

Balancing Yields and Sustainability: An Eco-Friendly Approach to Losartan Synthesis Using Green Palladium Nanoparticles

 , , and
, , and
Abstract

1. Introduction
2. Results
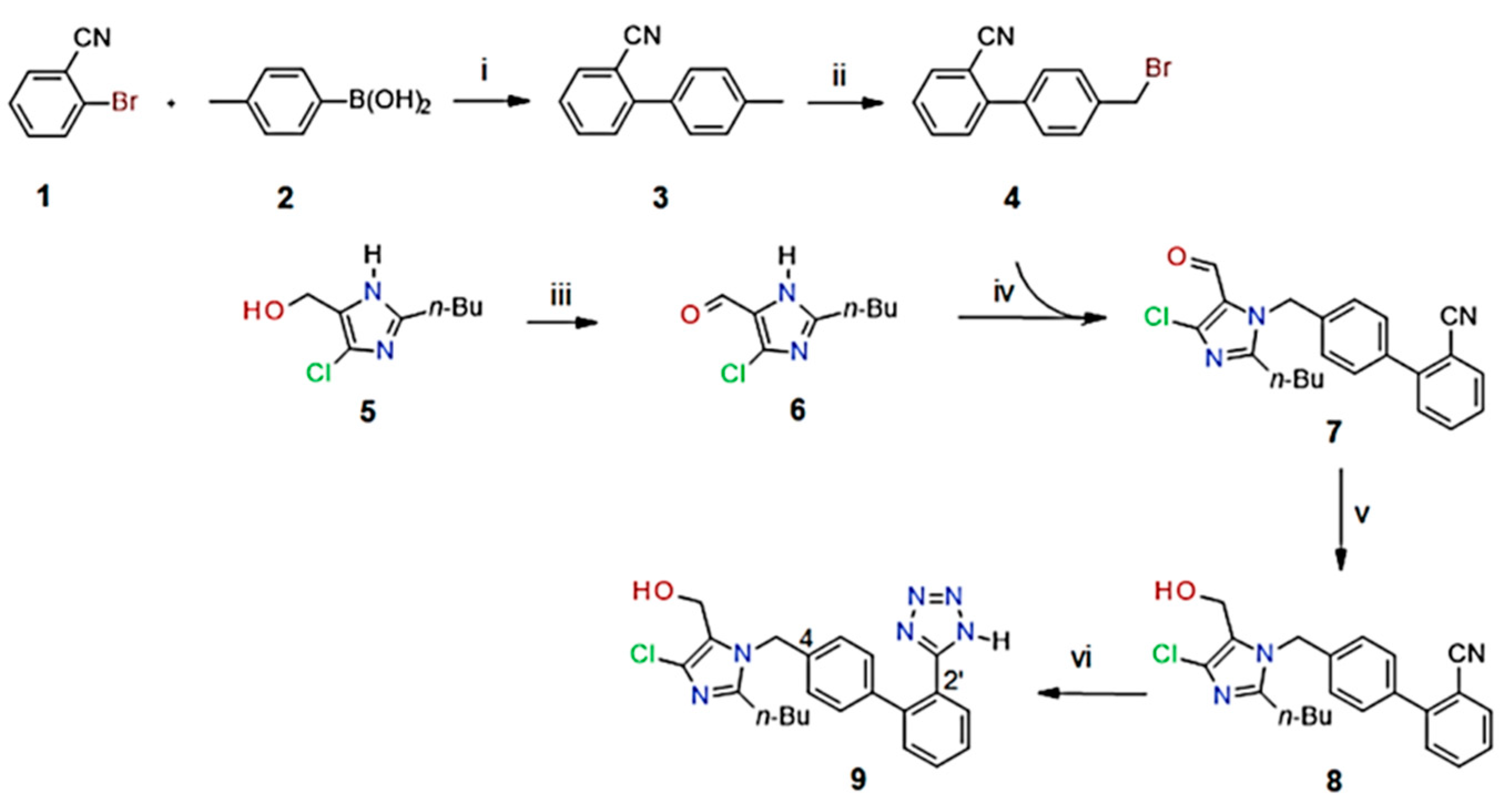
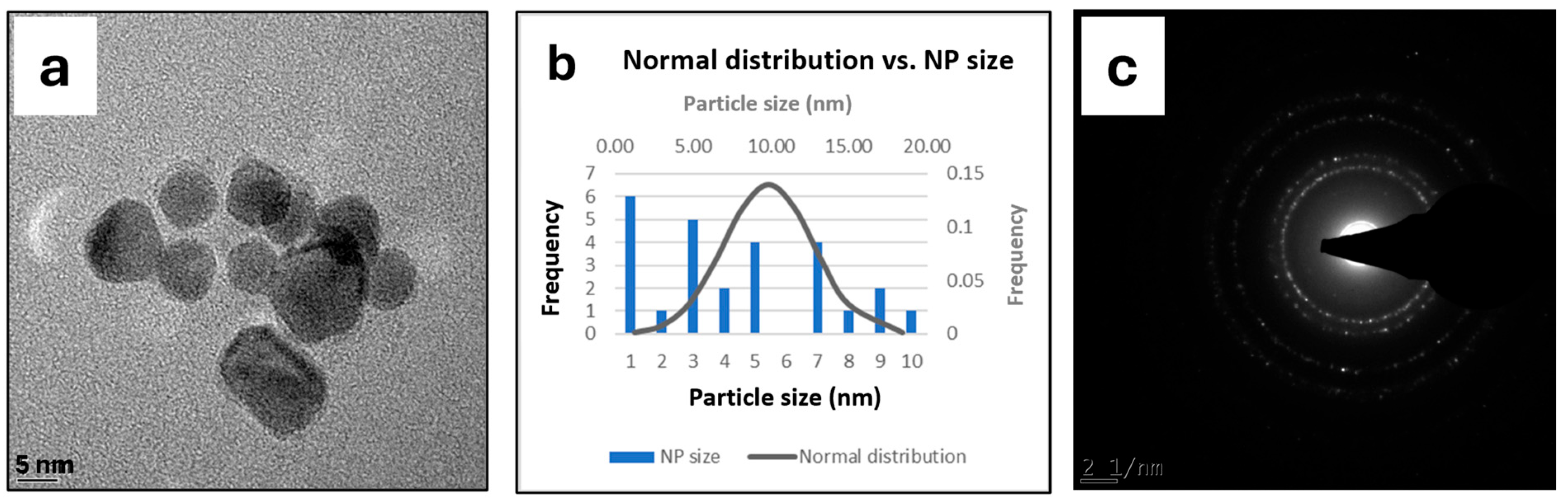
2.1. The Synthesis and Characterization of the PdNP Nanocatalyst
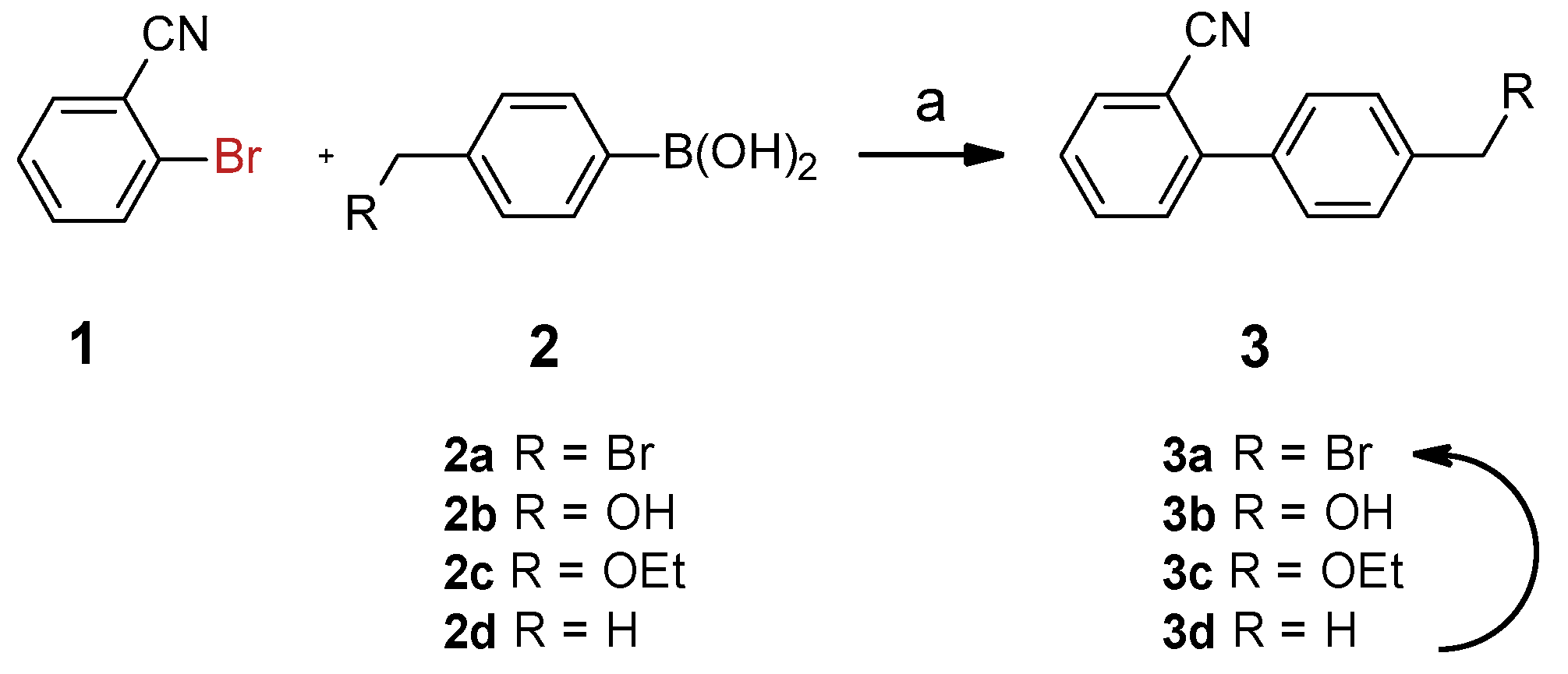
2.2. Synthesis of Biphenylcarbonitrile (3)
2.2.1. Reaction Kinetics
2.2.2. Catalyst Recyclability
2.3. Synthesis of 2-(4-Bromomethylphenyl)benzonitrile (4)
