, 241–251. [Google Scholar] [CrossRef]
- Behera, A.; Rai, D.; Kushwaha, D.; Kulkarni, S.S. Total Synthesis of Trisaccharide Repeating Unit of O-Specific Polysaccharide of Pseudomonas fluorescens BIM B-582. Org. Lett. 2018, 20, 5956–5959. [Google Scholar] [CrossRef]
- López-Prados, J.; Cuevas, F.; Reichardt, N.-C.; de Paz, J.-L.; Morales, E.Q.; Martín-Lomas, M. Design and synthesis of inositolphosphoglycan putative insulin mediators. Org. Biomol. Chem. 2005, 3, 764–786. [Google Scholar] [CrossRef]
- Herradón, B.; Cueto, S.; Morcuende, A.; Valverde, S. Regio- and enantioselective esterifications of polyoxygenated compounds catalyzed by lipases. Tetrahedron Asymmetry 1993, 4, 845–864. [Google Scholar] [CrossRef]
- Simas, A.B.C.; da Silva, A.A.T.; Filho, T.J.D.S.; Barroso, P.T.W. Direct selective and controlled protection of multiple hydroxyl groups in polyols via iterative regeneration of stannylene acetals. Tetrahedron Lett. 2009, 50, 2744–2746. [Google Scholar] [CrossRef]
- Ligthart, N.A.M.; de Geus, M.A.R.; van de Plassche, M.A.T.; García, D.T.; Isendoorn, M.M.E.; Reinalda, L.; Ofman, D.; van Leeuwen, T.; van Kasteren, S.I. A Lysosome-Targeted Tetrazine for Organelle-Specific Click-to-Release Chemistry in Antigen Presenting Cells. J. Am. Chem. Soc. 2023, 145, 12630–12640. [Google Scholar] [CrossRef]
- Häner, M.; Hammelev, C.H.; Pedersen, C.M. Conformational Distortion Using a Molecular Lever: Synthesis and Conformational Studies of Galactoside Derivatives. Eur. J. Org. Chem. 2018, 2018, 5532–5537. [Google Scholar] [CrossRef]
- Lv, J.; Luo, T.; Zou, D.; Dong, H. Using DMF as Both a Catalyst and Cosolvent for the Regioselective Silylation of Polyols and Diols. Eur. J. Org. Chem. 2019, 2019, 6383–6395. [Google Scholar] [CrossRef]
- Pan, D.; Sun, J.; Jin, H.; Li, Y.; Li, L.; Wu, Y.; Zhang, L.; Yang, Z. Supramolecular assemblies of novel aminonucleoside phospholipids and their bonding to nucleic acids. Chem. Commun. 2015, 51, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Bernardoni, B.; Di Terlizzi, L.; Galathri, E.M.; Kokotos, C.G.; Fagnoni, M.; Protti, S. Visible photons for the regioselective nucleophilic ring opening of epoxides. Green Chem. 2024, 26, 9833–9839. [Google Scholar] [CrossRef]
- Batovska, D.I.; Tsubota, S.; Kato, Y.; Asano, Y.; Ubukata, M. Lipase-mediated desymmetrization of glycerol with aromatic and aliphatic anhydrides. Tetrahedron Asymmetry 2004, 15, 3551–3559. [Google Scholar] [CrossRef]
- Sutter, M.; Dayoub, W.; Métay, E.; Raoul, Y.; Lemaire, M. 1-O-Alkyl (di)glycerol ethers synthesis from methyl esters and triglycerides by two pathways: Catalytic reductive alkylation and transesterification/reduction. Green Chem. 2013, 15, 786. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| R1 | R2 | R3 | α/β | |
| 1a | H | H | H | 3:1 |
| 1b | H |  | H | 1:1 |
| 1c | H | 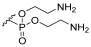 | H | 1:1 |
| 1d |  | H | H | 1:0 |
| 1e | H | H |  | 1:1 |
| 1f | H |  | H | 1:10 |
| 1g | H |  | H | 1.3:1 |
| 1h | 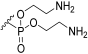 | H |  | 1:2 |
| Compounds | Luminol | APF | Amplex Red |
|---|---|---|---|
| 1a | 34 ± 7% 100µM | 27 ± 6% 50µM | <20% 50µM |
| 1b | 36 ± 9% 25µM | 29 ± 10% 25µM | <20% 25µM |
| 1c | <20% 100µM | 22 ± 8% 50µM | <20% 50µM |
| 1d | <20% 100µM | 21 ± 6% 50µM | 30 ± 9% 50µM |
| 1e | 83 ± 7 μM | <20% 50µM | <20% 50µM |
| 1f | <20% 100µM | <20% 50µM | <20% 50µM |
| 1g | <20% 100µM | <20% 50µM | <20% 50µM |
| 1h | 27 ± 15% 25µM | <20% 25µM | <20% 25µM |
| Quercetin | 1.2 ± 0.2 μM | 2.4 ± 0.2 μM | 4.1 ± 0.3 μM |
| DPI | 0.08 ± 0.01 μM | 0.030 ± 0.003 μM | 0.060 ± 0.004 μM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinheiro, L.; Cipriano, C.; Santos, F.; Máximo, P.; Fernandes, E.; Freitas, M.; Branco, P.S. Floridoside Phosphotriester Derivatives: Synthesis and Inhibition of Human Neutrophils’ Oxidative Burst. Molecules 2025, 30, 2850. https://doi.org/10.3390/molecules30132850
Pinheiro L, Cipriano C, Santos F, Máximo P, Fernandes E, Freitas M, Branco PS. Floridoside Phosphotriester Derivatives: Synthesis and Inhibition of Human Neutrophils’ Oxidative Burst. Molecules. 2025; 30(13):2850. https://doi.org/10.3390/molecules30132850
Chicago/Turabian StylePinheiro, Luís, Catarina Cipriano, Filipe Santos, Patrícia Máximo, Eduarda Fernandes, Marisa Freitas, and Paula S. Branco. 2025. "Floridoside Phosphotriester Derivatives: Synthesis and Inhibition of Human Neutrophils’ Oxidative Burst" Molecules 30, no. 13: 2850. https://doi.org/10.3390/molecules30132850
APA StylePinheiro, L., Cipriano, C., Santos, F., Máximo, P., Fernandes, E., Freitas, M., & Branco, P. S. (2025). Floridoside Phosphotriester Derivatives: Synthesis and Inhibition of Human Neutrophils’ Oxidative Burst. Molecules, 30(13), 2850. https://doi.org/10.3390/molecules30132850