3.1. Chemistry
All non-aqueous reactions were carried out under an atmosphere of nitrogen in dried glassware unless otherwise noted. Solvents were dried and distilled according to standard protocols. Analytical thin-layer chromatography was performed with Silica gel 60PF254 (Merck KGaA, Darmstadt, Germany). Silica gel column chromatography was performed with Silica gel 60 (70–230 mesh, Kanto Co. Lit., Tokyo, Japan). All melting points were determined on micro-melting point apparatus MP-500D (Yanaco Technical Science Co., Ltd., Tokyo, Japan) and are uncorrected. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on JEOL JNM-ECZ400S (JEOL Ltd., Tokyo, Japan). Chemical shifts are reported relative to Me4Si (δ 0.00). Multiplicity is indicated by one or more of the following: s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); and br (broad). Carbon nuclear magnetic resonance (13C-NMR) spectra were recorded on a JEOL JNM-ECZ400S at 100 MHz. Chemical shifts are reported relative to CDCl3 (δ 77.0) and DMSO-d6 (δ 39.7). Infrared spectra were recorded with ATR method using a Shimadzu FTIR-8000 spectrophotometer (Shimadzu corporation, Kyoto, Japan) and DuraScop (Sensir Technologies, NC, USA). Low and high-resolution mass spectra were recorded on JEOL JMS-700 spectrometers (JEOL Ltd., Tokyo, Japan) by direct inlet system. The microwave assisted reaction was carried out at 180 W and 2450 MHz with Discover (CEM corporation, NC, USA).
3.1.1. 2-(3-Hydroxymethylquinolin-2-yl)ethynyl-3-[(methoxymethoxy)methyl]benzaldehyde (14a)
To a solution of 2-iodoquinoline 12 (95 mg, 0.33 mmol), CuI (6 mg, 0.033 mmol), Pd2(dba)3 (13 mg, 0.013 mmol) and iPr2NH (0.3 mL, 2.31 mmol) in THF (1.5 mL), a solution of 2-ethynylbenzaldehyde 13 (81 mg, 0.40 mmol) in THF (1.5 mL) was added. The reaction mixture was stirred at 100 °C for 10 min. After cooling to ambient temperature, the reaction mixture was filtered through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give the 2-alkynylbenzaldehyde 14a (124 mg, 86%) as a white solid. mp was 59–61 °C (EtOAc-hexane). IR (ATR) ν = 1685, 2190, and 3629 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.41 (s, 3H), 3.52 (br s, 1H), 4.79 (s, 2H), 5.00 (s, 2H), 5.06 (s, 2H), 7.54–7.60 (m, 2H), 7.74 (t, J = 7.8 Hz, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 8.25 (s, 1H), and 10.70 (s, 1H). 13C-NMR (400 MHz, CDCl3) δ 55.6, 62.8, 67.1, 85.6, 95.9, 98.2, 123.7, 127.5 (2C), 127.8, 127.9, 129.1, 129.4, 130.1, 133.8, 135.0, 135.2, 136.8, 141.8, 142.0, 147.6, and 191.5. MS m/z: 361 (M+). HRMS (EI): calcd for C22H19NO4 361.1314; found 361.1332.
3.1.2. 2-{[3-(tert-Butyldimethylsilyloxy)methyl]quinolin-2-yl}ethynyl-3-[(methoxymethoxy)methyl]benzaldehyde (14b)
A solution of alcohol 14a (120 mg, 0.33 mmol) in CH2Cl2 (2 mL) was dropwise added to a suspension of imidazole (56 mg, 0.82 mmol) in CH2Cl2 (1 mL) under ice cooling. After stirring at same temperature for 15 min, TBSCl (123 mg, 0.82 mmol) in CH2Cl2 (1 mL) was added to the reaction mixture and then was stirred at rt for 12 h. After quenching with H2O, the reaction mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give the silyl ether 14b (123 mg, 78%) as a yellow solid. mp was 74–75 °C (EtOAc-hexane). IR (ATR) ν = 1697, 2168 cm−1. 1H-NMR (400 MHz, CDCl3) δ 0.19 (s, 6H), 1.00 (s, 9H), 3.42 (s, 3H), 4.82 (s, 2H), 5.00 (s, 2H), 5.14 (s, 2H), 7.54–7.61 (m, 2H), 7.73 (t, J = 7.8 Hz, 1H), 7.84–7.88 (m, 2H), 7.95 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 8.33 (s, 1H), and 10.77 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ -5.3 (2C), 18.4, 25.9 (3C), 55.6, 62.2, 67.0, 85.7, 96.3, 97.9, 123.7, 126.7, 127.6 (2C), 127.7, 129.1, 129.4, 129.6, 132.7, 133.0, 135.9, 136.7, 140.3, 142.0, 147.3, and 191.2. MS m/z: 475 (M+). HRMS (EI): calcd for C28H33NO4Si 475.2179; found 475.2181.
3.1.3. 2-[3-(Acetoxymethyl)quinolin-2-yl]ethynyl-3-[(methoxymethoxy)methyl]benzaldehyde (14c)
A solution of alcohol 14a (30 mg, 0.083 mmol) in CH2Cl2 (1 mL) was dropwise added to a suspension of DMAP (12 mg, 0.10 mmol) in CH2Cl2 (1 mL) under ice cooling. After stirring at same temperature for 15 min, Ac2O (9 μL, 0.10 mmol) was added to the reaction mixture and then was stirred at rt for 1 h. After quenching with H2O, the reaction mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give the compound 14c (25 mg, 75%) as a yellow solid. mp was 110–111 °C (EtOAc-hexane). IR (ATR) ν = 1685, 1736, and 2198 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.17 (s, 3H), 3.43 (s, 3H), 4.84 (s, 2H), 5.01 (s, 2H), 5.53 (s, 2H), 7.57 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.78 (t, J = 7.8 Hz, 1H), 7.85–7.87 (m, 2H), 7.96 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.27 (s, 1H), and 10.77 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.9, 55.6, 63.5, 67.0, 85.7, 96.4, 97.9, 123.5, 126.6, 127.2, 127.7, 128.0, 129.3, 129.5, 130.3, 130.5, 132.9, 136.2, 136.8, 142.0, 142.2, 147.9, 170.6, and 191.4. MS m/z: 403 (M+). HRMS (EI): calcd for C24H21NO5 403.1420; found 403.1411.
3.1.4. 2-(3-Hydroxymethylquinolin-2-yl)ethynyl-3-[(methoxymethoxy)methyl]benzaldehyde oxime (15a)
A mixture of benzaldehyde 14a (100 mg, 0.25 mmol), NH2OH·HCl (34 mg, 0.50 mmol), and AcONa (40 mg, 0.50 mmol) in EtOH (5 mL) was stirred at rt for 2 h. After removal of solvent, the residue was diluted with H2O and then filtered off to give the oxime 15a (65 mg, 63%) as a yellow solid. Mp was 220–221 °C (EtOAc-hexane). IR (ATR) ν = 1736, 2191, 3444, and 3610 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 4.76 (s, 2H), 4.85 (s, 2H), 4.91 (d, J = 5.5 Hz, 3H), 5.66 (t, J = 5.5 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.78 (t, J = 7.8 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 8.03–8.06 (m, 2H), 8.44 (s, 1H), 8.68 (s, 1H), and 11.72 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 55.0, 60.3, 67.0, 86.7, 95.8, 96.7, 119.1, 123.9, 127.2, 127.7, 127.9, 128.4, 128.7, 129.7, 129.8, 133.0, 134.8, 136.9, 140.9, 141.2, 146.0, and 146.6. MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1441.
