
Catalyst-Free Spontaneous Aza-Mannich/Lactamization Cascade Reaction: Easy Access to Polycyclic δ-Lactams



Abstract

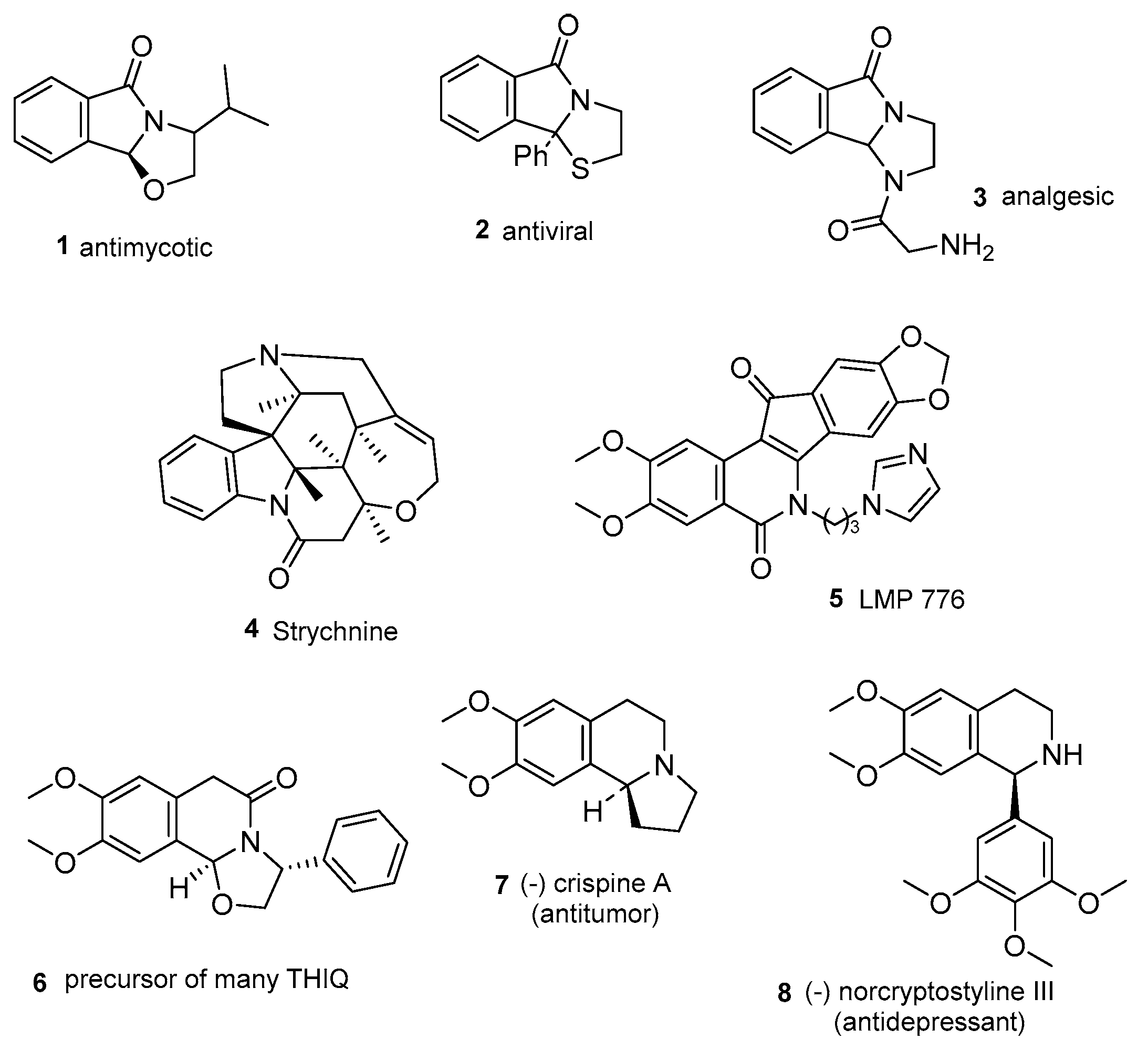
1. Introduction
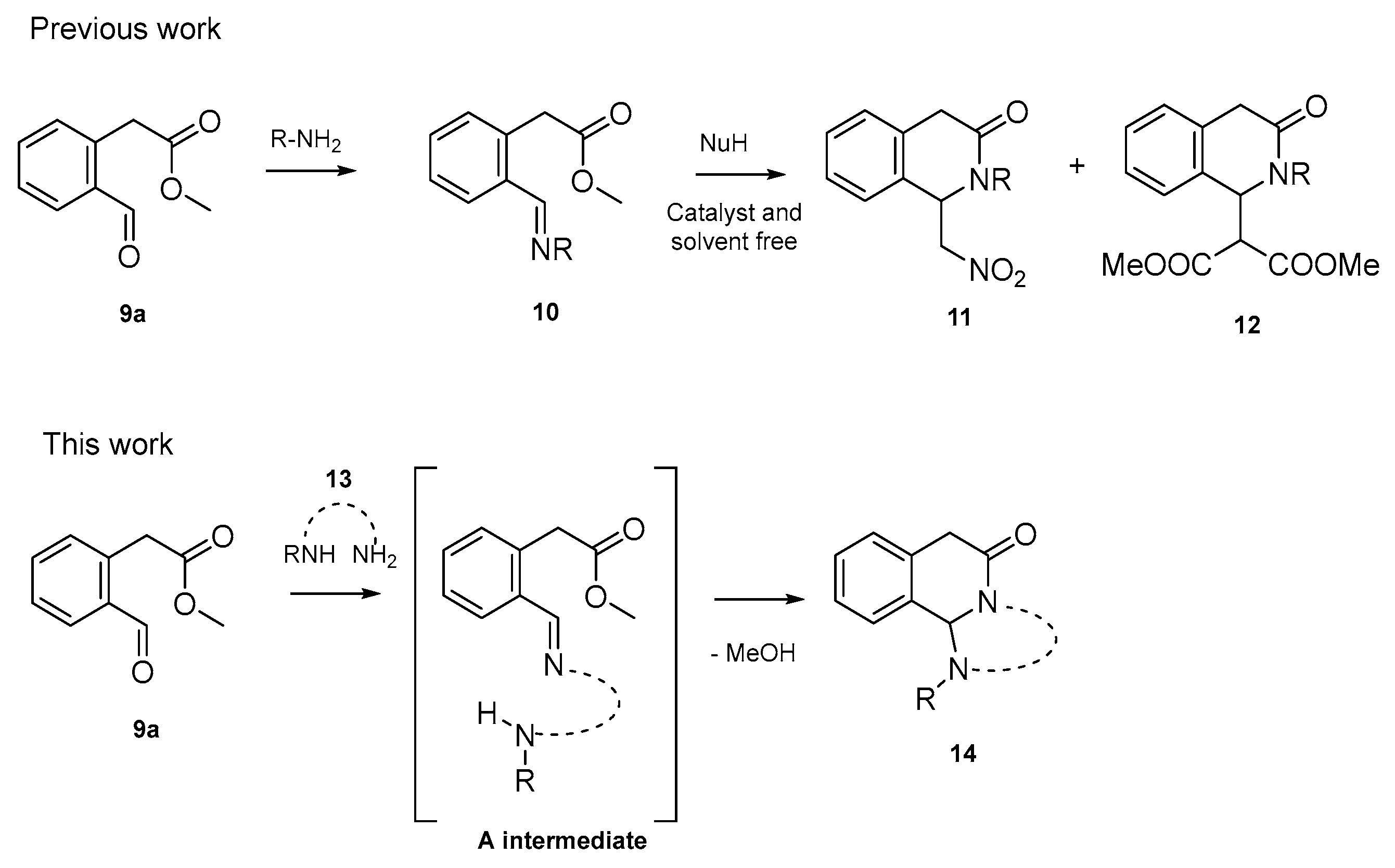
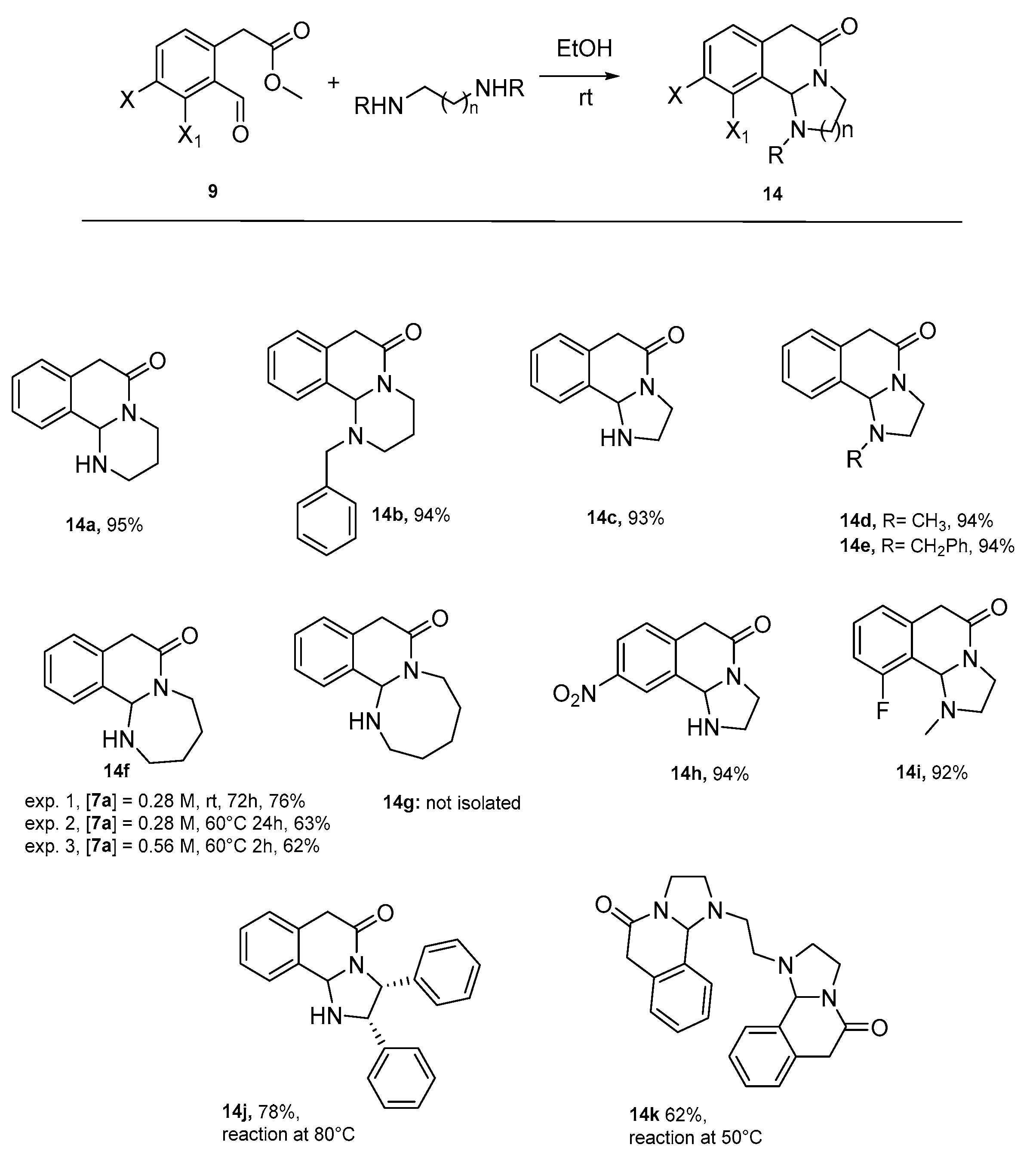
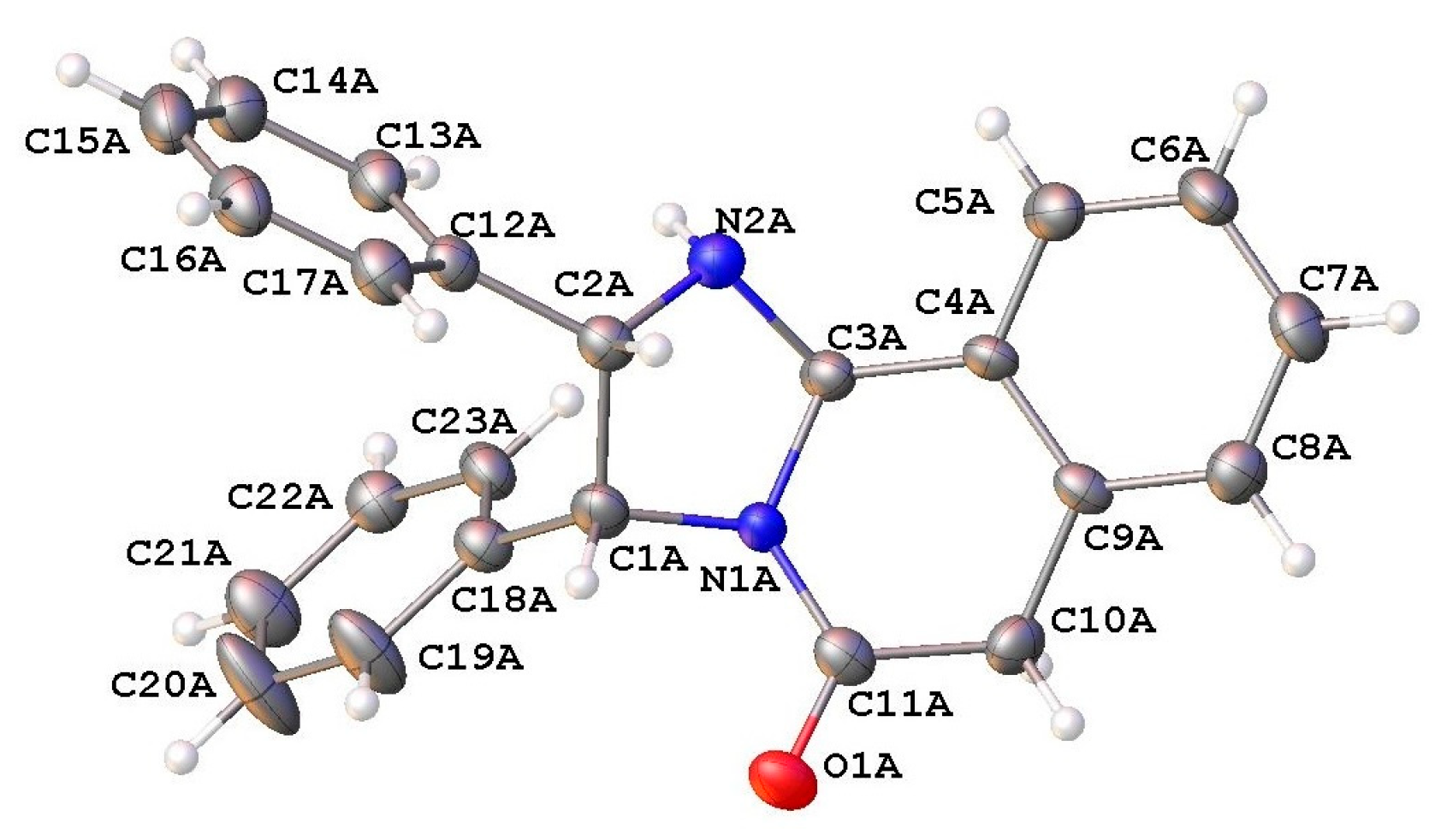
2. Results
3. Materials and Methods
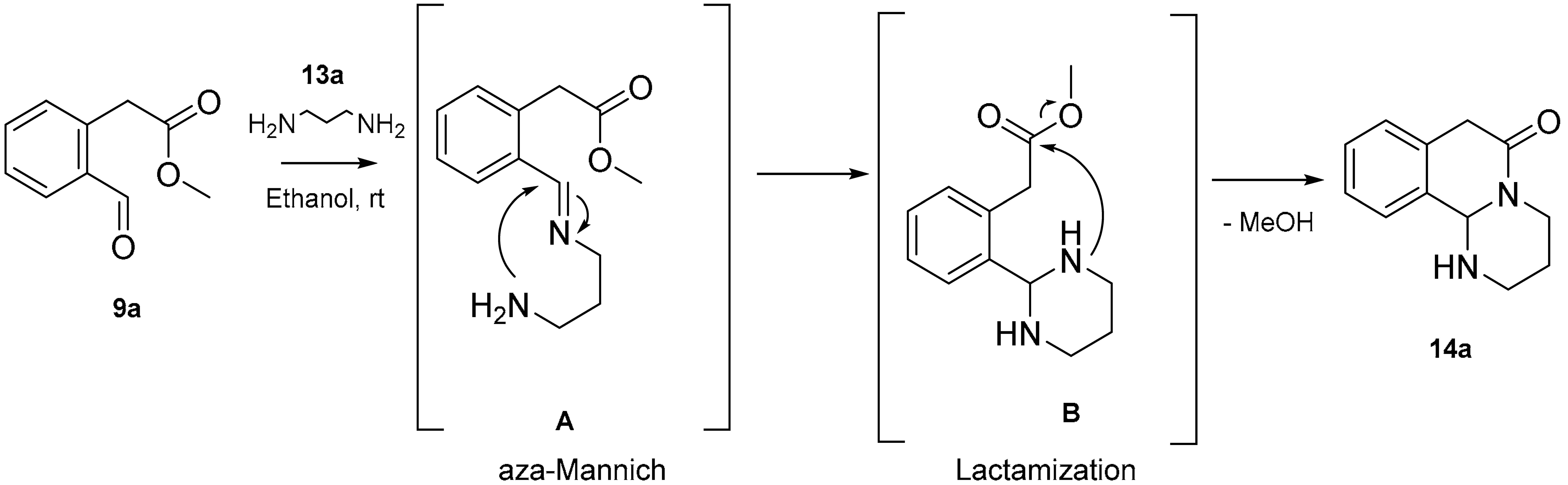
3.1. General Procedure for the Synthesis of Tricyclic Lactams 14a–14k
- 1,2,3,4,7,11b-hexahydro-6H-pyrimido [2,1-a]isoquinolin-6-one 14a
- 1-benzyl-1,2,3,4,7,11b-hexahydro-6H-pyrimido [2,1-a]isoquinolin-6-one 14b
- 2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14c
- 1-methyl-2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14d
- 1-benzyl-2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14e
- 2,3,4,5,8,12b-hexahydroazepino [2,1-a]isoquinolin-7(1H)-one 14f
- 9-nitro-2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14h
- 10-fluoro-1-methyl-2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14i
- 2,3-diphenyl-2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14j
- 1,1′-(ethane-1,2-diyl)bis(2,3,6,10b-tetrahydroimidazo [2,1-a]isoquinolin-5(1H)-one 14k
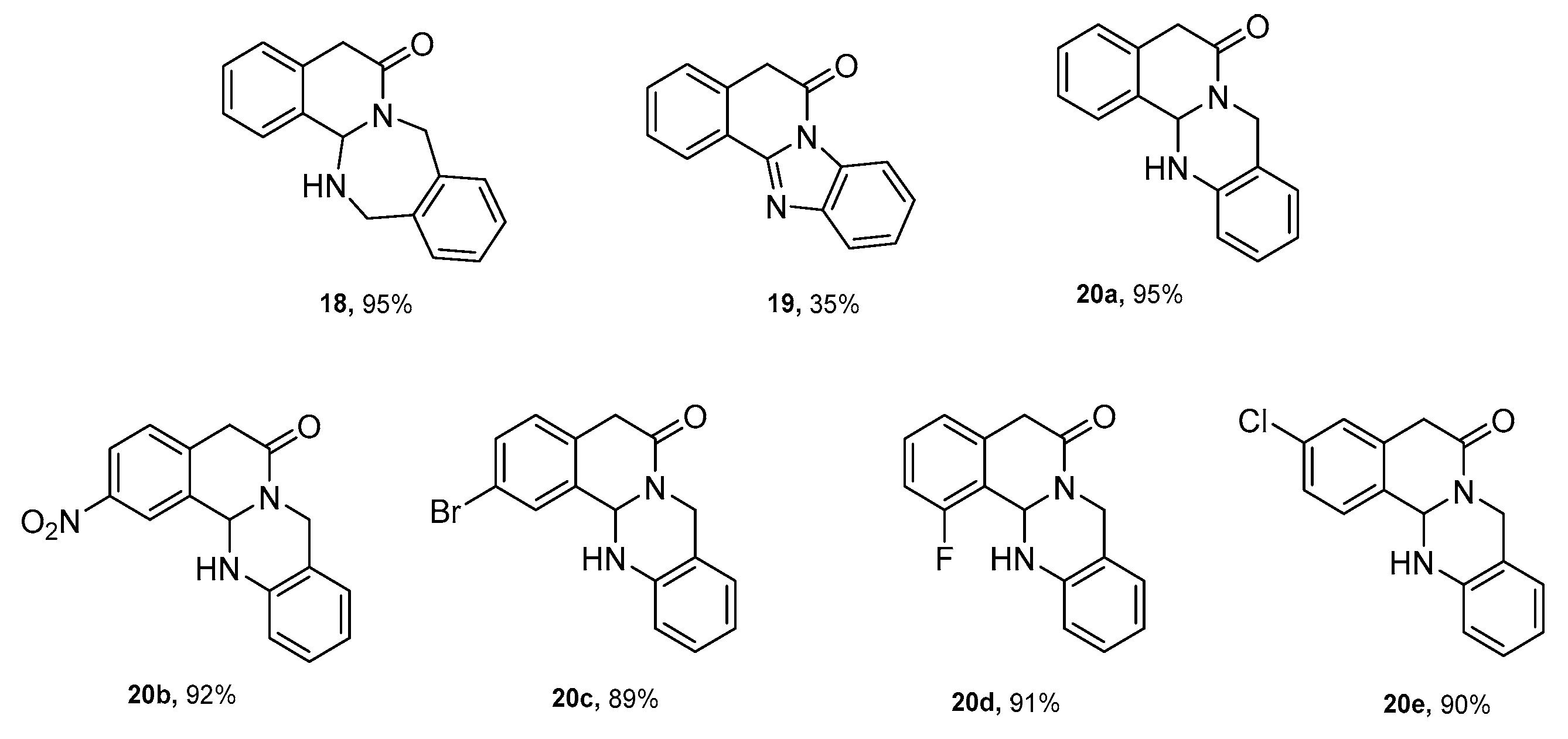
