

Evaluation of the Antitumor and Antiproliferative Potential of Synthetic Peptides Derived from IsCT1, Associated with Cisplatin, in Squamous Cell Carcinoma of the Oral Cavity

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Enhanced Cytotoxicity of Cisplatin Combined with AC-AFPK-IsCT Peptide in Tumor Cells
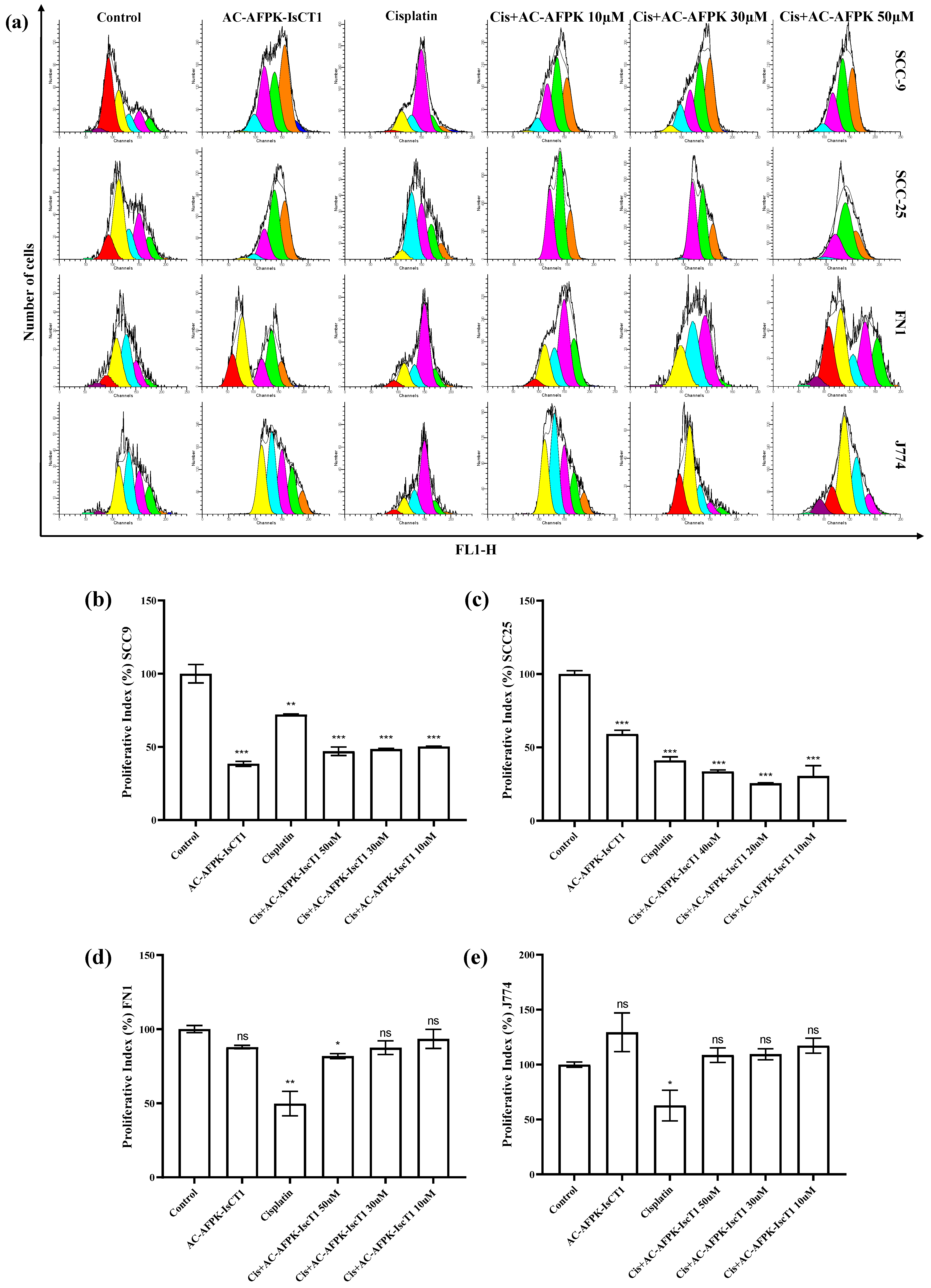