3.1.5. 2-[3-(Acetoxymethyl)quinolin-2-yl]ethynyl-3-[(methoxymethoxy)methyl]benzaldehyde oxime (15c)
The same procedure as above was carried out with 2-arylethynylbenzaldehyde 14c (96 mg, 0.23 mmol) to give the oxime 15c (78 mg, 81%) as a pink solid. mp was 138–139 °C (EtOAc-hexane). IR (ATR) ν = 1751, 2233, and 3629 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 2.12 (s, 3H), 3.29 (s, 3H), 4.74 (s, 2H), 4.84 (s, 2H), 5.47 (s, 2H), 7.53 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.82–7.87 (m, 2H), 8.05–8.08 (m, 2H), 8.49 (s, 1H), 8.65 (s, 1H), and 11.72 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 20.7, 55.0, 63.0, 67.0, 86.7, 95.7, 96.6, 118.8, 123.9, 126.8, 128.1 (2C), 128.5, 128.6, 129.9, 130.5, 130.8, 135.0, 136.1, 141.3, 141.8, 145.9, 147.1, and 170.3. MS m/z: 418 (M+). HRMS (EI): calcd for C24H22N2O5 418.1529; found 418.1533.
3.1.6. 3-(3-Hydroxymethylquinolin-2-yl)-5-[(methoxymethoxy)methyl]isoquinoline N-oxide (16a)
A solution of oxime 15a (65 mg, 0.16 mmol) in 1,2-dichlorobenzene (4 mL) was stirred at 80 °C for 20 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the N-oxide 16a (43 mg, 72%) as a white solid. mp was 239–240 °C (EtOAc-hexane). IR (ATR) ν = 1230, 3610 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.36 (s, 3H), 4.57 (s, 2H), 4.71 (s, 2H), 5.02 (s, 2H), 5.61 (br s, 1H), 7.63–7.67 (m, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.78 (t, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 7.8 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.27 (s, 1H), 8.40 (s, 1H), and 8.99 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 55.7, 63.7, 66.2, 95.8, 123.2, 125.5, 127.7, 127.8 (2C), 128.7, 129.4, 129.7, 130.0, 130.1 (2C), 133.8, 134.2, 137.8, 138.1, 146.4, 147.6, and 151.9 MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1417.
3.1.7. 2-{3-[(tert-Butyldimethylsilyloxy)methyl]quinolin-2-yl}-5-[(methoxymethoxy)methyl]isoquinoline N-oxide (16b)
A mixture of benzaldehyde 15b (43 mg, 0.090 mmol), NH2OH·HCl (12 mg, 0.18 mmol), and AcONa (11 mg, 0.14 mmol) in EtOH (3 mL) was stirred at rt for 2 h. The resulting mixture was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried with Na2SO4, and evaporated in vacuo. Next, 1,2-dichlorobenzene (2 mL) was added to the residue, and the mixture was stirred at 80 °C for 12 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the N-oxide 16b (28 mg, 63%) as a black oil. IR (ATR) ν = 1254 cm−1. 1H-NMR (400 MHz, CDCl3) δ –0.07 (s, 3H), 0.02 (s, 3H), 0.83 (s, 9H), 3.36 (s, 3H), 4.70 (s, 2H), 4.77 (d, J = 13.7 Hz, 1H), 4.95 (d, J = 11.9 Hz, 1H), 5.02 (d, J = 11.9 Hz, 1H), 5.11 (d, J = 13.7 Hz, 1H), 7.59–7.67 (m, 3H), 7.71–7.76 (m, 2H), 7.93 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.19 (s, 1H), 8.37 (s, 1H), and 8.90 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ -5.5 (2C), 18.2, 25.8 (3C), 55.6, 62.2, 66.2, 95.7, 122.6, 124.9, 127.4, 127.6 (2C), 128.3, 129.0, 129.3 (3C), 130.2, 133.9, 134.0, 135.3, 136.5, 146.5, 146.9, and 150.9. MS m/z: 490 (M+). HRMS (EI): calcd for C28H34N2O4Si 490.2288; found 490.2276.
3.1.8. 2-(3-Acetoxymethylquinolin-2-yl)-5-[(methoxymethoxy)methyl]isoquinoline N-oxide (16c)
The N-oxide 16c (yield 72%) was prepared according to a synthetic method for 16a as a black solid. mp was 169–171 °C (EtOAc-hexane). IR (ATR) ν = 1230, 1743 cm−1. 1H-NMR (400 MHz, CDCl3) δ 1.94 (s, 3H), 3.37 (s, 3H), 4.71 (s, 2H), 4.97 (d, J = 11.9 Hz, 1H), 5.05 (d, J = 11.9 Hz, 1H), 5.27 (d, J = 13.3 Hz, 1H), 5.45 (d, J = 13.3 Hz, 1H), 7.61–7.67 (m, 3H), 7.76 (t, J = 7.8 Hz, 2H), 7.93 (d, J = 7.8 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.26 (s, 1H), 8.33 (s, 1H), and 8.91 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.7, 55.6, 63.5, 66.2, 95.7, 122.9, 125.0 (2C), 127.6, 127.7, 127.9, 129.2, 129.4, 130.0 (2C), 130.4, 134.0, 135.6 (2C), 136.7, 146.0, 147.3, 151.4, and 170.3. MS m/z: 418 (M+). HRMS (EI): calcd for C24H22N2O5 418.1529; found 418.1535.
3.1.9. 3-{3-[(tert-Butyldimethylsilyloxy)methyl]quinolin-2-yl}-5-[(methoxymethoxy)methyl]isoquinolin-1-one (17b)
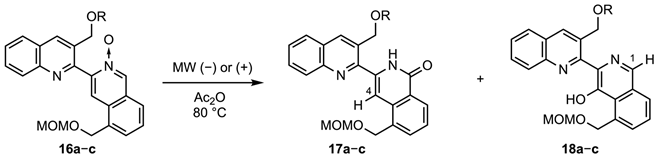
A solution of N-oxide 16b (25 mg, 0.051 mmol) in Ac2O (1 mL) was heated at 80 °C under microwave irradiation for 2 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 2:3 v/v) to give the isoquinolone 17b (9 mg, 36%) as a yellow solid. mp was 114–115 °C (EtOAc-hexane). IR (ATR) ν = 1651, 3305 cm−1. 1H-NMR (400 MHz, CDCl3) δ 0.21 (s, 6H), 1.00 (s, 9H), 3.40 (s, 3H), 4.73 (s, 2H), 4.97 (s, 2H), 5.20 (s, 2H), 7.43 (s, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.78 (t, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 8.16 (d, J = 7.8 Hz, 1H), 8.49 (d, J = 7.8 Hz, 2H), and 10.73 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ-5.1 (2C), 18.4, 25.9 (3C), 55.5, 63.1, 66.7, 95.2, 105.1, 127.2, 127.3, 127.5, 127.7 (2C), 127.8, 129.2, 130.3, 132.4, 133.3, 133.6, 136.2, 136.4, 136.6, 146.2, 148.3, and 162.6. MS m/z: 490 (M+). HRMS (EI): calcd for C28H34N2O4Si 490.2288; found 490.2284.