2.4. Synthesis of Losartan Intermediates 6–8
2.5. Synthesis of Tetrazole (Losartan, 9)
3. Discussion
4. Materials and Methods
4.1. General Experimental Details
4.2. Synthetic Procedures
4.2.1. Collection of Sargassum incisifolium and Preparation and Characterization of Aqueous Extract
4.2.2. Synthesis of Palladium Nanoparticles
4.2.3. The General Procedure for the Suzuki–Miyaura Coupling Reactions (Scheme 2: 3a–3d)
- Compound 3a: 1H NMR (400 MHz, CDCl3) δ 7.76 (dd, J = 7.7, 1.4 Hz, 1H), 7.64 (td, J = 7.7, 1.4 Hz, 2H), 7.57–7.48 (m, 7H), 7.45 (td, J = 7.6, 1.3 Hz, 2H), 4.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 144.6, 138.2, 138.1, 133.7, 132.9, 129.9, 129.4, 129.1, 127.8, 118.5, 111.1, 32.8; MS m/z (TOF MS): 192.08 [(M-HBr)+]. Yield: 97.1%.
- Compound 3b: 1H NMR (400 MHz, CDCl3) δ 7.72 (dd, J = 7.7, 1.4 Hz, 1H), 7.61 (td, J = 7.7, 1.4 Hz, 1H), 7.54–7.37 (m, 7H), 4.67 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 145.1, 141.5, 137.0, 133.6, 132.8, 129.86, 128.7, 127.4, 127.0, 118.7, 110.8, 64.2, 50.2, 15.0; MS m/z (TOF MS): 192.08 [(M-HOH)+]. Yield: 96.0%.