3.2. General Procedure for the Synthesis of Tetracyclic Lactams 18, 19, 20a–20e
- 8,13,14,14a-tetrahydrobenzo [5,6][1,3]diazepino [2,1-a]isoquinolin-6(5H)-one 18
- benzo [4,5]imidazo [2,1-a]isoquinolin-6(5H)-one 19
- 5,8,13,13a- tetrahydro -6H-isoquinolino [1,2-b]quinazolin-6-one 20a
- 2-nitro-5,8,13,13a-tetrahydro-6H-isoquinolino [1,2-b]quinazolin-6-one 20b
- 2-bromo-5,8,13,13a-tetrahydro-6H-isoquinolino [1,2-b]quinazolin-6-one 20c
- 1-fluoro-5,8,13,13a-tetrahydro-6H-isoquinolino [1,2-b]quinazolin-6-one 20d
- 3-chloro-5,8,13,13a-tetrahydro-6H-isoquinolino [1,2-b]quinazolin-6-one 20e
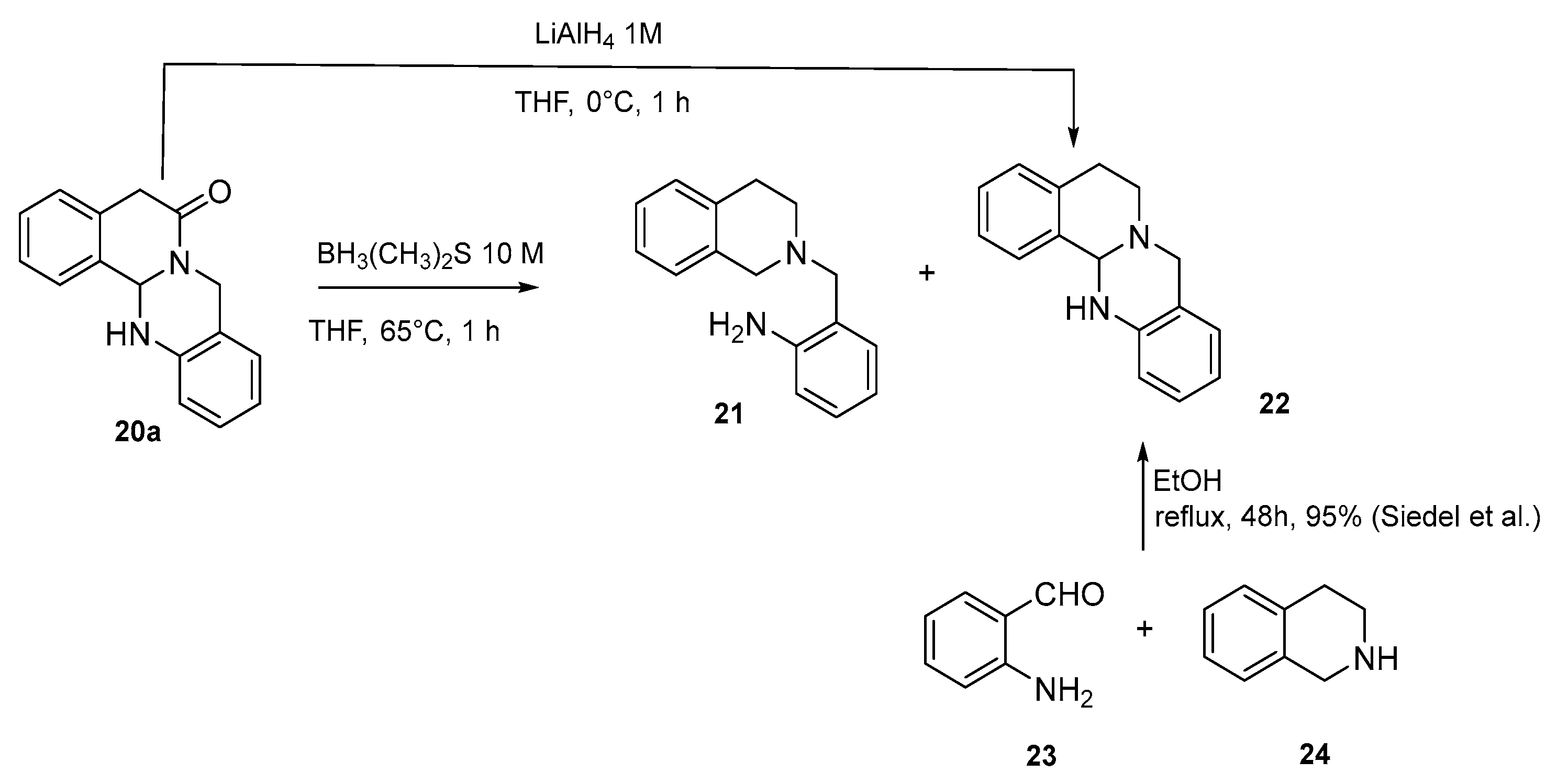
3.3. Follow-Up Chemistry
3.3.1. Reduction Using BH3(CH3)2S
- 2-((3,4-dihydroisoquinolin-2(1H)-yl)methyl)aniline 21
3.3.2. Reduction Using LiAlH4
- 5,8,13,13a-tetrahydro-6H-isoquinolino [1,2-b]quinazoline 22 [30]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Schyf, C.J.; Geldenhuys, W.J. Polycyclic Compounds: Ideal Drug Scaffolds for the Design of Multiple Mechanism Drugs? Neurotherapeutics 2009, 6, 175–186. [Google Scholar] [CrossRef]
- Blumer, M. Polycyclic Aromatic Compounds in Nature. Sci. Am. 1976, 234, 34–45. [Google Scholar] [CrossRef]
