
Intramolecular Versus Intermolecular Bonding in Drug Gemcitabine and Nucleobases: A Computational Study


Abstract

1. Introduction
2. Computational Methods
3. Results and Discussion
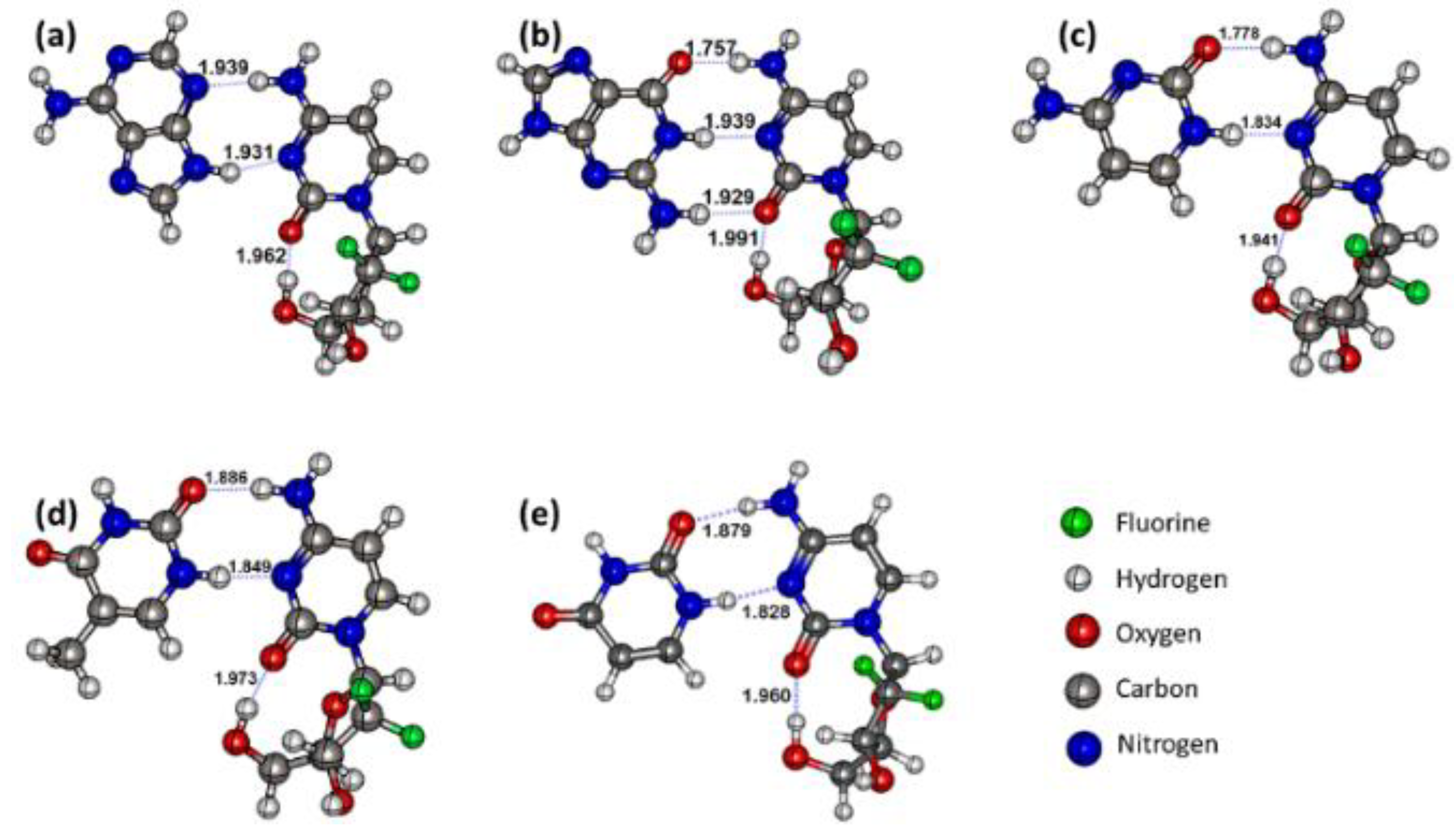
3.1. Structures and Geometric Parameters of the Gemcitabine–Nucleobase Complexes
3.2. Energetics
3.3. Frontier Molecular Orbitals and Conceptual DFT
3.4. Nature of Interactions
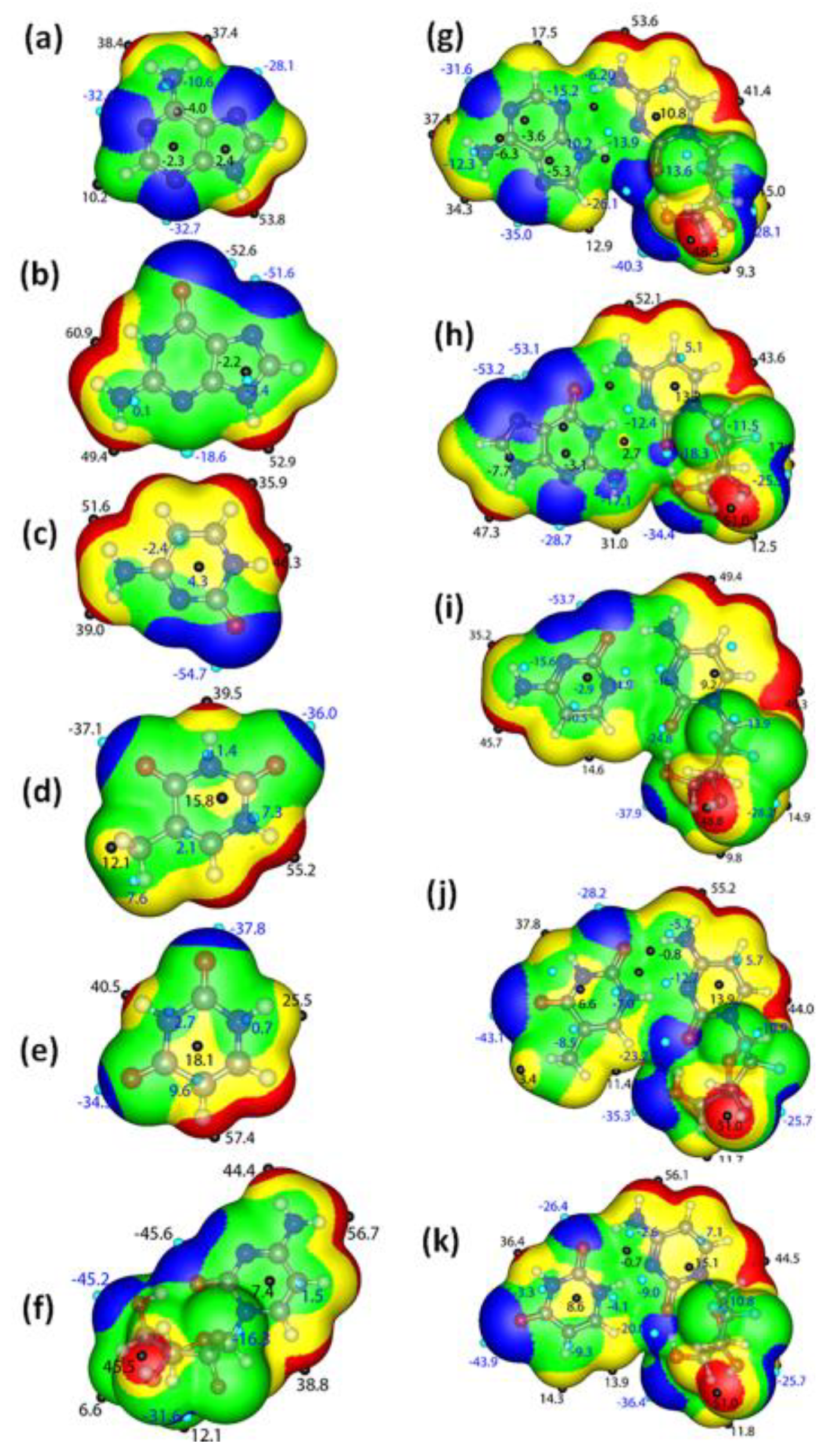
3.4.1. π-Electron Delocalization Indicators and Molecular Electrostatic Potential
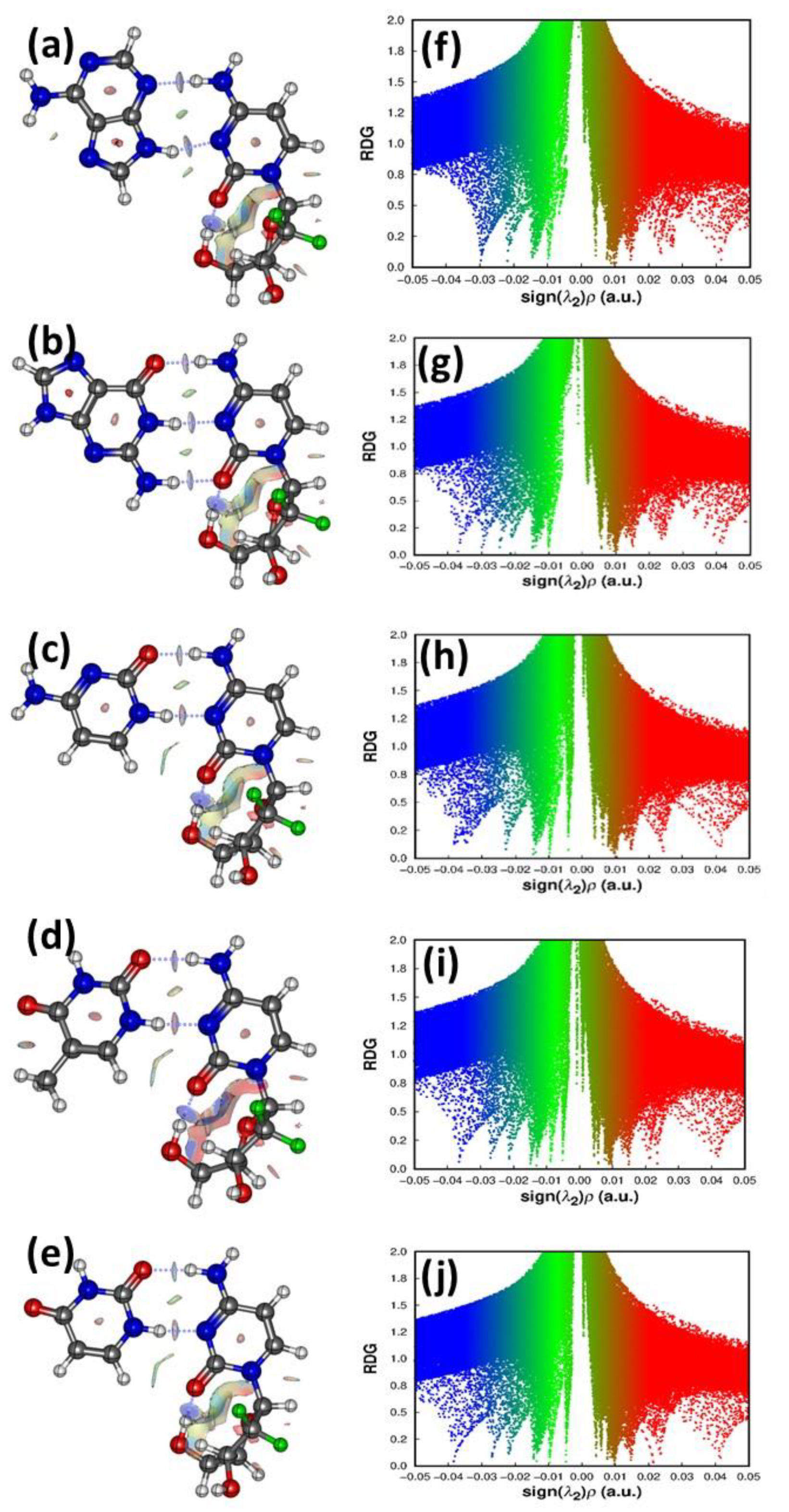
3.4.2. Atom in Molecule Analysis and Non-Covalent Interaction Analysis
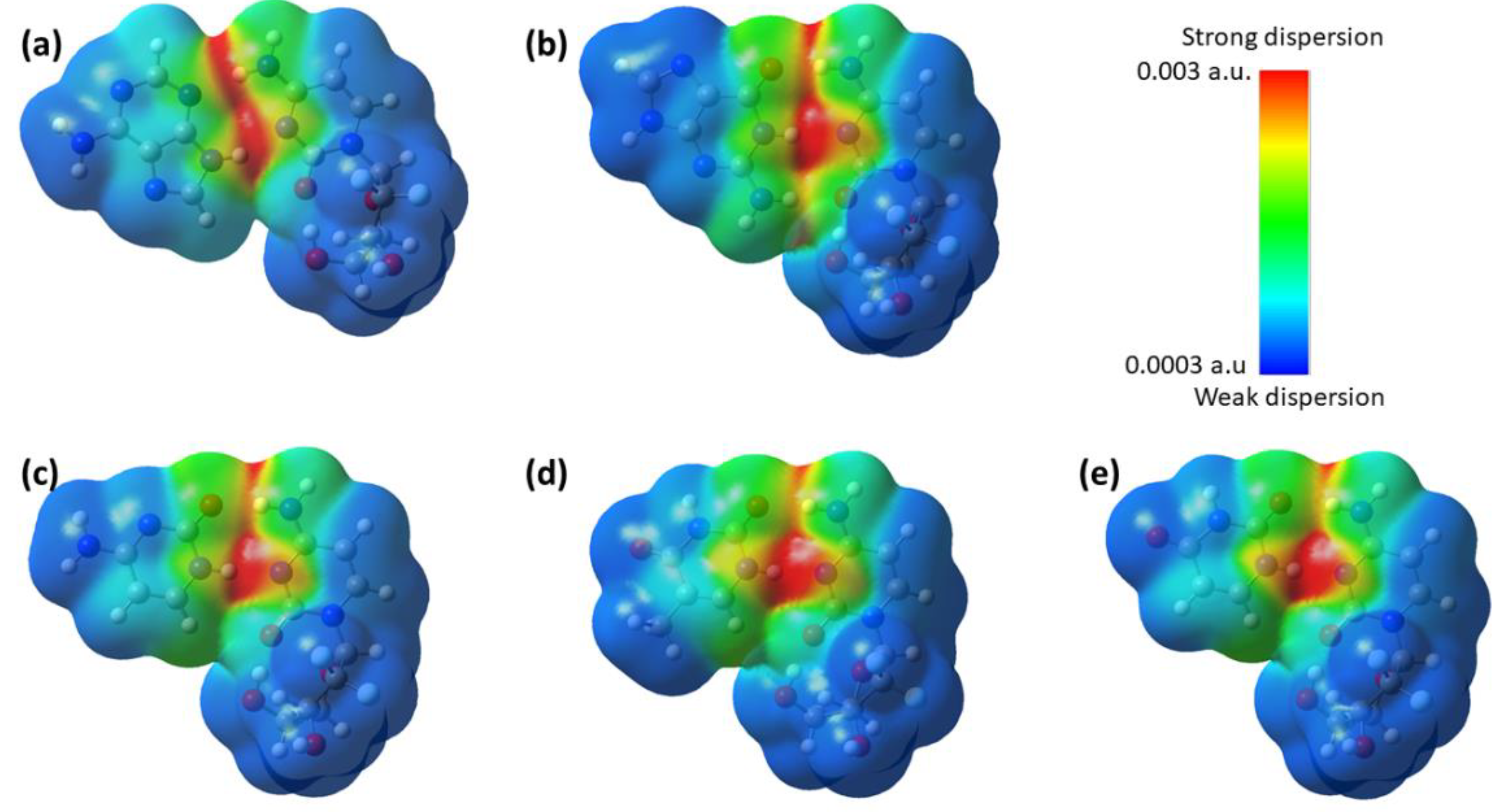
3.4.3. Dispersion Interaction Density (DID) and Local Energy Decomposition (LED) Analyses
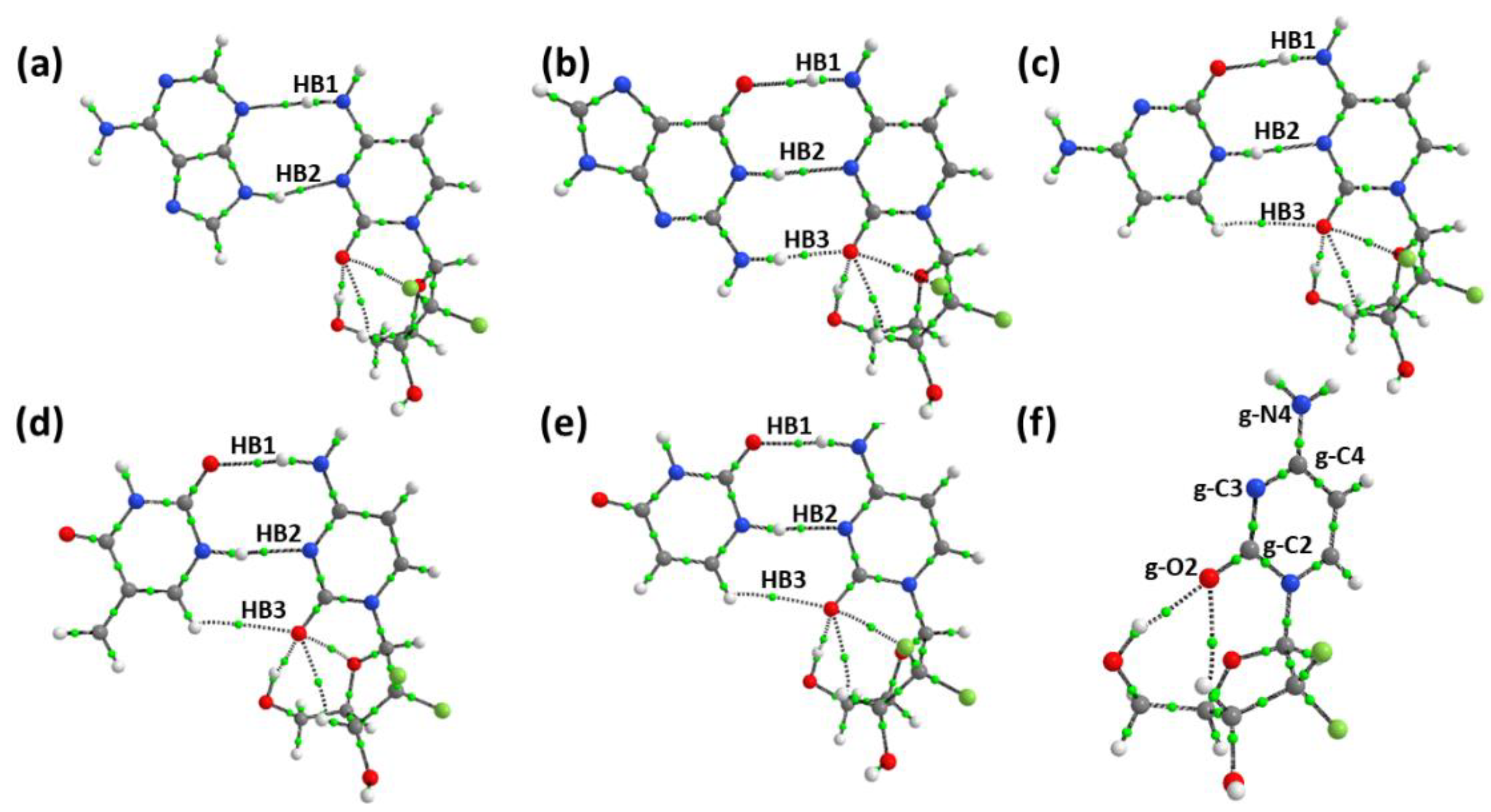
3.5. Role of Intramolecular Hydrogen Bond of Gemcitabine in Stabilizing the Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCSD(T) | Coupled Cluster Singles Doubles with perturbative Triples |
| DFT | Density Functional Theory |
| DLPNO | Domain-Based Local Pair Natural Orbital |
| HOMA | Harmonic Oscillator Measure of Aromaticity |
| NICS | Nucleus-Independent Chemical Shift |
| NCI–RDG | Non-Covalent Interaction–Reduced Density Gradient |
| QTAIM | Quantum Theory of Atoms in Molecules |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2022, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef]
- Cha, H.-M.; Kim, U.I.; Ahn, S.B.; Lee, M.K.; Lee, H.; Bang, H.; Jang, Y.; Kim, S.S.; Bae, M.A.; Kim, K.; et al. Evaluation of Antiviral Activity of Gemcitabine Derivatives against Influenza Virus and Severe Acute Respiratory Syndrome Coronavirus 2. ACS. Infect. Dis. 2023, 9, 1033–1045. [Google Scholar] [CrossRef]
