
Nanoporous Carbons from Hydrothermally Treated Alga: Role in Batch and Continuous Capacitive Deionization (CDI)


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
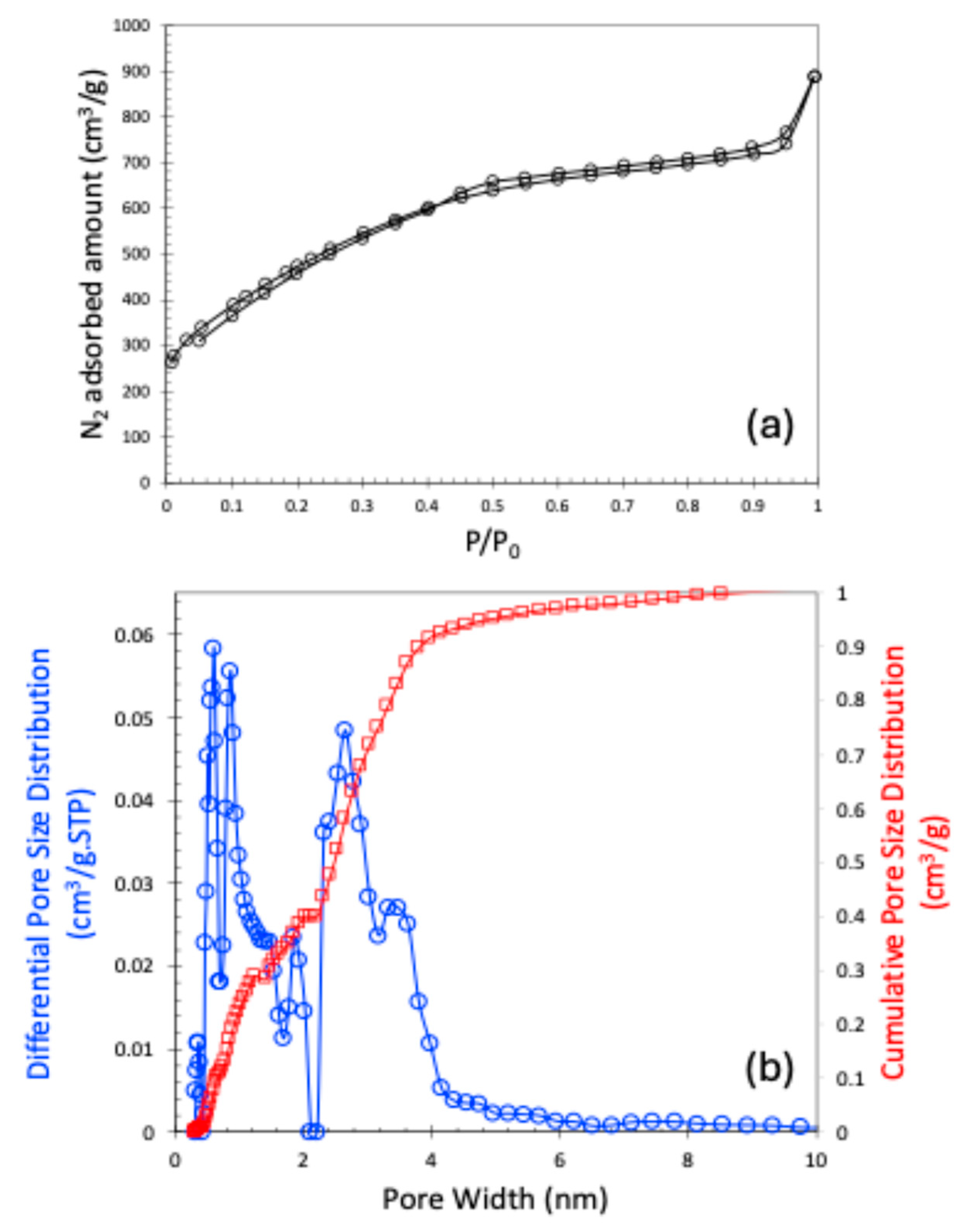