- Breytenbach, J.C.; Van Dyk, S.; Van den Heever, I.; Allin, S.M.; Hodkinson, C.C.; Northfield, C.C.; Page, M.I. synthesis and Antimicrobial Activity of Some Isoindolin-1-ones Derivatives. Bioorg. Med. Chem. Lett. 2000, 10, 1629–1631. [Google Scholar] [CrossRef]
- Mertens, A.; Zilch, H.; König, B.; Schäfer, W.; Poll, T.; Wolfgang, K.; Seidel, H.; Leser, U.; Leinert, H. Selective Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. New 2,3-Dihydrothiazolo[2,3-a]isoindol-5(9bH)-ones and Related Compounds with Anti-HIV-Activity. J. Med. Chem. 1993, 36, 2526–2535. [Google Scholar] [CrossRef] [PubMed]
- Allin, S.M.; Vaidya, D.G.; Page, M.I.; Slawin, A.M.Z. Highly diastereoselective synthesis of 2,3-dihydro-9bH- thiazolo[2,3- a]isoindolin-5-ones. ARKIVOC 2000, ii, 151–157. [Google Scholar] [CrossRef]
- Katritzky, A.R.; He, H.-Y.; Verma, A.K. Stereoselective syntheses of chiral (3S,9bS)-1,2,3,9b-tetrahydro-5H-imidazo[2,1-a]isoindol-5-ones. Tetrahedron Asymmetry 2002, 13, 933–938. [Google Scholar] [CrossRef]
- Sharma, R.K. Spinal Poisons. In Concise Textbook of Forensic Medicine and Toxicology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 306–308. [Google Scholar]