- Larson, A.C.; Doty, K.R.; Solheim, J.C. The double life of a chemotherapy drug: Immunomodulatory functions of gemcitabine in cancer. Cancer Med. 2024, 13, e7287. [Google Scholar] [CrossRef]
- Alsarayreh, N.; Abdelghany, S.; Alqudah, D.; Dbuarqoub, D.; Alshaer, W. Gemcitabine-loaded niosomes: Optimization, characterization, and in vitro efficacy against invasive malignancies. J. Drug. Deliv. Sci. Technol. 2024, 95, 105617. [Google Scholar] [CrossRef]
- Hruba, L.; Das, V.; Hajduch, M.; Dzubak, P. Nucleoside-based anticancer drugs: Mechanism of action and drug resistance. Biochem. Pharmacol. 2023, 215, 115741. [Google Scholar] [CrossRef]
- Chai, X.; Meng, Y.; Ge, W.; Wang, J.; Li, F.; Wang, X.J.; Wang, X. A novel synthesized prodrug of gemcitabine based on oxygen-free radical sensitivity inhibited the growth of lung cancer cells. J. Biomed. Res. 2023, 37, 355–366. [Google Scholar] [CrossRef]
- Natu, J.; Nagaraju, G.P. Gemcitabine effects on tumor microenvironment of pancreatic ductal adenocarcinoma: Special focus on resistance mechanisms and metronomic therapies. Cancer Lett. 2023, 573, 216382. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, A.; Dehghani, M.; Setayesh, M.; Tavakkoli, M.; Rostamipour, H.A.; Ghorbani, M.; Ramzi, M.; Omidvari, S.; Moosavi, F.; Firuzi, O. Cytidine deaminase enzyme activity is a predictive biomarker in gemcitabine-treated cancer patients. Cancer Chemother. Pharmacol. 2023, 92, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Alniss, H.Y.; Al-Jubeh, H.M.; Msallam, Y.A.; Siddiqui, R.; Makhlouf, Z.; Ravi, A.; Hamdy, R.; Soliman, S.S.M.; Khan, N.A. Structure-based drug design of DNA minor groove binders and evaluation of their antibacterial and anticancer properties. Eur. J. Med. Chem. 2024, 271, 116440. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Sameti, M.; Borojeni, Z.I. Interaction of 5-fluorouracil anticancer drug with nucleobases: Insight from DFT, TD-DFT, and AIM calculations. J. Biomol. Struct. Dyn. 2023, 41, 5882–5893. [Google Scholar] [CrossRef]