2.2. Impact of the Cisplatin and AC-AFPK-IsCT1 on the Proliferative Activity of Tongue Squamous Cell Carcinoma and Normal Cells
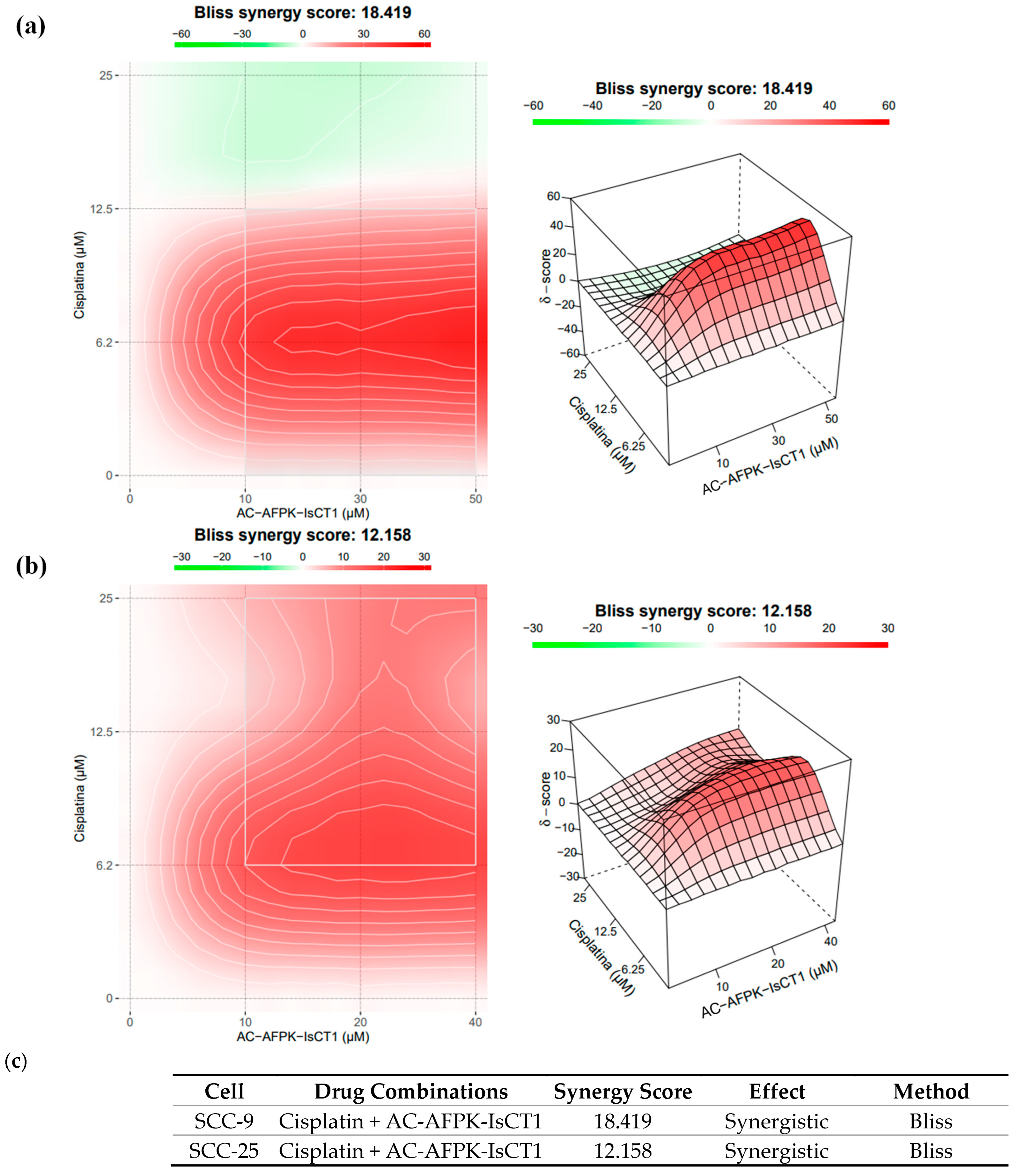