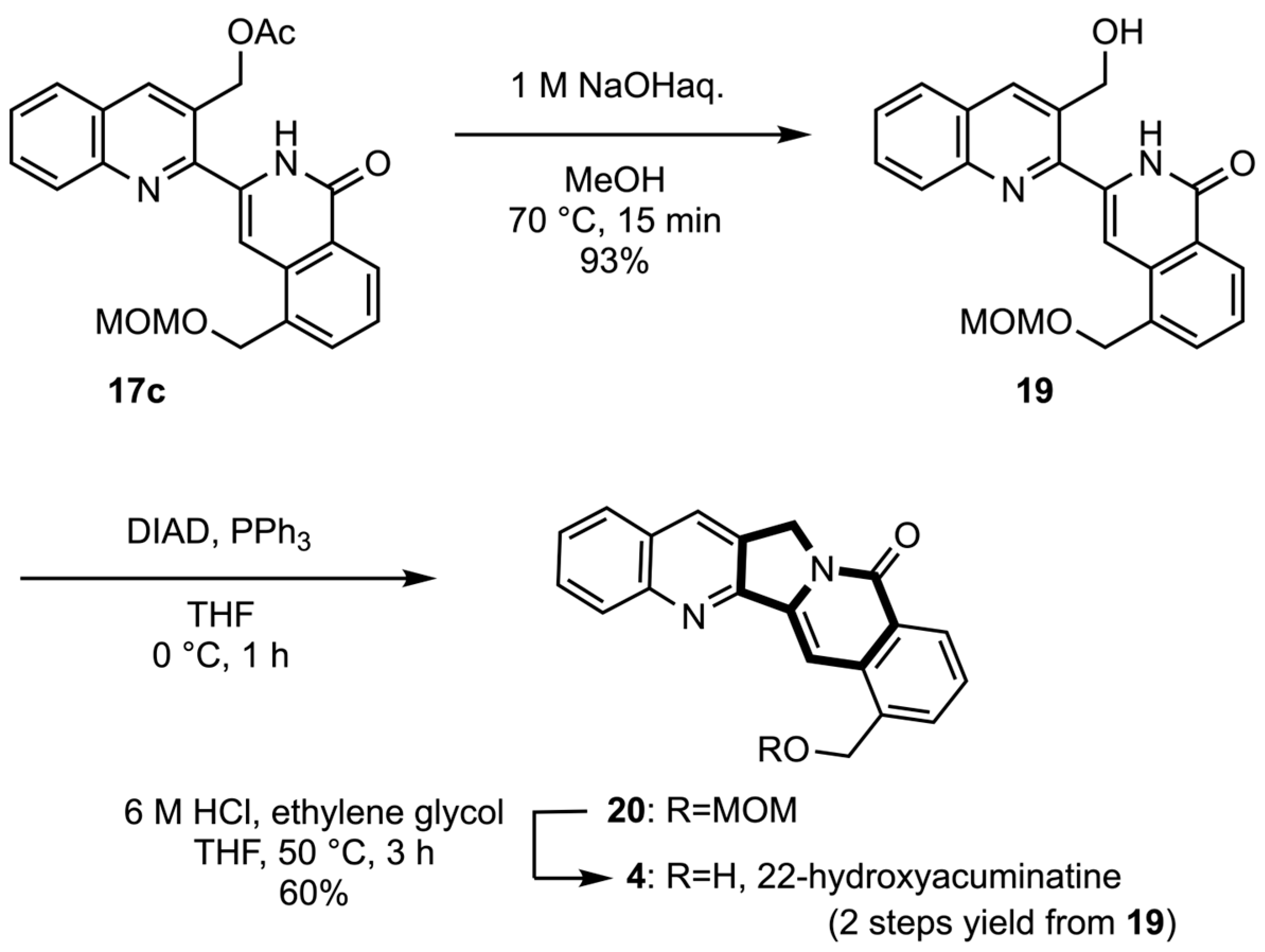
3.1.10. 3-(3-Acetoxymethylquinolin-2-yl)-5-[(methoxymethoxy)methyl]isoquinolin-1-one (17c) and 3-[3-(Acetoxymethyl)quinolin-2-yl]-4-hydroxy-3-[(methoxymethoxy)methyl]isoquinoline (18c)
A solution of N-oxide 16c (30 mg, 0.071 mmol) in Ac2O (2 mL) was heated at 80 °C under MW irradiation for 5 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the isoquinolone 17c (15 mg, 50%) and the 4-acetoxyisoquinoline 18c (11 mg, 37%).
17c: yellow solid, mp 149–150 °C (EtOAc-hexane). IR (ATR) ν = 1643, 1743, and 3286 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 3.39 (s, 3H), 4.71 (s, 2H), 4.95 (s, 2H), 5.54 (s, 2H), 7.54 (t, J = 7.8 Hz, 1H), 7.57 (s, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 8.17 (d, J = 7.8 Hz, 1H), 8.45 (s, 1H), 8.50 (d, J = 7.8 Hz, 1H), and 10.36 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 21.0, 55.5, 64.1, 66.6, 95.1, 105.3, 126.5, 127.2, 127.3, 127.4, 127.5, 127.8, 128.1, 129.2, 131.1, 133.4, 133.7, 135.3, 136.3, 140.7, 146.8, 149.7, 162.6, and 170.6. MS m/z: 418 (M+). HRMS (EI): calcd for C24H22N2O5 418.1529; found 418.1523.
18c: yellow solid, mp 108–109 °C (EtOAc-hexane). IR (ATR) ν = 1732 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.08 (s, 3H), 3.47 (s, 3H), 4.80 (s, 2H), 5.16 (s, 2H), 5.71 (s, 2H), 7.60 (t, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 8.36 (s, 1H), 8.70 (s, 1H), and 9.36 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 21.0, 55.7, 64.3, 66.5, 95.9, 117.7, 127.0, 127.4 (2C), 127.5, 127.8, 128.3, 128.9, 129.4, 129.8, 130.4, 133.9, 135.0, 136.0, 147.2, 151.8, 152.2, 156.3, and 170.7. MS m/z: 418 (M+). HRMS (EI): calcd for C24H22N2O5 418.1529; found 418.1518.