- Hokmabady, L.; Raissi, H.; Khanmohammadi, A. Interactions of the 5-fluorouracil anticancer drug with DNA pyrimidine bases: A detailed computational approach. Struct. Chem. 2016, 27, 487–504. [Google Scholar] [CrossRef]
- Nakhaei, E.; Nowroozi, A.; Ravari, F. The influence of 5-fluorouracil anticancer drug on the DNA base pairs; a quantum chemical study. J. Biomol. Struct. Dyn. 2018, 37, 1–19. [Google Scholar] [CrossRef]
- Qiu, Z.M.; Wang, G.L.; Wang, H.L.; Xi, H.P.; Hou, D.N. MP2 study on the hydrogen-bonding interaction between 5-fluorouracil and DNA bases: A, C, G, T. Struct. Chem. 2014, 25, 1465–1474. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Sahara, R. Unveiling the Intermolecular Interactions between Drug 5-Fluorouracil and Watson–Crick/Hoogsteen Base Pairs: A Computational Analysis. ACS Omega 2024, 9, 24831–24844. [Google Scholar] [CrossRef]
- Mazumdar, P.; Kashyap, A.; Choudhury, D. Investigation of hydrogen bonding in small nucleobases using DFT, AIM, NCI and NBO technique. Comput. Theor. Chem. 2023, 1226, 114188. [Google Scholar] [CrossRef]
- Sukumaran, S.; Zochedh, A.; Chandran, K.; Sultan, A.B.; Karthiresan, T. Exploring the co-activity of FDA approved drug gemcitabine and docetaxel for enhanced anti-breast cancer activity: DFT, docking, molecular dynamics simulation and pharmacophore studies. Int. J. Quantum Chem. 2024, 124, e27359. [Google Scholar] [CrossRef]