- Cinelli, M.A.; Reddy, P.V.N.; Lv, P.-C.; Liang, J.-H.; Chen, L.; Agama, K.; Pommier, Y.; van Breemen, R.B.; Cushman, M. Identification, Synthesis, and Biological Evaluation of Metabolites of the Experimental Cancer Treatment Drugs Indotecan (LMP400) and Indimitecan (LMP776) and Investigation of Isomerically Hydroxylated Indenoisoquinoline Analogues as Topoisomerase I Poisons. J. Med. Chem. 2012, 55, 10844–10862. [Google Scholar] [PubMed]
- Amat, M.; Elias, V.; Llor, N.; Subrizi, F.; Molins, E.; Bosch, J. A General Methodology for the Enantioselective Synthesis of 1-Substituted Tetrahydroisoquinoline Alkaloids. Eur. J. Org. Chem. 2010, 2010, 4017–4026. [Google Scholar] [CrossRef]
- Kim, A.N.; Ngamnithiporn, A.; Du, E.; Stoltz, B.M. Recent Advances in the Total Synthesis of the Tetrahydroisoquinoline Alkaloids (2002–2020). Chem. Rev. 2023, 123, 9447–9496. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Wu, L.; Ren, A.; Lu, P.; Wang, Y. Preparation of 4-Diazoisoquinolin-3-ones via Dimroth Rearrangement and Their Extension to 4-Aryltetrahydroisoquinolin-3-ones. Org. Lett. 2020, 22, 26–30. [Google Scholar] [CrossRef]
- Li, L.; Zhou, B.; Wang, Y.-H.; Shu, C.; Pan, Y.-F.; Lu, X.; Ye, L.-W. Zinc-Catalyzed Alkyne Oxidation/C-H Functionalization: Highly Site-Selective Synthesis of Versatile Isoquinolones and b-Carbolines. Angew. Chem. Int. Ed. 2015, 54, 8245–8249. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.E.; dos Santos, E.N. Tandem catalysis: A taxonomy and illustrative review. Coord. Chem. Rev. 2004, 248, 2365–2379. [Google Scholar] [CrossRef]
- Hayashi, Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. [Google Scholar] [CrossRef]
- Dutta, S.; Chatterjee, S.; Al-Thabaiti, S.A.; Bawaked, S.; Mokhtar, M.; Maiti, D. C–H activation: A strategic approach toward lactams using transition metals. Chem Catal. 2022, 5, 1046–1083. [Google Scholar] [CrossRef]
- Chen, C.; Ni, C.; Song, J.-H.; Ding, L.-Y.; Zhang, X.-X.; Guo, H.; Wang, K.; Chen, Z.; Zhu, B. Pd/NBE-Catalyzed One-Pot Modular Synthesis of Polycyclic Fused δ-Lactams and Investigation of Room-Temperature Phosphorescence. ACS Catal. 2024, 14, 12181–12191. [Google Scholar] [CrossRef]
- Niu, Y.-N.; Wang, K.-Y.; Han, F.-Y.; Xia, X.-F. Recent developments for the synthesis of the dihydroisoquinolin-1 (2H)-ones via cyclization of N-allylbenzamides. Tetrahedron 2025, 174, 134497. [Google Scholar] [CrossRef]
- Kant, K.; Naik, P.; Patel, C.K.; Some, S.; Banerjee, S.; Aljaar, N.; Atta, A.K.; Malakar, C.C. Organocatalytic Approaches Towards the Synthesis of Asymmetric Tetrahydroquinoline (THQ) Derivatives. Synthesis 2025, 57, 296–330. [Google Scholar]