3.1.11. 3-(3-Hydroxymethylquinolin-2-yl)-5-[(methoxymethoxy)methyl]isoquinolin-1-one (19)
To a solution of isoquinolone 17c (9 mg, 0.020 mmol) in MeOH (1 mL), 1 M NaOH aq. (0.1 mL) was added dropwise under ice cooling and was stirred at 70 °C for 15 min. After cooling at ambient temperature, the resulting mixture was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 3:7 v/v) to give the alcohol 19 (7 mg, 93%) as a white solid. mp was 199–200 °C (EtOAc-hexane). IR (ATR) ν = 1620, 3286, 3737 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 3.28 (s, 3H), 4.70 (s, 2H), 4.80 (d, J = 5.0 Hz, 2H), 4.85 (s, 2H), 5.78 (t, J = 5.0 Hz, 1H), 7.16 (s, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.77–7.83 (m, 2H), 8.08–8.12 (m, 2H), 8.26 (d, J = 7.8 Hz, 1H), 8.55 (s, 1H), and 11.48 (br s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 55.0, 60.5, 66.6, 95.5, 103.1, 126.2, 126.7, 126.8, 127.5 (2C), 127.6, 127.8, 128.8, 130.0, 133.0, 133.6, 133.9, 136.0, 138.0, 145.9, 151.3, and 161.9. MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1433.
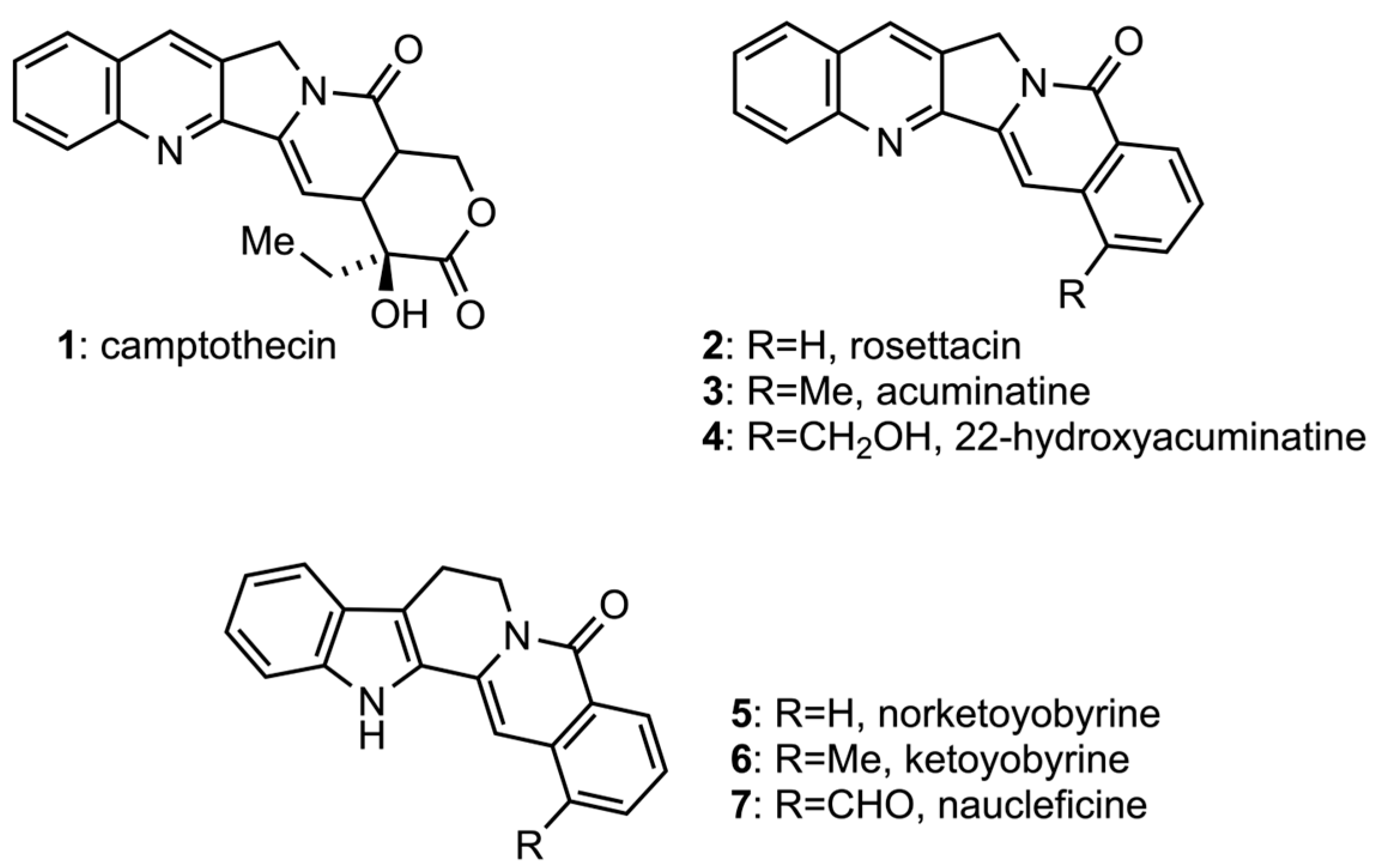
3.1.12. 22-Hydroxyacuminatine (4)
To a solution of alcohol
19 (8 mg, 0.021 mmol) and PPh
3 (8 mg, 0.032 mmol) in THF (1 mL), DIAD (1.9 M in toluene, 0.017 mL, 0.032 mmol) was added dropwise under ice cooling, and was stirred at 0 °C for 1 h. The resulting mixture was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried with Na
2SO
4, and evaporated in vacuo. To a suspension of the residue and ethylene glycol (0.1 mL) in THF (0.5 mL), 6 M HCl (0.5 mL) was added dropwise under ice cooling and then stirred at 50 °C for 3 h. After cooling to ambient temperature, the residue was alkalified with 1 M aqueous NaOH. The resulting mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried with Na
2SO
4, and evaporated in vacuo. The residue was filtered off to give 22-hydroxyacuminatine (
4) (4 mg, 60%) as a white solid. mp 255–256 °C (EtOAc-hexane lit. [
3] mp 258–260 °C). IR (ATR) ν = 1651, 3359 cm
−1.
1H-NMR (400 MHz, DMSO-
d6) δ 4.95 (d,
J = 5.5 Hz, 2H), 5.36 (s, 2H), 5.50 (t,
J = 5.5 Hz, 1H), 7.54–7.63 (m, 1H), 7.69 (t,
J = 7.8 Hz, 1H), 7.73 (s, 1H), 7.81 (d,
J = 7.8 Hz, 1H), 7.83–7.87 (m, 1H), 8.11 (d,
J = 7.8 Hz, 1H), 8.19 (d,
J = 7.8 Hz, 1H), 8.30 (d,
J = 7.8 Hz, 1H), and 8.66 (s, 1H).
13C-NMR (100 MHz, DMSO-
d6) δ 49.6, 61.2, 96.2, 125.9 (2C), 126.7, 127.3, 127.9, 128.5, 128.9, 129.7, 130.2, 131.1, 131.3, 135.3, 138.5, 140.1, 148.0, 153.4, and 159.9. MS
m/
z: 314 (M
+). HRMS (EI): calcd for C
20H
14N
2O
2 314.1055; found 314.1061.