- Ajeel, F.N.; Bardan, K.H.; Kareem, S.H.; Khudhair, A.M. Pd doped carbon nanotubes as a drug carrier for Gemcitabine anticancer drug: DFT studies. Chem. Phys. Impact 2023, 7, 100298. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Sahara, R.; Kawazoe, Y. Unveiling the gemcitabine drug complexation with cucurbit[n]urils (n = 6–8): A computational analysis. Struct. Chem. 2023, 34, 1869–1882. [Google Scholar] [CrossRef]
- Roohi, H.; Rouh, M.; Facehi, A. Assessing the performance of Al- and Ga-doped BNNTs for sensing and delivering Cytarabine and Gemcitabine anticancer drugs: A M06-2X study. Mol. Phys. 2023, 121, e2233639. [Google Scholar] [CrossRef]
- Bibi, S.; Ur-rehman, S.; Khalid, L.; Bhatti, I.A.; Bhatti, H.N.; Iqbal, J.; Bai, F.Q.; Zhang, H.-X. Investigation of the adsorption properties of gemcitabine anticancer drug with metal-doped boron nitride fullerenes as a drug-delivery carrier: A DFT study. RSC Adv. 2022, 12, 2873–2887. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Kumar, A.; Rayala, R.; Hindi, R.M.; Adhikary, A.; Wnuk, S.F.; Sevilla, M.D. One-Electron Oxidation of Gemcitabine and Analogs: Mechanism of Formation of C3′ and C2′ Sugar Radicals. J. Am. Chem. Soc. 2014, 136, 15646–15653. [Google Scholar] [CrossRef]
- Malekzadeh, M.; Heshmati, E.; Badalkhani-Khamseh, F.; Nojoumi, S.A. Investigation of conformational structures of gemcitabine and its 2′,2′-difluoro 2′-deoxy derivatives: A computational study. Comput. Theor. Chem. 2020, 1177, 112727. [Google Scholar] [CrossRef]
- Ali, M.S.; Muthukumaran, J.; Jain, M.; Al-Lohedan, H.A.; Farah, M.A.; Alsowilem, O.I. Experimental and computational investigation on the binding of anticancer drug gemcitabine with bovine serum albumin. J. Biomol. Struct. Dyn. 2022, 40, 9144–9157. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhang, J.; Dolg, M. Global optimization of clusters of rigid molecules using the artificial bee colony algorithm. Phys. Chem. Chem. Phys. 2016, 18, 3003–3010. [Google Scholar] [CrossRef]