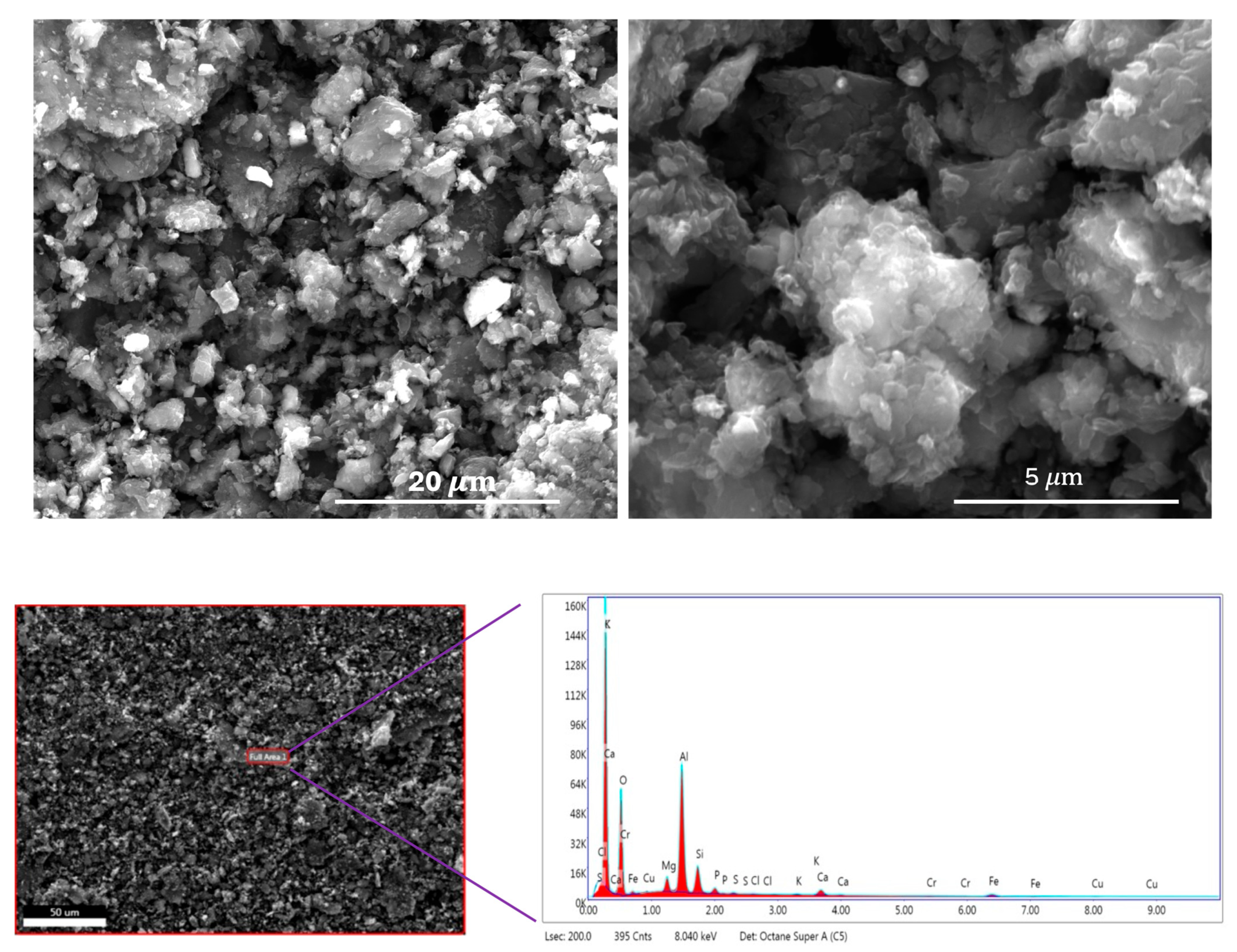
2.1. Materials’ Characteristics
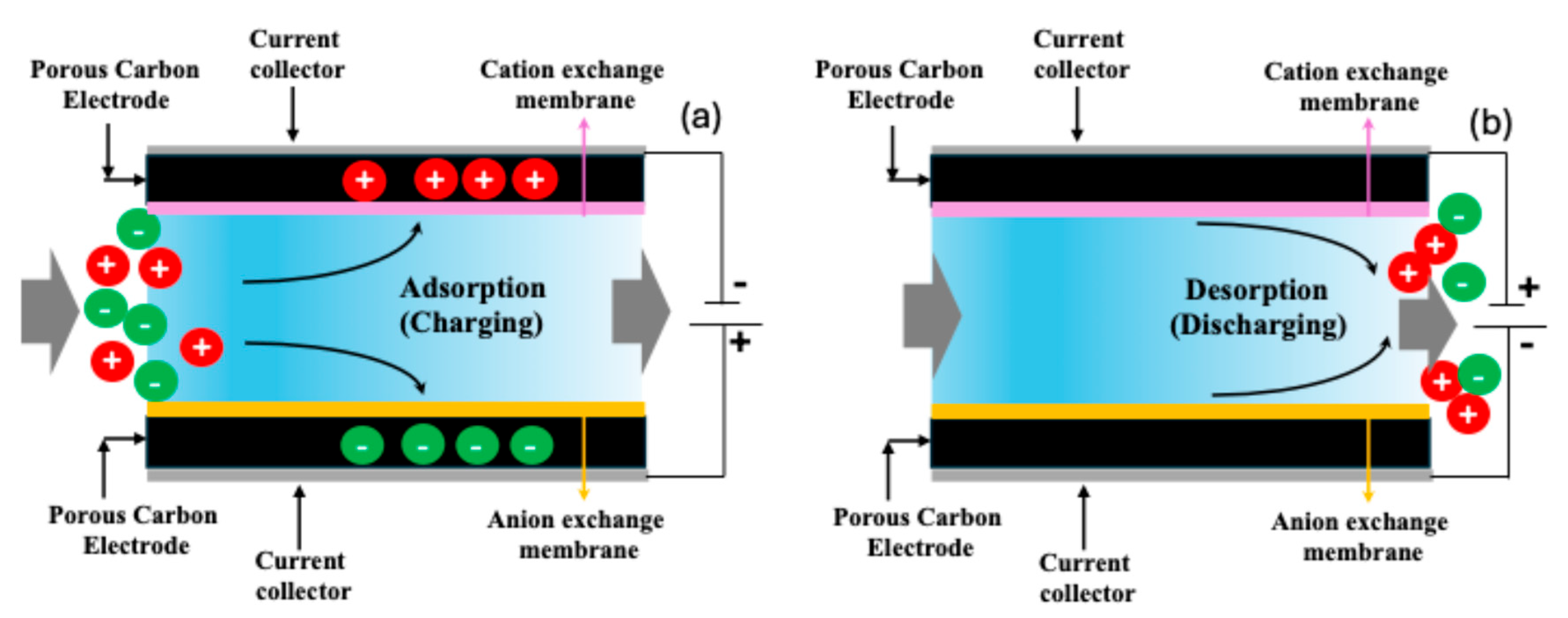
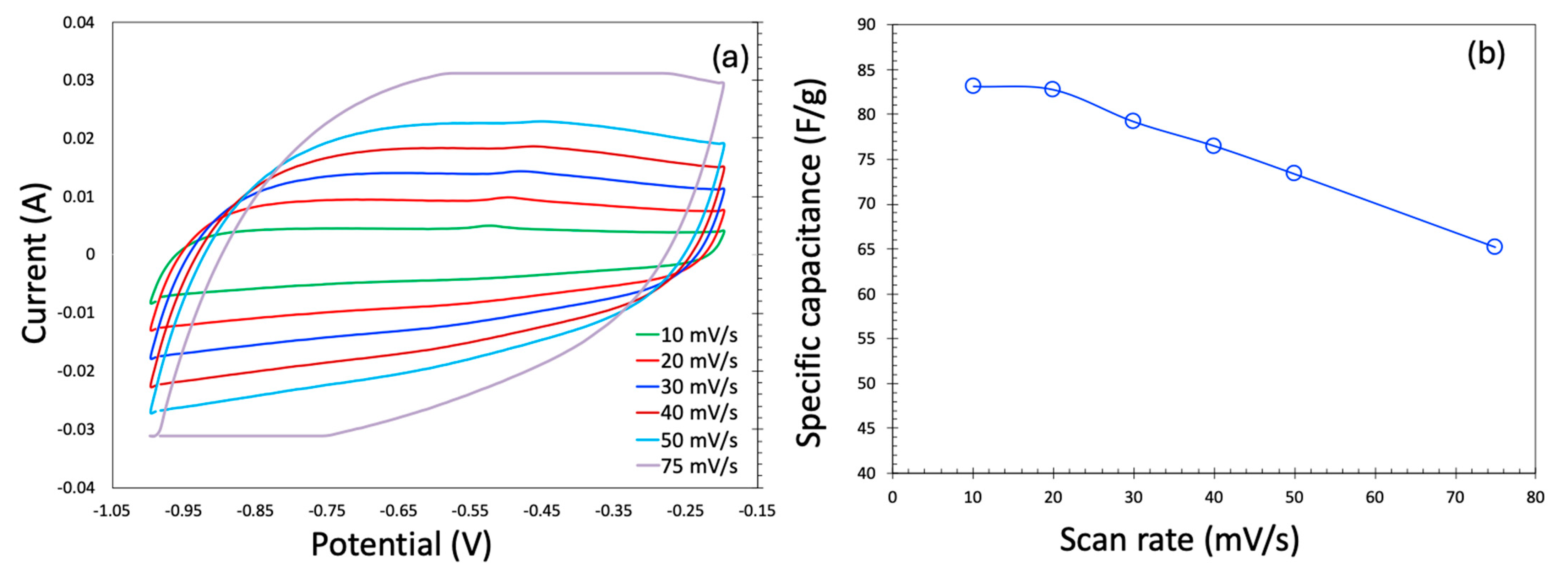
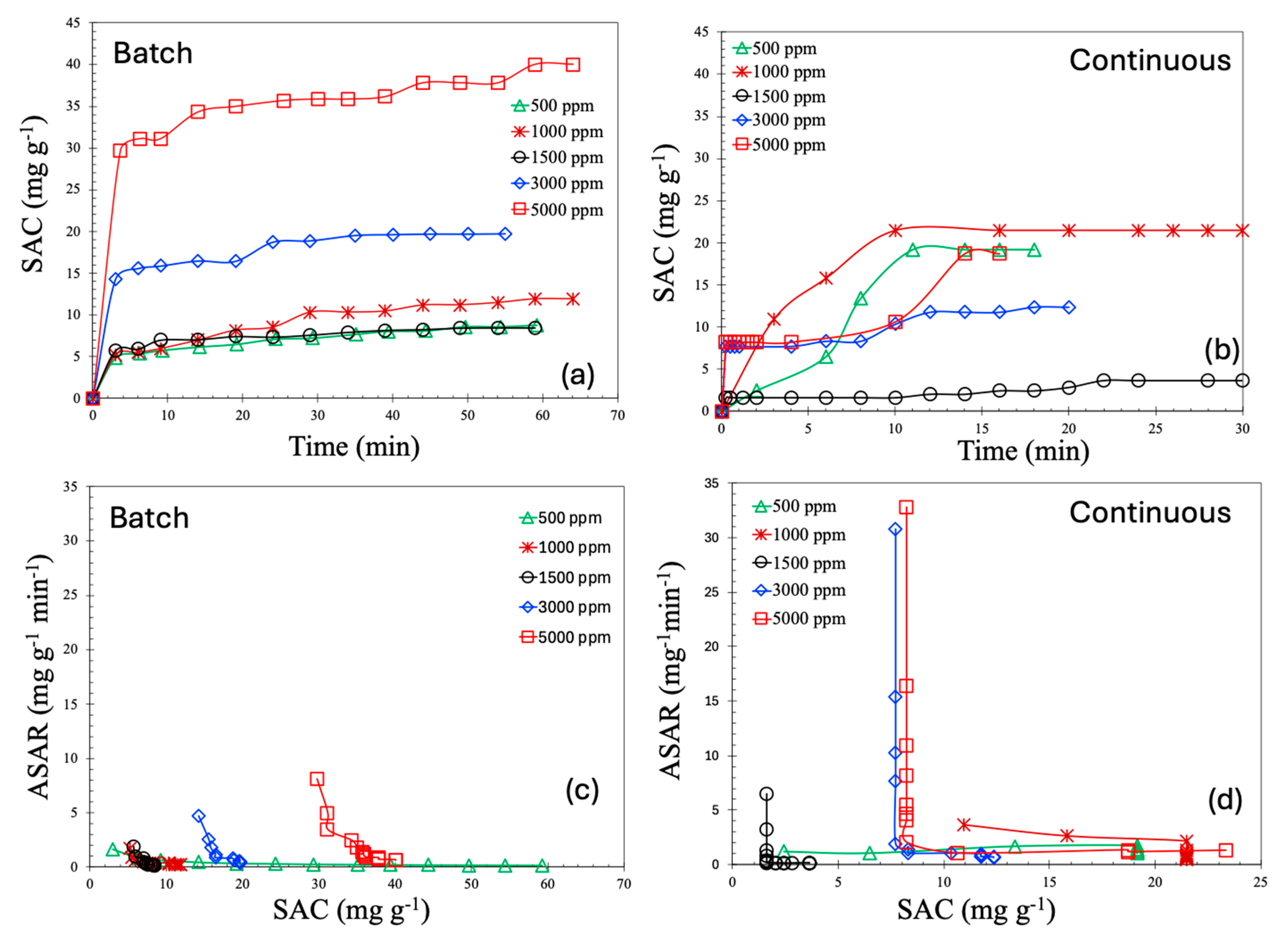
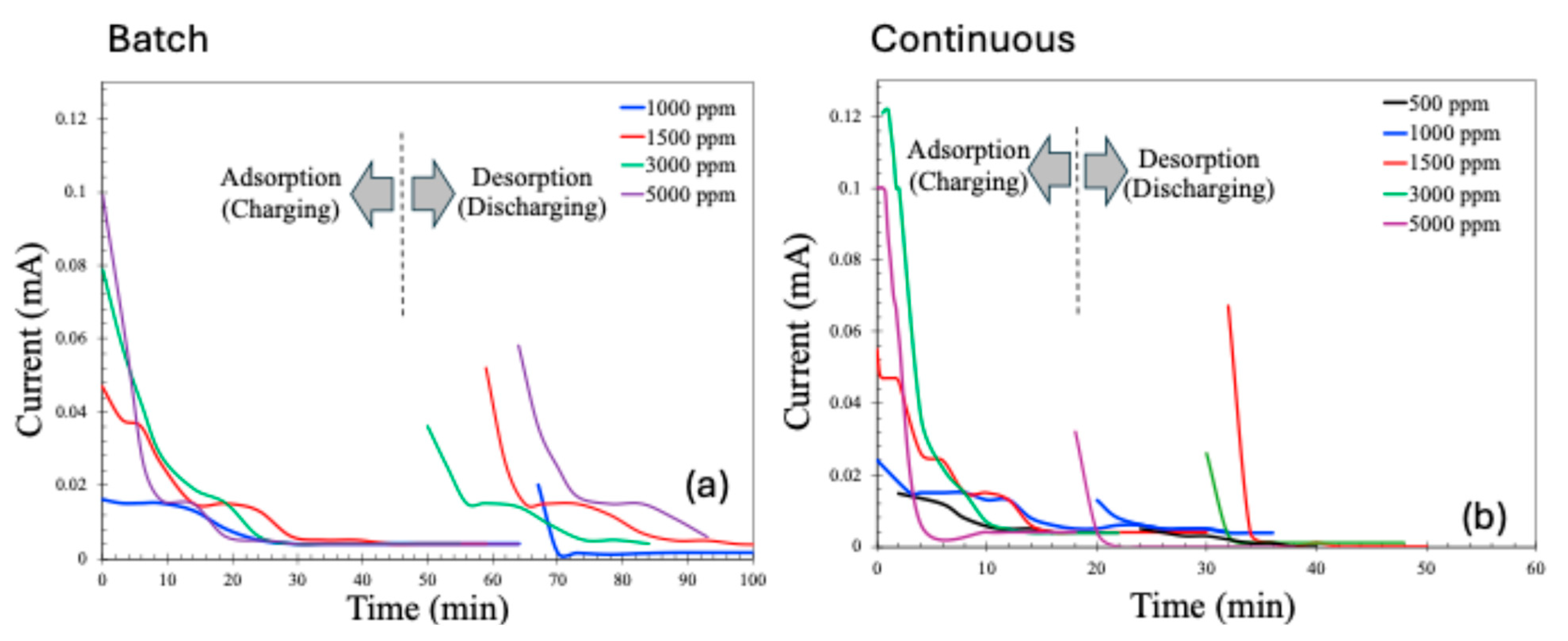
2.2. CDI Experiments
- (1)
- Stronger hydration shells due to a higher charge density: Divalent ions, such as Ca2+ and Mg2+ commonly present in hard water have higher charge densities, which result in stronger hydration shells. These shells are energetically difficult to remove, making it more challenging for the ions to pass through ion-exchange membranes and be adsorbed into the electrical double layers (EDLs) of the electrodes.