- Compound 3c: 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 7.8, 1.4 Hz, 1H), 7.62 (td, J = 7.7, 1.4 Hz, 1H), 7.57–7.37 (m, 6H), 4.56 (s, 2H), 3.58 (q, J = 7.0 Hz, 2H), 1.27 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 145.3, 139.3, 137.3, 133.8, 132.8, 130.1, 128.8, 128.1, 127.6, 118.8, 111.2, 72.3, 66.0, 15.3; MS m/z (TOF MS): 192.08 [(M-HOCH2CH3)+]. Yield: 98.2%.
- Compound 3d: 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 7.9, 1.4 Hz, 1H), 7.62 (td, J = 7.7, 1.4 Hz, 1H), 7.52–7.38 (m, 4H), 7.33–7.27 (m, 2H), 2.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 145.4, 138.6, 135.2, 133.6, 132.7, 129.9, 129.4, 128.5, 127.2, 118.8, 111.1, 21.2; MS m/z (TOF MS): 192.08 [(M-H)+]. Yield: 98.0%.
4.2.4. Recyclability Studies
4.2.5. Synthesis of 2-[4-(Bromomethyl)phenyl]benzonitrile (4)
4.2.6. Synthesis of 2-Butyl-4-chloro-1H-imidazole-5-carbaldehyde (6)
4.2.7. Synthesis of 2-[4-[(2-Butyl-4-chloro-5-formyl-imidazol-1-yl)methyl]phenyl]benzonitrile (7)
4.2.8. Synthesis of 2-N-butyl-4-chloro-l-[2′-cyanobiphenyl-4-yl) methyl]-5-(hydroxymethyl)-imidazole (8)
4.2.9. Synthesis of Losartan (9)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.C.; Reckelhoff, J.F. State-of-the-Art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension 1999, 34, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Strawn, W.B. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 2006, 98, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L., Jr.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T., Jr.; et al. Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003, 42, 1206–1252. [Google Scholar]
- Yang, R.; Smolders, I.; Dupont, A. Blood pressure and renal hemodynamic effects of angiotensin fragments. Hypertens. Res. 2011, 34, 674–683. [Google Scholar] [CrossRef]
- Xei, M.H.; Liu, F.Y.; Wong, P.C.; Timmermans, P.B.M.W.M.; Cogan, M.G. Proximal nephron and renal effects of DuP 753, a nonpeptide angiotensin II receptor antagonist. Kidney Int. 1990, 38, 473. [Google Scholar]
- Gallo, G.; Volpe, M.; Rubattu, S. Angiotensin receptor blockers in the management of hypertension: A real-world perspective and current recommendations. Vasc. Health Risk Manag. 2022, 18, 507–515. [Google Scholar] [CrossRef]
- Madasu, S.B.; Vekariya, N.A.; Koteswaramma, C.; Islam, A.; Sanasi, P.D.; Korupolu, R.B. An efficient, commercially viable, and safe process for preparation of losartan potassium, an angiotensin II receptor antagonist. Org. Process Res. Dev. 2012, 16, 2025–2030. [Google Scholar] [CrossRef]
- Hilgers, K.F.; Mann, J.F.E. ACE inhibitors versus AT(1) receptor antagonists in patients with chronic renal disease. J. Am. Soc. Nephrol. 2002, 13, 1100–1108. [Google Scholar] [CrossRef]
- Larsen, R.D.; King, A.O.; Chen, C.Y.; Corley, E.G.; Foster, B.S.; Roberts, F.E.; Yang, C.; Lieberman, D.R.; Reamer, R.A.; Tschaen, D.M.; et al. Efficient Synthesis of Losartan, A Nonpeptide Angiotensin II Receptor Antagonist. J. Org. Chem. 1994, 59, 6391–6394. [Google Scholar] [CrossRef]
- Smith, G.B.; Dezeny, G.C.; Hughes, D.L.; King, A.O.; Verhoeven, T.R. Mechanistic Studies of the Suzuki Cross-Coupling Reaction. J. Org. Chem. 1994, 59, 8151–8156. [Google Scholar] [CrossRef]