3.1.13. Methyl [2-Iodo-N-(methoxymethyl)indol-3-yl]acetate (22)
A solution of methyl indolylacetate 21 (470 mg, 1.49 mmol) in DMF (8 mL) was added dropwise to a suspension of NaH (71 mg, 1.79 mmol) in DMF (4 mL) under ice cooling. After stirring at same temperature for 30 min, MOMCl (0.13 mL, 1.79 mmol) was added to the reaction mixture and then was stirred at rt for 2 h. After quenching with H2O, the reaction mixture was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give the N-MOM-indolylacetate 22 (454 mg, 85%) as a yellow solid. mp was 83–85 °C (EtOAc-hexane). IR (ATR) ν = 1728 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.31 (s, 3H), 3.69 (s, 3H), 3.79 (s, 2H), 5.54 (s, 2H), 7.16 (t, J = 7.8 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), and 7.55 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 33.6, 52.1, 56.0, 77.5, 88.0, 110.1, 116.1, 118.5, 120.7, 122.7, 128.4, 138.6, and 171.3. MS m/z: 359 (M+). HRMS (EI): calcd for C13H14INO3 359.0018; found 359.0022.
3.1.14. Methyl [2-Trimethylsilylethynyl-N-(methoxymethyl)lindol-3-yl]acetate (23)
A solution of 2-iodoindole 22 (200 mg, 0.56 mmol), CuI (11 mg, 0.056 mmol), PdCl2(PPh3)2 (23 mg, 0.022 mmol), and TMS-acetylene (0.091 mL, 0.66 mmol) in Et3N (5 mL, 35.6 mmol) was stirred at rt for 23 h. Afterward, the reaction mixture was filtered through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:9 v/v) to give the TMS-acetylene 23 (143 mg, 78%) as a yellow oil. IR (ATR) ν = 1736, 2152 cm−1. 1H-NMR (400 MHz, CDCl3) δ 0.30 (s, 9H), 3.28 (s, 3H), 3.70 (s, 3H), 3.87 (s, 2H), 5.57 (s, 2H), 7.17 (t, J = 7.8 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), and 7.57 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ -0.14 (3C), 31.2, 52.0, 56.0, 75.1, 94.4, 105.3, 110.3, 115.1, 119.6, 121.0, 121.3, 124.1, 127.1, 136.6, and 171.6. MS m/z: 329 (M+). HRMS (EI): calcd for C18H23NO3Si 329.1447; found 329.1451.
3.1.15. Methyl [2-Ethynyl-N-(methoxymethyl)indol-3-yl]acetate (24)
A solution of TBAF (1.0 M in THF, 2.19 mL, 2.19 mmol) was added dropwise to a solution of TMS-acetylene 23 (600 mg, 1.82 mmol) in THF (20 mL) at 0 °C. After being stirred at rt for 10 min, the reaction mixture was quenched with water and then extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography using EtOAc-hexane (1:4, v/v) as an eluent to give the 2-ethynylindole 24 (430 mg, 92%) as a red solid. mp was 92–94 °C (EtOAc-hexane). IR (ATR) ν = 1732, 2102 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.29 (s, 3H), 3.68 (s, 1H), 3.70 (s, 3H), 3.90 (s, 2H), 5.59 (s, 2H), 7.19 (t, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), and 7.60 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.0, 52.1, 56.0, 73.9, 75.1, 86.9, 110.3, 115.8, 119.7, 120.1, 121.1, 124.3, 127.0, 136.7, and 171.4. MS m/z: 257 (M+). HRMS (EI): calcd for C15H15NO3 257.1052; found 257.1046.
3.1.16. Methyl {2-[2-(2-Formylphenyl)ethynyl]-N-(methoxymethyl)indol-3-yl}acetate (26a)
A solution of 2-ethynylindole 24 (81 mg, 0.32 mmol), CuI (2.7 mg, 0.014 mmol), PdCl2(PPh3)2 (6 mg, 0.0086 mmol), and 2-iodobenzaldehyde 25a (66 mg, 0.29 mmol) in Et3N (2.6 mL, 18 mmol) was stirred at 70 °C for 45 min. Afterward, the reaction mixture was filtered through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:9 v/v) to give the 2-alkynylbenzaldehyde 26a (83 mg, 80%) as a yellow solid. mp was 75–77 °C (EtOAc-hexane). IR (ATR) ν = 1689, 1736, and 2202 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.35 (s, 3H), 3.71 (s, 3H), 3.98 (s, 2H), 5.70 (s, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.61–7.66 (m, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), and 10.59 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.3, 52.2, 56.2, 75.3, 86.1, 95.1, 110.3, 116.3, 119.8, 120.5, 121.2, 124.6, 125.2, 127.3, 128.6, 129.0, 133.3, 133.8, 135.6, 137.2, 171.3, and 191.1. MS m/z: 361 (M+). HRMS (EI): calcd for C22H19NO4 361.1314; found 361.1322.
3.1.17. Methyl {2-[2-(2-Formyl-3-[(methoxymethoxy)methyl]phenyl)ethynyl]-N-(methoxymethylindol-3-yl}acetate (26b)
A solution of 2-ethynylindole 24 (266 mg, 1.04 mmol), CuI (20 mg, 0.10 mmol), PdCl2(PPh3)2 (36 mg, 0.052 mmol), and 2-iodobenzaldehyde 25b (377 mg, 1.25 mmol) in iPr2NH (8 mL) was stirred at 100 °C for 3 h. Afterward, the reaction mixture was filtered through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:9 v/v) to give the 2-alkynylbenzaldehyde 26b (410 mg, 91%) as a yellow solid. mp was 76–78 °C (EtOAc-hexane). IR (ATR) ν = 1693, 1724, 2198 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.34 (s, 3H), 3.42 (s, 3H), 3.71 (s, 3H), 4.00 (s, 2H), 4.80 (s, 2H), 4.98 (s, 2H), 5.71 (s, 2H), 7.21 (d, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), and 10.64 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.3, 52.2, 55.5, 56.0, 66.8, 75.3, 91.3, 92.2, 96.1, 110.3, 116.6, 119.8, 120.3, 121.2, 123.5, 124.7, 127.3, 127.7, 128.7, 132.9, 135.9, 137.4, 141.2, 171.2, and 191.2. MS m/z: 435 (M+). HRMS (EI): calcd for C25H25NO6 435.1682; found 435.1696.