- Zhang, J.; Dolg, M. ABCluster: The artificial bee colony algorithm for cluster global optimization. Phys. Chem. Chem. Phys. 2015, 17, 24173–24181. [Google Scholar] [CrossRef]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A. 2013, 117, 12590–12600. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the Performance of the M05−2X and M06−2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Thanthiriwatte, K.S.; Hohenstein, E.G.; Burns, L.A.; Sherrill, C.D. Assessment of the performance of DFT and DFT-D methods for describing distance dependence of hydrogen-bonded interactions. J. Chem. Theory Comput. 2011, 7, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS. 2016. Available online: http://aim.tkgristmill.com (accessed on 25 October 2024).
- Venkataramanan, N.S.; Suvitha, A.; Vijayaraghavan, A.; Thamotharan, S. Investigation of inclusion complexation of acetaminophen with pillar[5]arene: UV–Vis, NMR and quantum chemical study. J. Mol. Liq. 2017, 241, 782–791. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2016, 16, 1679–1691. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Sirajunnisa, A.R.; Sahara, R. Intermolecular hydrogen bond interactions in water clusters of zwitterionic glycine: DFT, MESP, AIM, RDG, and molecular dynamics analysis. J. Mol. Liq. 2024, 396, 123932. [Google Scholar]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Schrenckenbach, G.; Ziegler, T. Calculation of NMR Shielding Tensors Using Gauge-Including Atomic Orbitals and Modern Density Functional Theory. J. Phys. Chem. 1995, 99, 606–611. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Version 1.8, Build 682. Available online: https://www.chemcraftprog.com (accessed on 10 February 2025).