- Demko, Z.P.; Sharpless, K.B. Preparation of 5-Substituted 1H-Tetrazoles from Nitriles in Water. J. Org. Chem. 2001, 66, 7945–7950. [Google Scholar] [CrossRef] [PubMed]
- Carini, D.J.; Duncia, J.V.; Aldrich, P.E.; Chiu, A.T.; Johnson, A.L.; Pierce, M.E.; Price, W.A.; Santella III, J.B.; Wells, G.J.; Wexler, R.R.; et al. Nonpeptide angiotensin II receptor antagonists: The discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives. J. Med. Chem. 1991, 34, 2525–2547. [Google Scholar] [CrossRef]
- Pandarus, V.; Desplantier-Giscard, D.; Gingras, G.; Béland, R.; Ciriminna, F.; Pagliaro, M. Greening the Valsartan Synthesis: Scale-up of Key Suzuki–Miyaura Coupling over Silia Cat DPP-Pd. Org. Process Res. Dev. 2013, 17, 1492–1497. [Google Scholar] [CrossRef]
- Himo, F.; Demko, Z.P.; Noodleman, L.; Sharpless, K.B. Mechanisms of tetrazole formation by addition of Azide to nitriles. J. Am. Chem. Soc. 2002, 124, 12210–12216. [Google Scholar] [CrossRef]
- Georgiou, N.; Gkalpinos, V.K.; Katsakos, S.D.; Vassiliou, S.; Tzakos, A.G.; Mavromoustakos, T. Rational design and synthesis of AT1R antagonists. Molecules 2021, 26, 2927. [Google Scholar] [CrossRef]
- Shuangxia, F.; Zheng, G.; Yelv, T.; Hui, L.; Guofang, J. An efficient and green synthetic route to losartan. J. Chem. Res. 2015, 39, 451–454. [Google Scholar] [CrossRef]
- European Medicines Agency. Scientific Review of the Risk of Nitrosamine Impurities in Human Medicines. 9 July 2020. Available online: https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/referral-procedures-human-medicines/nitrosamine-impurities/scientific-review-risk-nitrosamine-impurities-human-medicines (accessed on 11 March 2025).
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford Academic: Oxford, UK, 2000; pp. 11–20. [Google Scholar]
- Sheldon, R.A. The E Factor: 25 years on. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Kadam, S.U.; Tiwari, B.K.; O’Donnell, C.P. Extraction, structure and biofunctional activities of laminarin from brown algae. Int. J. Food Sci. Technol. 2015, 50, 24–31. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd metal catalysts for cross-couplings and related reactions in the 21st century: A critical review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gelbaum, C.; Heaner IV, W.L.; Fisk, J.; Jaganathan, A.; Holden, B.; Pollet, P.; Liotta, C.L. Palladium-catalyzed Suzuki reactions in water with no added ligand: Effects of reaction scale, temperature, pH of aqueous phase, and substrate structure. Org. Process Res. Dev. 2016, 20, 1489–1499. [Google Scholar] [CrossRef]
- Mmola, M.; Roes-Hill, M.L.; Durrell, K.; Bolton, J.J.; Sibuyi, N.; Meyer, M.E.; Beukes, D.R.; Antunes, E. Enhanced Antimicrobial and Anticancer Activity of Silver and Gold Nanoparticles Synthesised Using Sargassum incisifolium Aqueous Extracts. Molecules 2016, 21, 1633. [Google Scholar] [CrossRef] [PubMed]
- Mubaiwa, B.; Lerata, M.S.; Sibuyi, N.R.S.; Meyer, M.; Samaai, T.; Bolton, J.J.; Antunes, E.M.; Beukes, D.R. Green Synthesized sAuNPs as a Potential Delivery Platform for Cytotoxic Alkaloids. Materials 2023, 16, 1319. [Google Scholar] [CrossRef]
- Seki, M. An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012, 44, 3231–3237. [Google Scholar] [CrossRef]