3.1.18. Methyl {2-[22-Hydroxyiminophenyl)ethynyl]-N-(methoxymethyl)indol-3-yl}acetate (27a)
A mixture of benzaldehyde 26a (184 mg, 0.51 mmol), NH2OH·HCl (70 mg, 1.02 mmol), and AcONa (83 mg, 1.02 mmol) in EtOH (4 mL) was stirred at rt for 1.5 h. After removal of solvent, the residue was diluted with H2O and then filtered off to give the oxime 27a (170 mg, 88%) as a yellow solid. mp was 110–113 °C (EtOAc-hexane). IR (ATR) ν = 1701, 1732, 2218, and 3062 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.36 (s, 3H), 3.73 (s, 3H), 4.03 (s, 2H), 5.68 (s, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.38–7.40 (m, 2H), 7.49 (d, J = 7.8 Hz, 1H), 7.60–7.62 (m, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.70–7.72 (m, 1H), 8.46 (br s, 1H), and 8.59 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.5, 52.4, 56.2, 75.3, 84.4, 97.0, 110.2, 115.3, 119.8, 121.1, 121.2, 121.2, 124.3, 127.2, 127.4, 128.9, 129.3, 132.8, 133.0, 137.1, 148.8, and 172.3. MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1427.
3.1.19. Methyl {2-[2-(2-Hydroxyimino-3-[(methoxymethoxy)methyl]phenyl)ethynyl]-N-methoxymethylindol-3-yl}acetate (27b)
The same procedure as above was carried out with 2-arylethynylbenzaldehyde 26b (450 mg, 1.03 mmol) to give the oxime 27b (414 mg, 89%) as a white solid. Mp was 94–95 °C (EtOAc-hexane). IR (ATR) ν = 1736, 2164, and 3302 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.35 (s, 3H), 3.42 (s, 3H), 3.71 (s, 3H), 4.03 (s, 2H), 4.78 (s, 2H), 4.92 (s, 2H), 5.68 (s, 2H), 7.21 (t, J = 7.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.66–7.70 (m, 2H), 8.45 (s, 1H), and 8.68 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.4, 52.3, 55.4, 56.0, 67.4, 75.3, 89.8, 93.9, 95.9, 110.2, 115.6, 119.9 (2C), 121.0, 121.1, 124.4, 126.0, 127.4, 128.7, 128.7, 133.5, 137.3, 140.6, 149.0, and 172.0. MS m/z: 450 (M+). HRMS (EI): calcd for C25H26N2O6 450.1791; found 450.1799.
3.1.20. 3-[3-(2-Methoxy-2-oxoethyl)-1-(methoxymethyl)indol-2-yl]isoquinoline N-oxide (28a)
A solution of oxime 27a (45 mg, 0.12 mmol) in 1,2-dichlorobenzene (3 mL) was stirred at 120 °C for 17 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the N-oxide 28a (37 mg, 82%) as a brown oil. IR (ATR) ν = 1315, 1736 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.09 (s, 3H), 3.55 (d, J = 15.6 Hz, 1H), 3.69 (s, 3H), 3.83 (d, J = 15.6 Hz, 1H), 5.35 (d, J = 11.4 Hz, 1H), 5.67 (d, J = 11.4 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.62–7.71 (m, 3H), 7.79 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), and 8.94 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 30.9, 52.1, 56.0, 76.4, 110.4, 111.1, 119.7, 120.5, 123.8, 124.6, 127.2, 127.6, 128.5, 129.0, 129.2, 129.7, 129.8, 129.9, 137.0, 137.4, 138.2, and 172.1. MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1441.
3.1.21. 3-[3-(2-Methoxy-2-oxoethyl)-1-(methoxymethyl)indol-2-yl)-5-[(methoxymethoxy)methyl]isoquinoline N-oxide (28b)
The same procedure as above was carried out with the oxime 27b (30 mg, 0.067 mmol) to give the N-oxide 28b (21 mg, 69%) as a brown oil. IR (ATR) ν = 1323, 1731 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.10 (s, 3H), 3.36 (s, 3H), 3.54 (d, J = 15.6 Hz, 1H), 3.70 (s, 3H), 3.84 (d, J = 15.6 Hz, 1H), 4.73 (s, 2H), 4.94 (d, J = 12.3 Hz, 1H), 5.05 (d, J = 12.3 Hz, 1H), 5.37 (d, J = 11.0 Hz, 1H), 5.67 (d, J = 11.0 Hz, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.62–7.68 (m, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 8.43 (s, 1H) and 8.94 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 31.1, 52.1, 55.6, 56.0, 66.3, 95.9, 110.5, 111.2, 119.8 (2C), 120.6, 123.9, 124.9, 126.1, 127.1, 127.7, 129.3, 129.7, 129.9, 130.4, 134.3, 137.2, 137.5, 138.1 and 172.0. MS m/z: 450 (M+). HRMS (EI): calcd for C25H26N2O6 450.1791; found 450.1795.
3.1.22. Methyl 2-[1-(methoxymethyl)-2-(1-oxo-1,2-dihydroisoquinolin-3-yl)indol-3-yl]acetate (29a) and Methyl 2-[2-(4-acetoxyisoquinolin-3-yl)-1-(methoxymethyl)indol-3-yl]acetate (31a)
A solution of N-oxide 28a (15 mg, 0.040 mmol) in Ac2O (1 mL) was heated at 80 °C under MW irradiation for 6 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the isoquinolone 29a (9 mg, 60%) and the 4-acetoxyisoquinoline 31a (3 mg, 18%).
29a: White solid, mp was 150–151 °C (EtOAc-hexane). IR (ATR) ν = 1624, 1739, and 3649 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.46 (s, 3H), 3.79 (s, 3H), 3.86 (s, 2H), 5.48 (s, 2H), 6.90 (s, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.51–7.57 (m, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.67–7.73 (m, 2H), 8.47 (d, J = 7.8 Hz, 1H), and 9.98 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 30.9, 52.6, 56.5, 74.9, 109.2, 109.9, 110.5, 119.6, 121.3, 124.4, 125.9, 126.8, 127.4 (2C), 127.7, 129.6, 132.3, 132.9, 137.4, 137.6, 162.8, and 172.8. MS m/z: 376 (M+). HRMS (EI): calcd for C22H20N2O4 376.1423; found 376.1431.
31a: Brown oil. IR (ATR) ν = 1732, 1774 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.16 (s, 3H), 3.16 (s, 3H), 3.63 (s, 3H), 3.64–3.75 (m, 2H), 5.34 (d, J = 11.0 Hz, 1H), 5.56 (d, J = 11.0 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 8.11 (d, J = 7.8 Hz, 1H), and 9.30 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.4, 30.7, 51.8, 55.9, 75.5, 109.7, 110.8, 119.6, 120.5, 121.1, 123.1, 127.8, 128.0, 128.5, 129.4, 130.5, 131.4, 132.8, 136.5, 137.7, 142.2, 150.3, 168.5, and 172.0. MS m/z: 418 (M+). HRMS (EI): calcd for C24H22N2O5 418.1529; found 418.1527.