2.3. Synergistic Interaction of Cisplatin and AC-AFPK-IsCT1 in SCC-9 and SCC-25 Cells
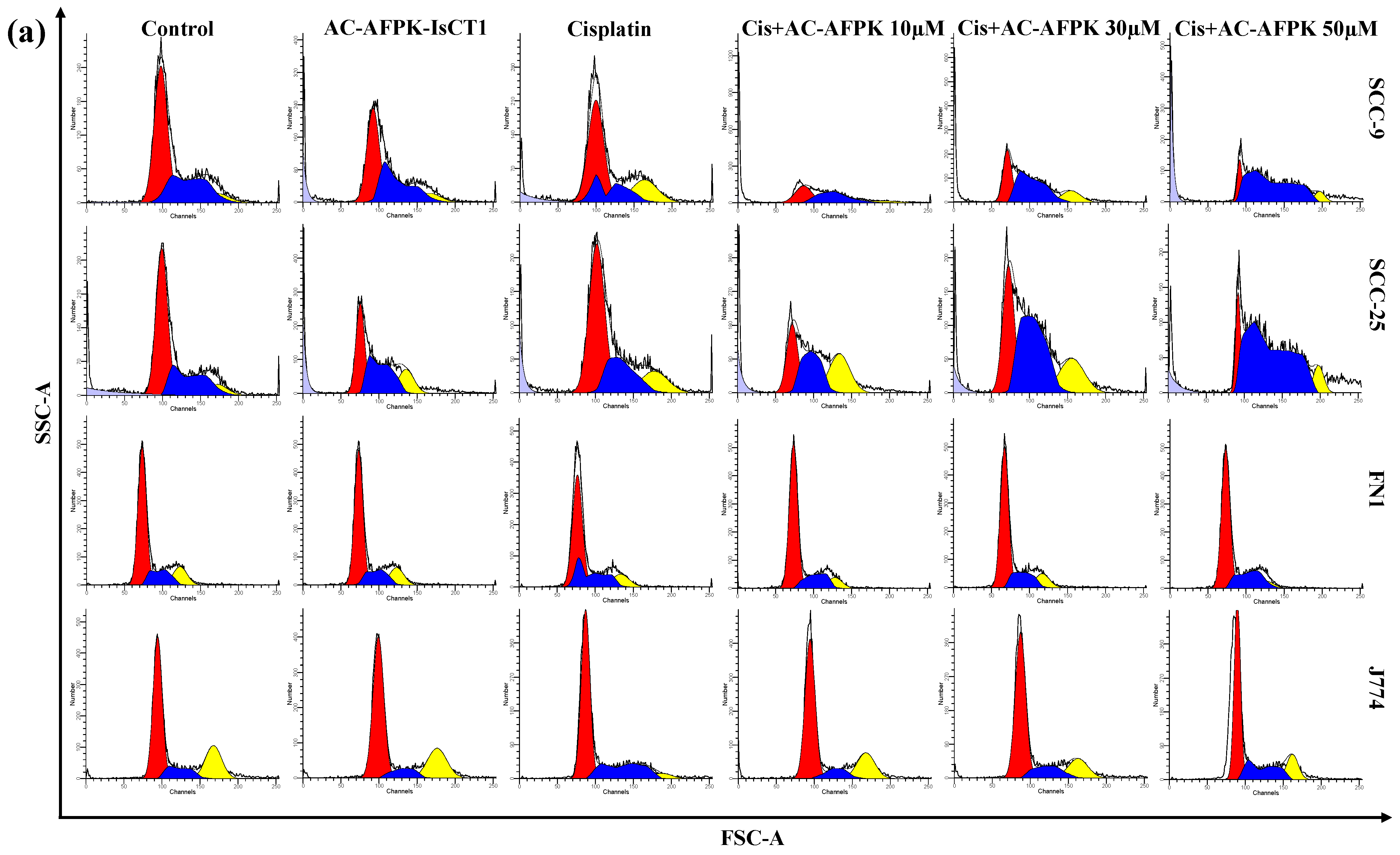
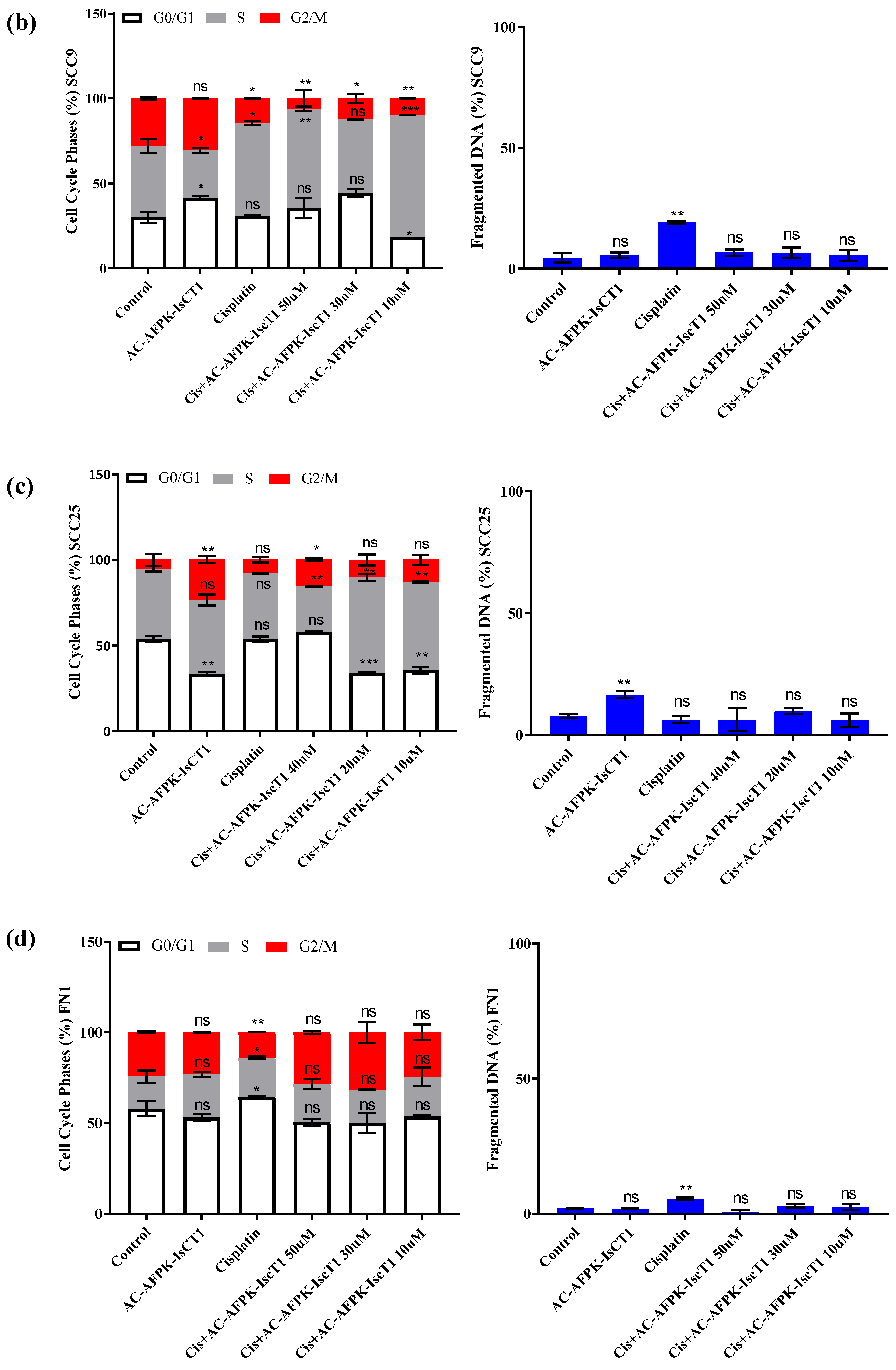