- Di Mola, A.; Sadeq Mousavi, M.; Simeone, L.; Pierri, G.; Massa, A. Regio- and Diastereoselective One-pot Double Cascade Hybrid Formation/[3+3] Spirocyclization for the Synthesis of 3-Spiropiperidines Under Phase Transfer Conditions. Adv. Synth. Cat. 2025, 366, e202401230. [Google Scholar] [CrossRef]
- Di Mola, A.; Nicastro, G.; Serusi, L.; Filosa, R.; Waser, M.; Massa, A. Scalable (Enantioselective) Syntheses of Novel 3-Methylated Analogs of Pazinaclone, (S)-PD172938 and Related Biologically Relevant Isoindolinones. Molecules 2022, 27, 5647. [Google Scholar] [CrossRef] [PubMed]
- Serusi, L.; Palombi, L.; Pierri, G.; Di Mola, A.; Massa, A. Asymmetric cascade Aza-Henry/lactamization reaction in the highly enantioselective organocatalytic synthesis of 3-(nitromethyl)isoindolin-1-ones from α-amido sulfones. J. Org. Chem. 2022, 87, 8420–8428. [Google Scholar] [CrossRef]
- Di Mola, A.; De Piano, F.; Serusi, L.; Pierri, G.; Palombi, L.; Massa, A. Asymmetric Organocatalytic Mannich Reaction in the Synthesis of Hybrid Isoindolinone-Pyrazole and Isoindolinone-Aminal from Functionalized α-Amidosulfone. Int. J. Mol. Sci. 2023, 24, 5783. [Google Scholar] [CrossRef] [PubMed]