- (2)
- Lower ionic mobility: Divalent ions exhibit lower mobility in aqueous solutions due to their larger hydrated radii. This reduces their diffusion rate toward the electrodes, thus limiting the efficiency of ion removal.
- (3)
- Electrostatic competition and charge screening: The presence of divalent ions increases the ionic strength of the solution, leading to charge screening effects that can diminish the local electric field near electrode surfaces, thereby reducing ion adsorption efficiency.
- (4)
- Membrane selectivity: In membrane capacitive deionization, the selective transport properties of the membranes may further hinder divalent ion migration due to steric hindrance, thereby reducing their contribution to overall deionization.
- (1)
- Shorter residence time: In the continuous flow mode, the residence time of water within the cell is shorter, reducing the time available for ions to migrate to the EDLs or pass through the membranes.
- (2)
- Limited control over charge/discharge cycles: The batch process allows for optimized operation with controlled flow and charging/discharging cycles, enhancing ion removal. Continuous flow lacks this level of control, potentially leading to the incomplete regeneration of electrodes.
- (3)
- Flow-induced mass transfer limitations: While flow enhances ion delivery, higher flow rates can lead to boundary layer effects near electrode surfaces that hinder ion transport to adsorption sites. In contrast, the batch process benefits from diffusion-dominated transport, which may be more effective under quiescent conditions.
3. Experimental Procedure
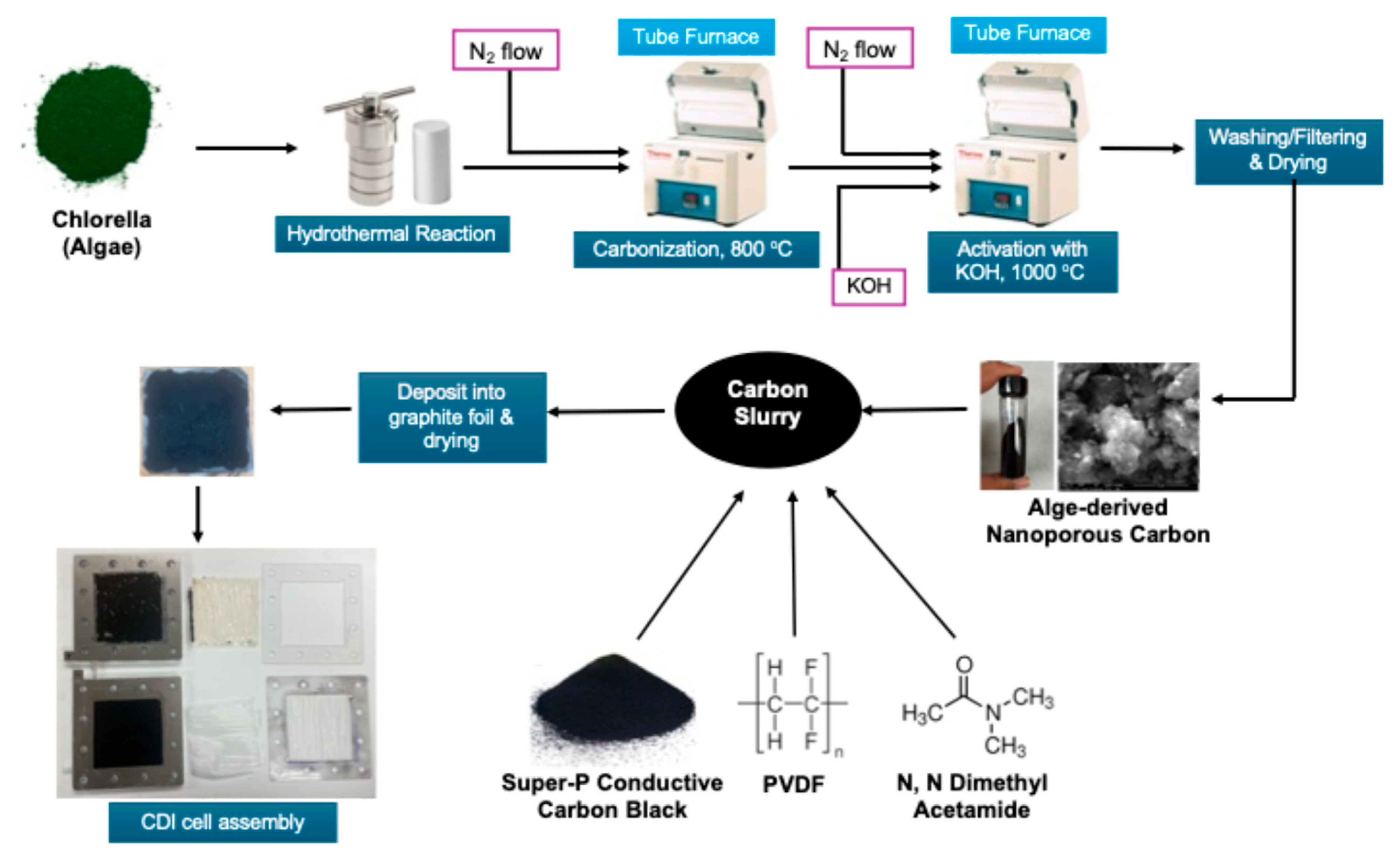
3.1. Synthesis of Activated Carbon from Alga
3.2. Characterization of Activated Carbon
3.3. Fabrication of CDI Electrode Compacting CDI Cell
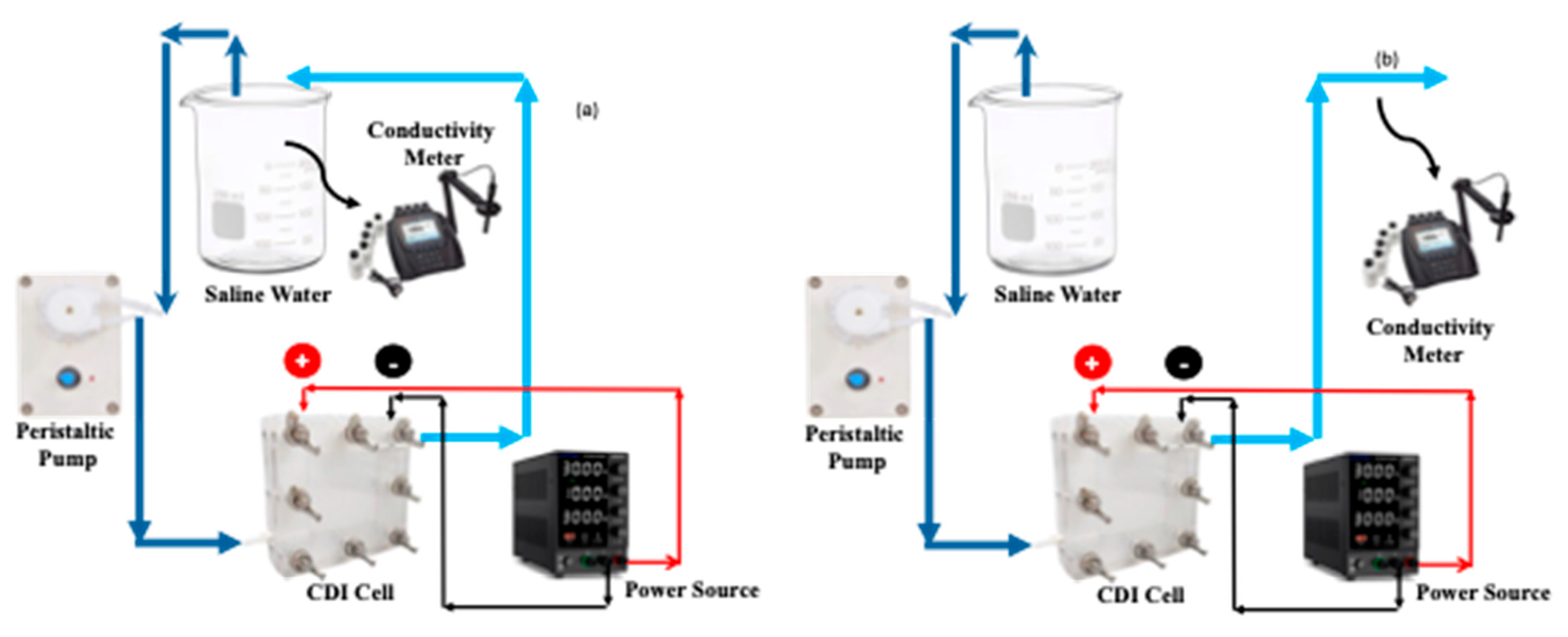
3.4. CDI Experiments with Saline and Hard Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salehi, M. Global water shortage and potable water safety; Today’s concern and tomorrow’s crisis. Environ. Int. 2022, 158, 106936. [Google Scholar] [CrossRef]
- Klaysom, C.; Cath, T.Y.; Depuydt, T.; Vankelecom, I.F.J. Forward and pressure retarded osmosis: Potential solutions for global challenges in energy and water supply. Chem. Soc. Rev. 2013, 42, 6959–6989. [Google Scholar] [CrossRef]
- Logan, B.E.; Elimelech, M. Membrane-based processes for sustainable power generation using water. Nature 2012, 488, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Strathmann, H. Electrodialysis, a mature technology with a multitude of new applications. Desalination 2010, 264, 268–288. [Google Scholar] [CrossRef]