- Marcos, C.F.; Neo, A.G.; Díaz, J.; Martínez-Caballero, S. A Safe and Green Benzylic Radical Bromination Experiment. J. Chem. Ed. 2020, 97, 582–585. [Google Scholar] [CrossRef]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Visible light induced ‘on water’ benzylic bromination with N-bromosuccinimide. Tetrahedron Lett. 2006, 47, 1097–1099. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Liu, N.; Qiu, J. A simple and efficient approach for the palladium-catalyzed ligand-free Suzuki reaction in water. Green Chem. 2012, 14, 2999–3003. [Google Scholar] [CrossRef]
- Siamaki, A.R.; Khder, A.E.R.S.; Abdelsayed, V.; El-Shall, M.S.; Gupton, B.F.J. Microwave-assisted synthesis of palladium nanoparticles supported on graphene: A highly active and recyclable catalyst for carbon–carbon cross-coupling reactions. J. Catal. 2011, 279, 1–11. [Google Scholar] [CrossRef]
- Remuzzi, G. Use of an Angiotensin II Receptor Antagonist for the Preparation of Drugs to Increase the Survival Rate of Renal Transplant Patients. U.S. Patent 6,576,652, 10 June 2003. [Google Scholar]
- Miller, R.A.; Hoerrner, R.S. Iodine as a Chemoselective Reoxidant of TEMPO: Application to the Oxidation of Alcohols to Aldehydes and Ketones. Org. Lett. 2003, 5, 285–287. [Google Scholar] [CrossRef]
- Topiwala, S.; Fan, W.; Hines, C.J.; Folk, W.R.; Ercal, N. Antioxidative potential of Sutherlandia frutescens and its protective effects against oxidative stress in various cell cultures. BMS Complement. Altern. Med. 2014, 14, 271. [Google Scholar] [CrossRef] [PubMed]
- Buskes, M.J.; Blanco, M.-J. Impact of Cross-Coupling Reactions in Drug Discovery and Development. Molecules 2020, 25, 3493. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Biaryl synthesis via Ullmann-type coupling reactions. Chem. Sci. 2008, 1, 13–31. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Time/h | Conversion, % a | TON b |
|---|---|---|---|
| PdNPs (0.3 mol%) | 24 | 94 | 37,170 |
| PdNPs (1 mol%) | 3 | 91 | 8897 |
| PdNPs (1 mol%) * | 102 * | 100 * | 10,877 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antunes, E.M.; Adegoke, Y.A.; Mgwigwi, S.; Bolton, J.J.; Malan, S.F.; Beukes, D.R. Balancing Yields and Sustainability: An Eco-Friendly Approach to Losartan Synthesis Using Green Palladium Nanoparticles. Molecules 2025, 30, 2314. https://doi.org/10.3390/molecules30112314
Antunes EM, Adegoke YA, Mgwigwi S, Bolton JJ, Malan SF, Beukes DR. Balancing Yields and Sustainability: An Eco-Friendly Approach to Losartan Synthesis Using Green Palladium Nanoparticles. Molecules. 2025; 30(11):2314. https://doi.org/10.3390/molecules30112314
Chicago/Turabian StyleAntunes, Edith M., Yusuf A. Adegoke, Sinazo Mgwigwi, John J. Bolton, Sarel F. Malan, and Denzil R. Beukes. 2025. "Balancing Yields and Sustainability: An Eco-Friendly Approach to Losartan Synthesis Using Green Palladium Nanoparticles" Molecules 30, no. 11: 2314. https://doi.org/10.3390/molecules30112314
APA StyleAntunes, E. M., Adegoke, Y. A., Mgwigwi, S., Bolton, J. J., Malan, S. F., & Beukes, D. R. (2025). Balancing Yields and Sustainability: An Eco-Friendly Approach to Losartan Synthesis Using Green Palladium Nanoparticles. Molecules, 30(11), 2314. https://doi.org/10.3390/molecules30112314