3.1.23. Methyl 2-{2-[5-[(Methoxymethoxy)methyl]-1-oxo-1,2-dihydroisoquinolin-3-yl]-1-(methoxymethyl)indol-3-yl}acetate (29b), 1-Acetoxy-3-[3-methoxycarbonylmethyl-N-(methoxymethyl)indol-2-yl]-5-[(methoxymethoxy)methyl]isoquinoline (30b) and 4-Acetoxy-3-[3-methoxycarbonylmethyl-N-(methoxymethyl)indol-2-yl]-5-[(methoxymethoxy)methyl]isoquinoline (31b)
The same procedure as above was carried out with the oxime 28b (32 mg, 0.071 mmol) to give the isoquinolone 29b (12 mg, 38%), the 1-acetoxyisoquinoline 30b (7 mg, 20%), and the 4-acetoxyisoquinoline 31b (11 mg, 32%).
29b: Orange oil. IR (ATR) ν = 1643, 1736, and 3672 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.39 (s, 3H), 3.49 (s, 3H), 3.79 (s, 3H), 3.87 (s, 2H), 4.73 (s, 2H), 4.89 (s, 2H), 5.49 (s, 2H), 7.17 (s, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.50–7.54 (m, 2H), 7.69 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 8.47 (d, J = 7.8 Hz, 1H), and 10.11 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 30.9, 52.6, 55.5, 56.5, 66.8, 74.9, 95.8, 105.7, 109.9, 110.7, 119.7, 121.3, 124.4, 126.4, 126.9, 127.5, 127.8, 129.8, 132.5, 133.3, 133.4, 136.2, 137.7, 162.8, and 172.7. MS m/z: 450 (M+). HRMS (EI): calcd for C25H26N2O6 450.1791; found 450.1783.
30b: Orange oil. IR (ATR) ν = 1736, 1778 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.51 (s, 3H), 3.18 (s, 3H), 3.41 (s, 3H), 3.74 (s, 3H), 3.88 (s, 2H), 4.78 (s, 2H), 5.08 (s, 2H), 5.75 (s, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 7.8 Hz, 1H), 8.03 (d, J = 7.8 Hz, 1H), and 8.39 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 21.3, 31.3, 52.2, 55.6, 56.0, 66.6, 75.2, 96.0, 109.8, 110.6, 118.8, 119.7 (2C), 120.8, 123.6, 123.7, 127.9, 128.2, 131.4, 134.3, 136.5, 137.9, 137.9, 141.7, 155.7, 168.9, and 172.5. MS m/z: 492 (M+). HRMS (EI): calcd for C27H28N2O7 492.1897; found 492.1891.
31b: Yellow oil. IR (ATR) ν = 1736, 1774 cm−1. 1H-NMR (400 MHz, CDCl3) δ 1.99 (s, 3H), 3.28 (s, 3H), 3.36 (s, 3H), 3.63 (s, 3H), 3.64 (d, J = 16.5 Hz, 1H), 3.73 (d, J = 16.5 Hz, 1H), 4.61 (d, J = 6.9 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 5.02 (d, J = 13.3 Hz, 1H), 5.21 (d, J = 13.3 Hz, 1H), 5.26 (d, J = 11.0 Hz, 1H), 5.57 (d, J = 11.0 Hz, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), and 9.28 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 21.0, 30.8, 51.8, 55.4, 56.1, 67.7, 75.9, 94.7, 109.3, 111.0, 119.6 (2C), 120.4, 123.0, 128.0, 128.3, 129.5, 130.7, 132.0, 133.1, 133.2, 137.7, 138.8, 142.3, 150.8, 168.8, and 172.0. MS m/z: 492 (M+). HRMS (EI): calcd for C27H28N2O7 492.1897; found 492.1889.
3.1.24. 3-[3-(2-Hydroxyethyl)-N-(methoxymethyl)indol-2-yl]isoquinolin-1-one (32a)
A solution of the isoquinolone 29a (25 mg, 0.066 mmol) in THF (2 mL) was added dropwise to a suspension of LiAlH4 (4 mg, 0.10 mmol) in THF (2 mL) under ice cooling and then stirred at 70 °C for 30 min. After quenching with H2O, the reaction mixture was filtered through Celite pad and washed with H2O and EtOAc, and the filtrate was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the alcohol 32a (22 mg, 95%) as a white solid. mp was 211–212 °C (EtOAc-hexane). IR (ATR) ν = 1732, 3351, 3629 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.12 (t, J = 5.5 Hz, 2H), 3.39 (s, 3H), 3.57 (br s, 1H), 4.12 (t, J = 5.5 Hz, 2H), 5.47 (s, 2H), 6.89 (s, 1H), 7.24 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.8 Hz, 1H), 7.48–7.54 (m, 2H), 7.61–7.63 (m, 2H), 7.69 (t, J = 7.8 Hz, 1H), 8.39 (d, J = 7.8 Hz, 1H), and 11.36 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 27.1, 56.2, 61.7, 74.9, 108.4, 110.4, 114.4, 119.2, 120.9, 124.0, 125.5, 126.7, 127.0, 127.4, 127.5, 130.4, 132.5, 132.7, 137.7, 138.3, and 163.3. MS m/z: 348 (M+). HRMS (EI): calcd for C21H20N2O3 348.1474; found 348.1468.
3.1.25. 3-[3-(2-Hydroxyethyl)-N-(methoxymethyl)indol-2-yl]-5-[(methoxymethoxy)methyl]isoquinolin-1-one (32b)
A solution of N-oxide 28b (25 mg, 0.056 mmol) in Ac2O (1 mL) was heated at 80 °C under MW irradiation for 4.5 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give a mixture of isoquinolone 29b and the 1-acetoxyisoquinoline 30b. A solution of the mixture in THF (1 mL) was added dropwise to a suspension of LiAlH4 (4 mg, 0.11 mmol) in THF (1 mL) under ice cooling and then stirred at 0 °C for 1 h. After quenching with H2O, the reaction mixture was filtered through Celite pad and washed with H2O and EtOAc, and the filtrate was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give the alcohol 32b (16 mg, 69%) as a white solid. mp was 175–176 °C (EtOAc-hexane). IR (ATR) ν = 1639, 3309, and 3529 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.11 (t, J = 5.5 Hz, 2H), 3.39 (s, 3H), 3.42 (s, 3H), 4.12 (t, J = 5.5 Hz, 2H), 4.73 (s, 2H), 4.87 (s, 2H), 5.46 (s, 2H), 7.16 (s, 1H), 7.23 (t, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 8.35 (d, J = 7.8 Hz, 1H), and 11.63 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 27.1, 55.5, 56.2, 61.7, 67.0, 75.0, 95.8, 104.9, 110.3, 114.8, 119.3, 120.9, 124.0, 126.0, 126.4, 127.5, 127.7, 130.7, 132.6, 132.9, 133.3, 136.5, and 138.4, 163.4. MS m/z: 422 (M+). HRMS (EI): calcd for C24H26N2O5 422.1842; found 422.1844.