- Greenlee, L.F.; Lawler, D.F.; Freeman, B.D.; Marrot, B.; Moulin, P. Reverse osmosis desalination: Water sources, technology, and today’s challenges. Water Res. 2009, 43, 2317–2348. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Cudero, A.L.; Palma, J. Capacitive deionization as an electrochemical means of saving energy and delivering clean water. Comparison to present desalination practices: Will it compete? Electrochim. Acta 2010, 55, 3845–3856. [Google Scholar] [CrossRef]
- Welgemoed, T.J.; Schutte, C.F. Capacitive Deionization Technology™: An alternative desalination solution. Desalination 2005, 183, 327–340. [Google Scholar] [CrossRef]
- AlMarzooqi, F.A.; Al Ghaferi, A.A.; Saadat, I.; Hilal, N. Application of Capacitive Deionisation in water desalination: A review. Desalination 2014, 342, 3–15. [Google Scholar] [CrossRef]
- Suss, M.E.; Porada, S.; Sun, X.; Biesheuvel, P.M.; Yoon, J.; Presser, V. Water desalination via capacitive deionization: What is it and what can we expect from it? Energy Environ. Sci. 2015, 8, 2296–2319. [Google Scholar] [CrossRef]
- Tang, W.; Liang, J.; He, D.; Gong, J.; Tang, L.; Liu, Z.; Wang, D.; Zeng, G. Various cell architectures of capacitive deionization: Recent advances and future trends. Water Res. 2019, 150, 225–251. [Google Scholar] [CrossRef]
- Porada, S.; Zhao, R.; van der Wal, A.; Presser, V.; Biesheuvel, P.M. Review on the science and technology of water desalination by capacitive deionization. Prog. Mater. Sci. 2013, 58, 1388–1442. [Google Scholar] [CrossRef]
- Wu, Q.; Liang, D.; Lu, S.; Wang, H.; Xiang, Y.; Aurbach, D.; Avraham, E.; Cohen, I. Advances and perspectives in integrated membrane capacitive deionization for water desalination. Desalination 2022, 542, 116043. [Google Scholar] [CrossRef]
- Song, Z.; Chen, Y.; Ren, N.; Duan, X. Recent advances in the fixed-electrode capacitive deionization (CDI): Innovations in electrode materials and applications. Environ. Funct. Mater. 2023, 2, 290–303. [Google Scholar] [CrossRef]
- Zhao, X.; Song, X.; Yang, S.; Hou, Y.; Wang, Y.; Yang, H.Y. Exploring ion-selective electrode materials for enhanced capacitive deionization. Green Energy Resour. 2023, 1. [Google Scholar] [CrossRef]
- Huang, Q.; Sheng, L.; Wu, T.; Huang, L.; Yan, J.; Li, M.; Chen, Z.; Zhang, H. Research progress on the application of carbon-based composites in capacitive deionization technology. Desalination 2025, 593. [Google Scholar] [CrossRef]
- Neme, I.; Gonfa, G.; Masi, C. Activated carbon from biomass precursors using phosphoric acid: A review. Heliyon 2022, 8, e11940. [Google Scholar] [CrossRef] [PubMed]