- Serusi, L.; Di Mola, A.; Massa, A. A facile access to 1-substituted and unsubstituted 3-isoquinolinones via Mannich or Sn2 initiated cascade reactions under catalyst-free conditions. RSC Adv. 2023, 13, 6557–6563. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; Suneja, A.; Bisai, V.; Singh, V.K. Approach to Isoindolinones, Isoquinolinones, and THIQs via Lewis Acid-Catalyzed Domino Strecker-Lactamization/Alkylations. Org. Lett. 2016, 18, 634–637. [Google Scholar] [CrossRef]
- Karmakar, R.; Suneja, A.; Bisai, V.; Singh, V.K. Ni(II)-Catalyzed Highly Stereo- and Regioselective Syntheses of Isoindolinones and Isoquinolinones from in Situ Prepared Aldimines Triggered by Homoallylation/Lactamization Cascade. Org. Lett. 2015, 17, 5650–5653. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, Y.-X.; Nie, X.-D.; Si, C.-M.; Sun, X.; Wei, B.-G. Approach to Chiral 1-Substituted Isoquinolone and 3-Substituted Isoindolin-1-one by Addition–Cyclization Process. J. Org. Chem. 2018, 83, 9879–9889. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, Z.; Zhao, C. Recent progress in the construction of eight-membered nitrogen-heterocycles. New J. Chem. 2024, 48, 4645–4669. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/structures (accessed on 18 June 2025).