3.1.26. N-Methoxymethyl-3,14,15,16,17,18,19,20-octadehydroyohimban-21-one (33a)
To a solution of alcohol 32a (21 mg, 0.060 mmol) and PPh3 (47 mg, 0.18 mmol) in THF (3 mL), DIAD (1.9 M in toluene, 0.94 mL, 0.18 mmol) was added dropwise under ice cooling and was stirred at 60 °C for 3 h. After cooling at ambient temperature, the resulting mixture was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:3 v/v) to give the indoloquinolizine 33a (19 mg, 95%) as a white solid. mp was 163–166 °C (EtOAc-hexane). IR (ATR) ν = 1732 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.10 (t, J = 6.2 Hz, 2H), 3.59 (s, 3H), 4.50 (t, J = 6.2 Hz, 2H), 5.62 (s, 2H), 7.24 (t, J = 7.8 Hz, 1H), 7.31 (s, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.59–7.66 (m, 3H), and 8.45 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.1, 40.4, 56.1, 75.1, 103.4, 109.9, 116.5, 119.4, 121.1, 124.5, 125.2, 125.4, 126.6, 128.0 (2C), 130.7, 131.1, 132.3, 136.4, 140.6, and 162.5. MS m/z: 330 (M+). HRMS (EI): calcd for C21H18N2O2 330.1368; found 330.1354.
3.1.27. N-Methoxymethyl-16-[(methoxymethoxy)methyl]-3,14,15,16,17,18,19,20-octadehydroyohimban-21-one (33b)
The same procedure as above was carried out with alcohol 32b (35 mg, 0.083 mmol) to give the indoloquinolizine 33b (30 mg, 90%) as a yellow solid. mp was 128–129 °C (EtOAc-hexane). IR (ATR) ν = 1736 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.10 (t, J = 6.4 Hz, 2H), 3.43 (s, 3H), 3.60 (s, 3H), 4.49 (t, J = 6.4 Hz, 2H), 4.79 (s, 2H), 4.90 (s, 2H), 5.63 (s, 2H), 7.24 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.58 (s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), and 8.46 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.1, 40.4, 55.4, 56.1, 66.9, 75.3, 95.9, 99.9, 110.0, 116.6, 119.4, 121.2, 124.6, 125.4, 125.7, 126.1, 128.3, 130.9, 131.3, 132.8, 132.9, 135.2, 140.7, and 162.5. MS m/z: 404 (M+). HRMS (EI): calcd for C24H24N2O4 404.1736; found 404.1744.
3.1.28. Norketoyobyrine (5)
To a suspension of indoloquinolizine
33a (23 mg, 0.070 mmol) and ethylene glycol (0.78 mg, 0.014 mmol) in THF (5 mL), 6 M HCl (3 mL) was added dropwise under ice cooling and then stirred at 60 °C for 15 h. After cooling to ambient temperature, the solvent was evaporated in vacuo. The residue was diluted with water and then was alkalified with 1 M aqueous NaOH. The resulting mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried with Na
2SO
4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:1
v/
v) to give norketoyobyrine (
5; 18 mg, 90%) as a yellow solid. mp was 298–299 °C (EtOAc-hexane lit. [
16] mp of 299–300 °C). IR (ATR) ν = 1732 cm
−1.
1H-NMR (400 MHz, DMSO-
d6) δ 3.08 (t,
J = 6.6 Hz, 2H), 4.39 (t,
J = 6.6 Hz, 2H), 7.06 (t,
J = 7.8 Hz, 1H), 7.06 (s, 1H), 7.21 (t,
J = 7.8 Hz, 1H), 7.42 (d,
J = 7.8 Hz, 1H), 7.45 (t,
J = 7.8 Hz, 1H), 7.55–7.63 (m, 2H), 7.70 (t,
J = 7.8 Hz, 1H), 8.23 (d,
J = 7.8 Hz, 1H), and 11.69 (br s, 1H).
13C-NMR (100 MHz, DMSO-
d6) δ 19.3, 40.4, 99.1, 111.7, 112.5, 119.1, 119.5, 123.5, 124.5, 125.6, 126.0, 126.2, 127.5, 128.2, 132.4, 132.7, 136.2, 138.0, and 161.3. MS
m/
z: 286 (M
+). HRMS (EI): calcd for C
19H
14N
2O 286.1106; found 286.1114.
3.1.29. 16-Hydroxymethyl-3,14,15,16,17,18,19,20-octadehydroyohimban-21-one (34)
The same procedure as above was carried out with alcohol
33b (40 mg, 0.099 mmol) to give alcohol
34 (28 mg, 90%) as a white solid. Mp was 283–284 °C (EtOAc-hexane lit. [
30] mp of 280–283 °C). IR (ATR) ν = 1747, 3317 cm
−1.
1H-NMR (400 MHz, DMSO-
d6) δ 3.08 (t,
J = 6.6 Hz, 2H), 4.39 (t,
J = 6.6 Hz, 2H), 4.87 (d,
J = 5.5 Hz, 2H), 5.40 (t,
J = 5.5 Hz, 1H), 7.06 (t,
J = 7.8 Hz, 1H), 7.19–7.23 (m, 2H), 7.40–7.45 (m, 2H), 7.58 (d,
J = 7.8 Hz, 1H), 7.74 (d,
J = 7.8 Hz, 1H), 8.15 (d,
J = 7.8 Hz, 1H), and 11.72 (br s, 1H).
13C-NMR (100 MHz, DMSO-
d6) δ 19.3, 40.4, 60.4, 95.6, 111.6, 112.5, 119.2, 119.5, 123.6, 124.7, 125.6 (2C), 126.2, 128.4, 130.2, 132.0, 133.6, 137.3, 138.0, and 161.4. MS
m/
z: 316 (M
+). HRMS (EI): calcd for C
20H
16N
2O
2 316.1212; found 316.1208.
3.1.30. Naucleficine (7)
A suspension of alcohol
34 (30 mg, 0.095 mmol) and active MnO
2 (165 mg, 1.9 mmol) in CH
2Cl
2 (2 mL) was stirred at reflux for 1 h. The reaction mixture was filtered through a Celite pad. The filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:1,
v/
v) to give naucleficine (
7; 21 mg, 70%) as an orange solid. mp was 276–277 °C (CHCl
3 lit. [
30] mp of 284–290 °C). IR (ATR) ν = 1592, 1643, and 3221 cm
−1.
1H-NMR (400 MHz, DMSO-
d6) δ 3.11 (t,
J = 6.6 Hz, 2H), 4.41 (t,
J = 6.6 Hz, 2H), 7.08 (t,
J = 7.8 Hz, 1H), 7.24 (t,
J = 7.8 Hz, 1H), 7.47 (d,
J = 7.8 Hz, 1H), 7.59–7.64 (m, 2H), 8.12 (s, 1H), 8.25 (d,
J = 7.8 Hz, 1H), 8.53 (d,
J = 7.8 Hz, 1H), 10.42 (s, 1H), 11.82 (br s, 1H).
13C-NMR (100 MHz, DMSO-
d6) δ 19.2, 40.5, 94.9, 112.0, 114.1, 119.4, 119.7, 124.1, 125.4, 125.5 (2C), 127.9, 129.6, 133.8, 135.1, 135.6, 138.5, 138.6, 160.8, and 192.8. MS
m/
z: 314 (M
+). HRMS (EI): calcd for C
20H
14N
2O
2 314.1055; found 314.1066.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}