- Abioye, A.M.; Ani, F.N. Recent development in the production of activated carbon electrodes from agricultural waste biomass for supercapacitors: A review. Renew. Sustain. Energy Rev. 2015, 52, 1282–1293. [Google Scholar] [CrossRef]
- Saha, D.; Orkoulas, G.; Bates, D. One-Step Synthesis of Sulfur-Doped Nanoporous Carbons from Lignin with Ultra-High Surface Area, Sulfur Content and CO2 Adsorption Capacity. Materials 2023, 16, 455. [Google Scholar] [CrossRef]
- Saha, D.; Kienbaum, M.J. Role of Oxygen, nitrogen and sulfur functionalities on the surface of nanoporous carbons in CO2 adsorption: A critical review. Micropor. Mesopor. Mat. 2019, 287, 29–55. [Google Scholar] [CrossRef]
- Saha, D.; Taylor, B.; Alexander, N.; Joyce, D.F.; Faux, G.I.; Lin, Y.; Shteyn, V.; Orkoulas, G. One-Step conversion of agro-wastes to nanoporous carbons: Role in separation of greenhouse gases. Bioresour. Technol. 2018, 256, 232–240. [Google Scholar] [CrossRef]
- Saha, D.; Thorpe, R.; Van Bramer, S.E.; Alexander, N.; Hensley, D.; Orkoulas, G.; Chen, J. Synthesis of Nitrogen and Sulfur Codoped Nanoporous Carbons from Algae: Role in CO2 Separation. ACS Omega 2018, 3, 18592–18602. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Bae, W.-S.; Choi, J.-H. Electrode reactions and adsorption/desorption performance related to the applied potential in a capacitive deionization process. Desalination 2010, 258, 159–163. [Google Scholar] [CrossRef]
- Porada, S.; Weinstein, L.; Dash, R.; van der Wal, A.; Bryjak, M.; Gogotsi, Y.; Biesheuvel, P.M. Water Desalination Using Capacitive Deionization with Microporous Carbon Electrodes. Acs. Appl. Mater. Inter. 2012, 4, 1194–1199. [Google Scholar] [CrossRef]
- Seo, S.-J.; Jeon, H.-J.; Lee, J.K.; Kim, G.-Y.; Park, D.; Nojima, H.; Lee, J.; Moon, S.-H. Investigation on removal of hardness ions by capacitive deionization (CDI) for water softening applications. Water. Res. 2010, 44, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Omosebi, A.; Gao, X.; Holubowitch, N.; Li, Z.; Landon, J.; Liu, K. Anion Exchange Membrane Capacitive Deionization Cells. J. Electrochem. Soc. 2017, 164, E242–E247. [Google Scholar] [CrossRef]
- Zhang, Y.; Srimuk, P.; Aslan, M.; Galei, M.; Presser, V. Polymer ion-exchange membranes for capacitive deionization of aqueous media with low and high salt concentration. Desalination 2020, 479, 114331. [Google Scholar] [CrossRef]