- Nesse, F. A perspective on the future of quantum chemical software: The example of the ORCA program package. Faraday Discuss. 2024, 254, 295–314. [Google Scholar] [CrossRef]
- Sathiyamoorthy, V.N.; Suvitha, A.; Sahara, R.; Kawazoe, Y. Intermolecular interactions in microhydrated ribonucleoside and deoxyribonucleoside: A computational study. Comput. Theor. Chem. 2021, 1204, 113422. [Google Scholar] [CrossRef]
- Suvitha, A.; Venkataramanan, N.S.; Sahara, R. The structure, stability, thermochemistry, and bonding in SO3-(H2O)n (n = 1–7) clusters: A computational analysis. Struct. Chem. 2023, 34, 225–237. [Google Scholar] [CrossRef]
- Balci, F.M.; Uras-Aytemiz, N. Isomeric forms of 1,4-dioxane in a microsolvation environment. Comput. Theor. Chem. 2024, 1236, 114587. [Google Scholar] [CrossRef]
- Jurečka, P.; Šponer, J.; Černy, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef]
- Chen, Z.; Shao, H. Exploring the in-situ conversion of intermolecular hydrogen bonds into static electricity through experimental and theoretical methods. J. Mol. Liq. 2024, 408, 125374. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Sahara, R. Structure, stability, and nature of bonding between high energy water clusters confined inside cucurbituril: A computational study. Comput. Theor. Chem. 2019, 1148, 44–54. [Google Scholar] [CrossRef]
- Nakhaei, E.; Nowroozi, A.; Ravari, F. The hydrogen-bonded complexes of the 5-fluorouracil with the DNA purine bases: A comprehensive quantum chemical study. Struct. Chem. 2018, 29, 69–80. [Google Scholar] [CrossRef]
- Meredith, N.Y.; Borsley, S.; Smolyar, I.V.; Nichol, G.S.; Baker, C.M.; Ling, K.B.; Cockroft, S.L. Dissecting Solvent Effects on Hydrogen Bonding. Angew. Chem. 2022, 61, e202206604. [Google Scholar] [CrossRef]
- Liu, C.; Gong, L.; Gong, B. Theoretical investigation of the electronic and second-order non-linear optical properties of [n] helicene derivatives. Comput. Theor. Chem. 2024, 1238, 114699. [Google Scholar] [CrossRef]
- Anan, J.; Adu Fosu, E.; Obuah, C.; Kojo Ainooson, M.; Aniagyei, A.; Hamenu, L.; Oppong, A.; Muller, A. A DFT and TD-DFT studies of the photosensitizing capabilities of thiophene-based dyes. Comput. Theor. Chem. 2024, 1237, 114633. [Google Scholar] [CrossRef]
- Pal, R.; Chattaraj, P.K. Electrophilicity index revisited. J. Comput. Chem. 2023, 44, 278–297. [Google Scholar] [CrossRef] [PubMed]
- Edet, H.O.; Louis, H.; Benjamin, I.; Gideon, M.; Unimuke, T.O.; Adalikwu, S.A.; Nwagu, A.D.; Adeyinka, A.S. Hydrogen storage capacity of C12X12 (X = N, P, and Si). Chem. Phys. Impact. 2022, 5, 100107. [Google Scholar] [CrossRef]
- Tegegn, D.F.; Zewude Belachew, H.; Salau, A.O. Investigation of substituent effects on the electronic structure and antiviral activity of favipiravir derivatives for COVID-19 treatment using DFT and molecular docking. Sci. Rep. 2024, 14, 8146. [Google Scholar] [CrossRef]
- Rajimon, K.J.; Sreelakshmi, N.; Rajendran Nair, D.S.; Fazulunnisa Begum, N.; Thomas, R. An in-depth study of the synthesis, electronic framework, and pharmacological profiling of 1-(anthracen-9-yl)-N-(4-nitrophenyl) methanimine: In vitro and in silico investigations on molecular docking, dynamics simulation, BSA/DNA binding and toxicity studies. Chem. Phys. Impact 2024, 8, 100462. [Google Scholar]
- Suvitha, A.; Venkataramanan, N.S.; Sahara, R.; Kawazoe, Y. A theoretical exploration of the intermolecular interactions between resveratrol and water: A DFT and AIM analysis. J. Mol. Model. 2019, 25, 56. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Non-covalent interaction via the reduced density gradient: Independent atom model vs experimental multipolar electron densities. Comput. Theor. Chem. 2012, 998, 148–163. [Google Scholar] [CrossRef]
- Baranac-Stojanović, M.; Aleksić, J.; Stojanović, M. Theoretical investigation of tautomerism of 2-and 4-pyridones: Origin, substituent and solvent effects. Org. Biomol. Chem. 2024, 22, 144–158. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, H.; Wu, W.; Mo, Y. Critical Check for the Role of Resonance in Intramolecular Hydrogen Bonding. Chem.-Eur.J. 2017, 23, 16885–16891. [Google Scholar] [CrossRef]
- Karuppasamy, J.; Zochedh, A.; Al-Asbahi, B.A.; Kumar, Y.A.; Shanmuganarayanan, A.; Sultan, A.B. Combined experimental and quantum computational studies on 5-Fluorouracil salicylic acid cocrystal: Assessment on anti-breast cancer potential through docking simulation. J. Mol. Struct. 2024, 1311, 138406. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Kumar, N.S.; El-Emam, A.A.; Venkataramanan, N.S.; Ghabbour, H.A.; Tamimi, A.-M.S.; Percino, J.; Thamotharan, S. Invesigation of potential anti-malarial lead candidate 2-(4-fluorobenzylthio)-5-(5-bormothiophene-2-yl)-1,3,4-oxadiazole: Insights from crystal structure, DFT, QTAIM and hybrid QM/MM binding energy analysis. J. Mol. Struct. 2019, 1175, 230–240. [Google Scholar] [CrossRef]