- Mai, S.; Yixin, L.; Xianyun, H.; Zhenghao, S.; Li, B.; Lu, Y.; Song, Q. Diversity-oriented synthesis of imidazo[2,1-a]isoquinolines. Chem. Commun. 2018, 54, 10240–10243. [Google Scholar] [CrossRef]
- Zhang, C.; Kanta De, C.; Mal, R.; Seidel, D. α-Amination of Nitrogen Heterocycles: Ring-Fused Aminals. J. Am. Chem. Soc. 2008, 130, 416–417. [Google Scholar] [CrossRef]
- Le Gall, E.; Malassenea, R.; Toupetb, L.; Hurvoisa, J.P.; Moinet, C. Synthesis of [1,2-b] and [2,1-b]Quinazoline Ring Systems via Suitably Substituted a-Cyanoamines. Synlett 1999, 9, 1383–1386. [Google Scholar] [CrossRef]
- Stambach, J.F.; Kanmacher, I.; Jung, L.; Schott, C.; Heitz, C.; Stoclet, J.C. 2-(Aminobenzyl)-1,2,3,4-tetrahydroisoquinolines: A new class of a2-adrenergic receptor antagonists. Eur. J. Med. Chem. 1993, 28, 427–432. [Google Scholar] [CrossRef]
- N’Ta Ambeu, C.; Le Guével, R.; Corlu, A.; Akhanovna Mamyrbekova, J.; Bazureau, J.P. A practical multi-step synthesis of ethyl N-functionalized β-amino benzimidazole acrylate derivatives as promising cytotoxic agents. Mol. Divers. 2018, 22, 685–708. [Google Scholar] [CrossRef] [PubMed]
- Gigant, N.; Claveau, E.; Bouyssou, P.; Gillaizeau, I. Diversity-oriented synthesis of polycyclic diazinic scaffolds. Org. Lett. 2012, 14, 844–847. [Google Scholar] [CrossRef] [PubMed]
- APEX3, version 2015.5-2; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT, version 8.34A; Bruker AXS Inc.: Madison, WI, USA, 2013.
- SADABS, version 2014/5; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Entry | [M] 9a | Time | Yield % a |
| 1 | 0.56 | 2 h | 50% b |
| 2 | 0.56 | 2 h | 52% c |
| 3 | 0.56 | 2 h | 73% d |
| 4 | 0.28 | 2 h | 74% d |
| 5 | 0.56 | 24 h | 95 % d |
| 6 | 0.28 | 24 h | 88% d |
| 7 | 0.14 | 24 h | 57% d |
| 8 | 0.56 | 2 h | 71% d,e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Mola, A.; Vietri, C.; Tedesco, C.; Massa, A. Catalyst-Free Spontaneous Aza-Mannich/Lactamization Cascade Reaction: Easy Access to Polycyclic δ-Lactams. Molecules 2025, 30, 2702. https://doi.org/10.3390/molecules30132702
Di Mola A, Vietri C, Tedesco C, Massa A. Catalyst-Free Spontaneous Aza-Mannich/Lactamization Cascade Reaction: Easy Access to Polycyclic δ-Lactams. Molecules. 2025; 30(13):2702. https://doi.org/10.3390/molecules30132702
Chicago/Turabian StyleDi Mola, Antonia, Caterina Vietri, Consiglia Tedesco, and Antonio Massa. 2025. "Catalyst-Free Spontaneous Aza-Mannich/Lactamization Cascade Reaction: Easy Access to Polycyclic δ-Lactams" Molecules 30, no. 13: 2702. https://doi.org/10.3390/molecules30132702
APA StyleDi Mola, A., Vietri, C., Tedesco, C., & Massa, A. (2025). Catalyst-Free Spontaneous Aza-Mannich/Lactamization Cascade Reaction: Easy Access to Polycyclic δ-Lactams. Molecules, 30(13), 2702. https://doi.org/10.3390/molecules30132702