- Yan, T.; Xu, B.; Zhang, J.; Shi, L.; Zhang, D. Ion-selective asymmetric carbon electrodes for enhanced capacitive deionization. RSC Adv. 2018, 5, 2490–2497. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Yang, Y.; Kim, T.; Shin, H.S.; Hong, S. Enhanced Electrochemical Stability of a Zwiterionic-Polymer-Functionalized Electrode for Capacitive Deionization. ACS Appl. Mater. Inter. 2018, 10, 6207–6217. [Google Scholar] [CrossRef]
- Kim, M.; Cero, M.D.; Hand, S.; Cusick, R.D. Enhancing capacitive deionization performance with charged structural polysaccharide electrode binders. Water Res. 2019, 148, 388–397. [Google Scholar] [CrossRef]
- Li, N.; An, J.; Wang, X.; Wang, H.; Lu, L.; Ren, Z.J. Resin-enhanced rolling activated carbon electrode for efficient capacitive deionization. Desalination 2017, 419, 20–28. [Google Scholar] [CrossRef]
- Bhat, A.P.; Reale, E.R.; Cerro, M.D.; Smith, K.C.; Cusick, R.D. Reducing impedance to ionic flux in capacitive deionization with Bi-tortuous activated carbon electrodes coated with asymmetrically charged polyelectrolytes. Water Res. 2019, 3, 100027. [Google Scholar] [CrossRef] [PubMed]
- Jo, K.; Back, Y.; Kim, S.; Hong, S.P.; Yoon, J. Evaluation of long-term stability in capacitive deionization using activated carbon electrodes coated with ion exchange polymers. Korean J. Chem. Eng. 2020, 37, 1199–1205. [Google Scholar] [CrossRef]
- Wang, L.; Liang, Y.; Zhang, L. Enhancing Performance of Capacitive Deionization with Polyelectrolyte-Infiltrated electrodes: Theory and experimental validation. Environ. Sci. Technol. 2020, 54, 5874–5883. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, D.; Schlosser, R.; Lapointe, L.; Comroe, M.L.; Samohod, J.; Whiting, E.; Young, D.S. Nanoporous Carbons from Hydrothermally Treated Alga: Role in Batch and Continuous Capacitive Deionization (CDI). Molecules 2025, 30, 2848. https://doi.org/10.3390/molecules30132848
Saha D, Schlosser R, Lapointe L, Comroe ML, Samohod J, Whiting E, Young DS. Nanoporous Carbons from Hydrothermally Treated Alga: Role in Batch and Continuous Capacitive Deionization (CDI). Molecules. 2025; 30(13):2848. https://doi.org/10.3390/molecules30132848
Chicago/Turabian StyleSaha, Dipendu, Ryan Schlosser, Lindsay Lapointe, Marisa L. Comroe, John Samohod, Elijah Whiting, and David S. Young. 2025. "Nanoporous Carbons from Hydrothermally Treated Alga: Role in Batch and Continuous Capacitive Deionization (CDI)" Molecules 30, no. 13: 2848. https://doi.org/10.3390/molecules30132848
APA StyleSaha, D., Schlosser, R., Lapointe, L., Comroe, M. L., Samohod, J., Whiting, E., & Young, D. S. (2025). Nanoporous Carbons from Hydrothermally Treated Alga: Role in Batch and Continuous Capacitive Deionization (CDI). Molecules, 30(13), 2848. https://doi.org/10.3390/molecules30132848