- Varadwaj, P.R. Halogen Bond via an Electrophilic π-Hole on Halogen in Molecules: Does It Exist? Int. J. Mol. Sci. 2024, 25, 4587. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Roos, G.; Murray, J.S. Probing intramolecular interactions using molecular electrostatic potentials: Changing electron density contours to unveil both attractive and repulsive interactions. Phys. Chem. Chem. Phys. 2024, 26, 7592–7601. [Google Scholar] [CrossRef]
- Thaynara Guimarães, M.; Nicolas Nascimento, C.; Murielly Fernanda Ribeiro, B.; Anna Karla dos Santos, P.; Grasiele Soares, C.; Douglas Henrique, P. Interactions between DNA and the acridine intercalator: A computational study. Comput. Biol. Chem. 2024, 109, 108029. [Google Scholar]
- Wolstenholme, D.J.; Stanley Cameron, T. Comparative study of weak interactions in molecular crystals: H− H bonds vs hydrogen bonds. J. Phys. Chem. A. 2006, 110, 8970–8978. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Palamarchuk, G.V.; Gorb, L.; Leszczynski, J. Intramolecular Hydrogen Bonds in Canonical 2 ‘-Deoxyribonucleotides: An Atoms in Molecules Study. J. Phys. Chem. B. 2006, 110, 4413–4422. [Google Scholar] [CrossRef]
- Gassoumi, B.; Mehri, A.; Hammami, H.; Castro, M.E.; Karayel, A.; Özkιnal, S.; Melendez, F.J.; Nouar, L.; Madi, F.; Ghalla, H.; et al. Spectroscopic characterization, host-guest charge transfer, Hirshfeld surfaces, AIM-RDG and ELF study of adsorption and chemical sensing of heavy metals with new derivative of Calix[4]quinone: A DFT-D3 computation. Mater. Chem. Phys. 2022, 278, 125555. [Google Scholar] [CrossRef]
- Bouchemela, H.; Madi, F.; Nouar, L. DFT investigation of host–guest interactions between α-Terpineol and β-cyclodextrin. J. Incl. Phenom. Macrocyclic Chem. 2022, 95, 247–258. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A. Encapsulation of sulfur, oxygen, and nitrogen mustards by cucurbiturils: A DFT study. J. Incl. Phenom. Macrocyclic Chem. 2015, 83, 387–400. [Google Scholar] [CrossRef]
- Wuttke, A.; Matta, R.A. Visualizing dispersion interactions through the use of local orbital spaces. J. Comput. Chem. 2017, 38, 15–23. [Google Scholar] [CrossRef]
- Bistoni, G. Finding chemical concepts in the Hilbert space: Coupled cluster analyses of noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1442. [Google Scholar] [CrossRef]
- Altun, A.; Izsák, R.; Bistoni, G. Local energy decomposition of coupled-cluster interaction energies: Interpretation, benchmarks, and comparison with symmetry-adapted perturbation theory. Int. J. Quantum Chem. 2021, 121, e26339. [Google Scholar] [CrossRef]
- Venkataramanan, N.S. Evaluating the drug delivery and sensing performance of XB23N24,(X = B, Al, Ga) nanocages for gemcitabine anticancer drug. Comput. Theor. Chem. 2025, 1248, 115222. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleobase | Binding Energy * (kcal mol−1) | Interaction Energy (kcal mol−1) | Deformation Energy (kcal mol−1) | ΔH (kcal mol−1) | ΔG (kcal mol−1) | |||
|---|---|---|---|---|---|---|---|---|
| Gas | Solution | Gas | Solution | Nucleobase | Gemcitabine | |||
| Adenine | −18.83 (−19.05) | −10.79 | −21.33 | −5.28 | 0.44 | 0.91 | −10.37 | 0.33 |
| Guanine | −26.51 (−27.26) | −14.84 | −31.25 | −17.49 | 1.18 | 1.54 | −14.39 | −3.14 |
| Cytosine | −21.82 (−22.25) | −11.69 | −25.53 | −13.44 | 1.04 | 1.19 | −11.27 | −0.61 |
| Thymine | −19.93 (−20.15) | −11.56 | −23.39 | −13.27 | 0.94 | 1.14 | −11.14 | −0.34 |
| Uracil | −20.68 (−20.88) | −11.63 | −23.66 | −13.35 | 0.84 | 0.89 | −11.21 | −0.55 |
| System | Egap | μ | η | ω | σ | ΔN |
|---|---|---|---|---|---|---|
| Gemcitabine | 7.827 | 4.558 | 7.827 | 1.327 | 0.128 | −0.582 |
| Adenine | 7.670 | 3.787 | 7.670 | 0.935 | 0.130 | −0.494 |
| Guanine | 6.929 | 3.791 | 6.929 | 1.037 | 0.144 | −0.547 |
| Cytosine | 7.772 | 4.150 | 7.772 | 1.108 | 0.129 | −0.534 |
| Thymine | 7.862 | 4.394 | 7.862 | 1.228 | 0.127 | −0.559 |
| Uracil | 8.129 | 4.641 | 8.129 | 1.325 | 0.123 | −0.571 |
| Gemcitabine–Adenine | 6.658 | 4.132 | 6.658 | 1.282 | 0.150 | −0.621 |
| Gemcitabine–Guanine | 5.959 | 3.888 | 5.959 | 1.268 | 0.168 | −0.652 |
| Gemcitabine–Cytosine | 7.025 | 4.234 | 7.025 | 1.276 | 0.142 | −0.603 |
| Gemcitabine–Thymine | 6.845 | 4.390 | 6.845 | 1.408 | 0.146 | −0.641 |
| Gemcitabine–Uracil | 7.150 | 4.579 | 7.150 | 1.466 | 0.140 | −0.640 |
| Complex | Gemcitabine | A/G | C/T/U | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HOMA | NICS(0) | NICS(1) | HOMA (6/5) | NICS(0) (6/5) | NICS(1) (6/5) * | HOMA | NICS(0) | NICS(1) | |
| Gemcitabine | 0.676 | −1.488 | −3.668 | - | - | - | - | - | - |
| Adenine | - | - | - | 0.981/0.881 | −6.304/−11.455 | −8.946/−10.199 | - | - | - |
| Guanine | - | - | - | 0.700/0.883 | −2.628/−11.481 | −3.719/−9.721 | - | - | - |
| Cytosine | - | - | - | - | - | - | 0.672 | −0.541 | −3.010 |
| Thymine | - | - | - | - | - | - | 0.466 | −1.079 | −2.226 |
| Uracil | - | - | - | - | - | - | 0.499 | −0.661 | −1.814 |
| Gemcitabine–Adenine | 0.693 | −1.376 | −3.254 | 0.983/0.912 | −6.346/−11.727 | −8.703/−10.675 | - | - | - |
| Gemcitabine–Guanine | 0.703 | −1.568 | −3.396 | 0.824/0.875 | −3.166/−11.001 | −4.141/−9.385 | - | - | - |
| Gemcitabine–Cytosine | 0.693 | −1.549 | −3.377 | - | - | - | 0.794 | −1.441 | −3.833 |
| Gemcitabine–Thymine | 0.701 | −1.616 | −3.372 | - | - | - | 0.540 | −1.315 | −2.422 |
| Gemcitabine–Uracil | 0.706 | −1.658 | −3.480 | - | - | - | 0.574 | −0.951 | −2.187 |
| BCP | Bond Length | ρ | ∇2ρ | λ1 | λ2 | λ3 | V | G | H | EHB | Ellipticity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gemcitabine–Adenine complex | |||||||||||
| HB1 | 1.939 | 0.02981 | 0.08956 | −0.04274 | −0.04004 | 0.17233 | −0.02203 | 0.02221 | −0.00018 | 6.91 | 0.07 |
| HB2 | 1.931 | 0.02988 | 0.09520 | −0.04237 | −0.04013 | 0.17770 | −0.02290 | 0.02335 | −0.00045 | 7.18 | 0.06 |
| Gemcitabine–Guanine complex | |||||||||||
| HB1 | 1.757 | 0.03704 | 0.13148 | −0.05969 | −0.05781 | 0.24897 | −0.03392 | 0.03340 | 0.00053 | 10.64 | 0.03 |
| HB2 | 1.939 | 0.02992 | 0.09284 | −0.04263 | −0.03997 | 0.17543 | −0.02244 | 0.02282 | −0.00039 | 7.04 | 0.07 |
| HB3 | 1.929 | 0.02475 | 0.09970 | −0.03412 | −0.03222 | 0.16603 | −0.01977 | 0.02235 | −0.00258 | 6.20 | 0.06 |
| Gemcitabine–Cytosine complex | |||||||||||
| HB1 | 1.778 | 0.03512 | 0.12700 | −0.05529 | −0.05339 | 0.23568 | −0.03151 | 0.03163 | −0.00012 | 9.88 | 0.04 |
| HB2 | 1.834 | 0.03815 | 0.10525 | −0.06008 | −0.05632 | 0.22164 | −0.03214 | 0.02922 | 0.00291 | 10.08 | 0.07 |
| Gemcitabine–Thymine complex | |||||||||||
| HB1 | 1.886 | 0.02698 | 0.10566 | −0.03861 | −0.03682 | 0.18109 | −0.02193 | 0.02417 | −0.00224 | 6.87 | 0.05 |
| HB2 | 1.849 | 0.03667 | 0.10455 | −0.05674 | −0.05278 | 0.21407 | −0.03045 | 0.02829 | 0.00216 | 9.55 | 0.08 |
| Gemcitabine–Uracil complex | |||||||||||
| HB1 | 1.879 | 0.02719 | 0.10691 | −0.03916 | −0.03753 | 0.18360 | −0.02224 | 0.02448 | −0.00225 | 6.98 | 0.04 |
| HB2 | 1.828 | 0.03849 | 0.10526 | −0.06085 | −0.05704 | 0.22315 | −0.03259 | 0.02945 | 0.00314 | 10.23 | 0.07 |
| System | ΔEint | ΔEgeo-prep | ||||||
|---|---|---|---|---|---|---|---|---|
| Gemcitabine–Adenine | −17.68 | 0.92 | 76.44 | −81.51 | −12.62 | 5.45 | −5.33 | −0.93 |
| Gemcitabine–Guanine | −27.59 | 2.00 | 109.34 | −119.48 | −17.44 | 7.53 | −6.50 | −1.03 |
| Gemcitabine–Cytosine | −22.59 | 1.57 | 93.33 | −101.03 | −14.89 | 6.75 | −5.38 | −0.82 |
| Gemcitabine–Thymine | −19.96 | 1.44 | 79.74 | −86.60 | −13.11 | 6.60 | −5.40 | −0.81 |
| Gemcitabine–Uracil | −20.65 | 1.27 | 82.13 | −89.41 | −13.36 | 6.63 | −5.29 | −0.81 |
| Donor NBO (i) | Acceptor NBO (j) | E(2) kcal mol−1 | F(I,j) a.u. | EHB |
|---|---|---|---|---|
| GEM | ||||
| LP(1) O29 | BD*(1) O22-H23 | 3.03 | 0.06 | 5.46 |
| LP(1) O29 | BD*(1) C15-H16 | 0.53 | 0.02 | 3.10 |
| Gemcitabine–Adenine complex | ||||
| LP(1) O29 | BD*(1) O22-H23 | 2.92 | 0.06 | 5.54 |
| LP(1) O29 | BD*(1) C15-H16 | 0.50 | 0.22 | 2.97 |
| Gemcitabine–Guanine complex | ||||
| LP(1) O29 | RY*(1) H23 | 0.84 | 0.04 | 5.39 |
| LP(1) O29 | RY*(1) H23 | 0.60 | 0.04 | - |
| LP(1) O29 | BD*(1) C15-H16 | - | - | 2.98 |
| Gemcitabine–Cytosine complex | ||||
| LP(1) O29 | BD*(1) O22-H23 | 3.21 | 0.06 | 5.92 |
| LP(1) O29 | BD*(1) C15-H16 | - | - | 3.01 |
| Gemcitabine–Thymine complex | ||||
| LP(1) O29 | BD*(1) O22-H23 | 2.69 | 0.05 | 5.39 |
| LP(1) O29 | BD*(1) C15-H16 | - | - | 2.74 |
| Gemcitabine–Uracil complex | ||||
| BD*(2) C28-29 | BD*(1) O22-H23 | 0.69 | 0.04 | 5.62 |
| LP(1) O29 | BD*(1) C15-H16 | - | - | 2.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkataramanan, N.S.; Suvitha, A.; Sahara, R. Intramolecular Versus Intermolecular Bonding in Drug Gemcitabine and Nucleobases: A Computational Study. Molecules 2025, 30, 2732. https://doi.org/10.3390/molecules30132732
Venkataramanan NS, Suvitha A, Sahara R. Intramolecular Versus Intermolecular Bonding in Drug Gemcitabine and Nucleobases: A Computational Study. Molecules. 2025; 30(13):2732. https://doi.org/10.3390/molecules30132732
Chicago/Turabian StyleVenkataramanan, Natarajan Sathiyamoorthy, Ambigapathy Suvitha, and Ryoji Sahara. 2025. "Intramolecular Versus Intermolecular Bonding in Drug Gemcitabine and Nucleobases: A Computational Study" Molecules 30, no. 13: 2732. https://doi.org/10.3390/molecules30132732
APA StyleVenkataramanan, N. S., Suvitha, A., & Sahara, R. (2025). Intramolecular Versus Intermolecular Bonding in Drug Gemcitabine and Nucleobases: A Computational Study. Molecules, 30(13), 2732. https://doi.org/10.3390/molecules30132732