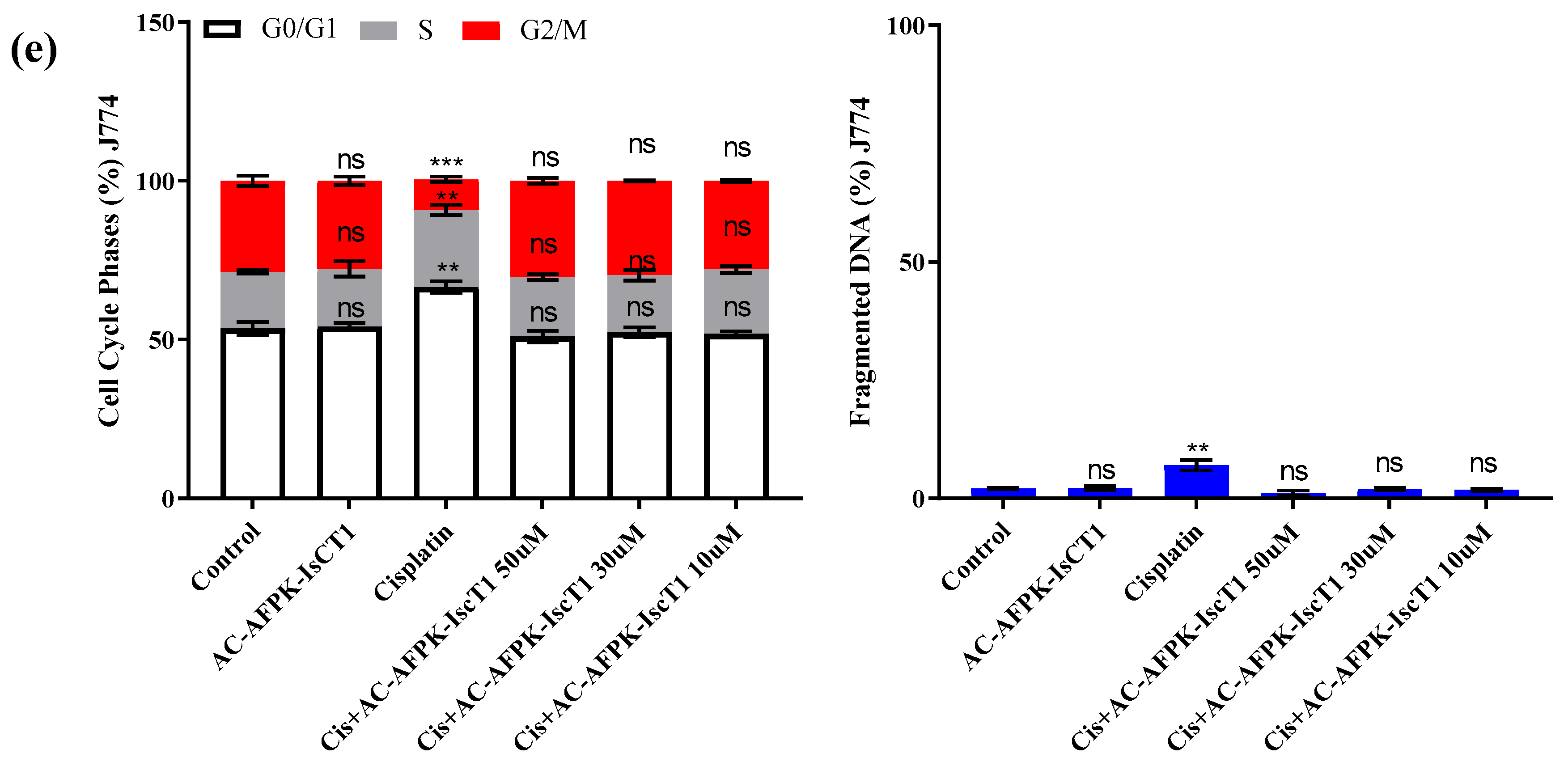
2.4. Cell Cycle Modulation and DNA Fragmentation
2.5. Impact of Cisplatin and AC-AFPK-IsCT1 on the Mitochondrial Membrane Electrical Potential (ΔΨm)
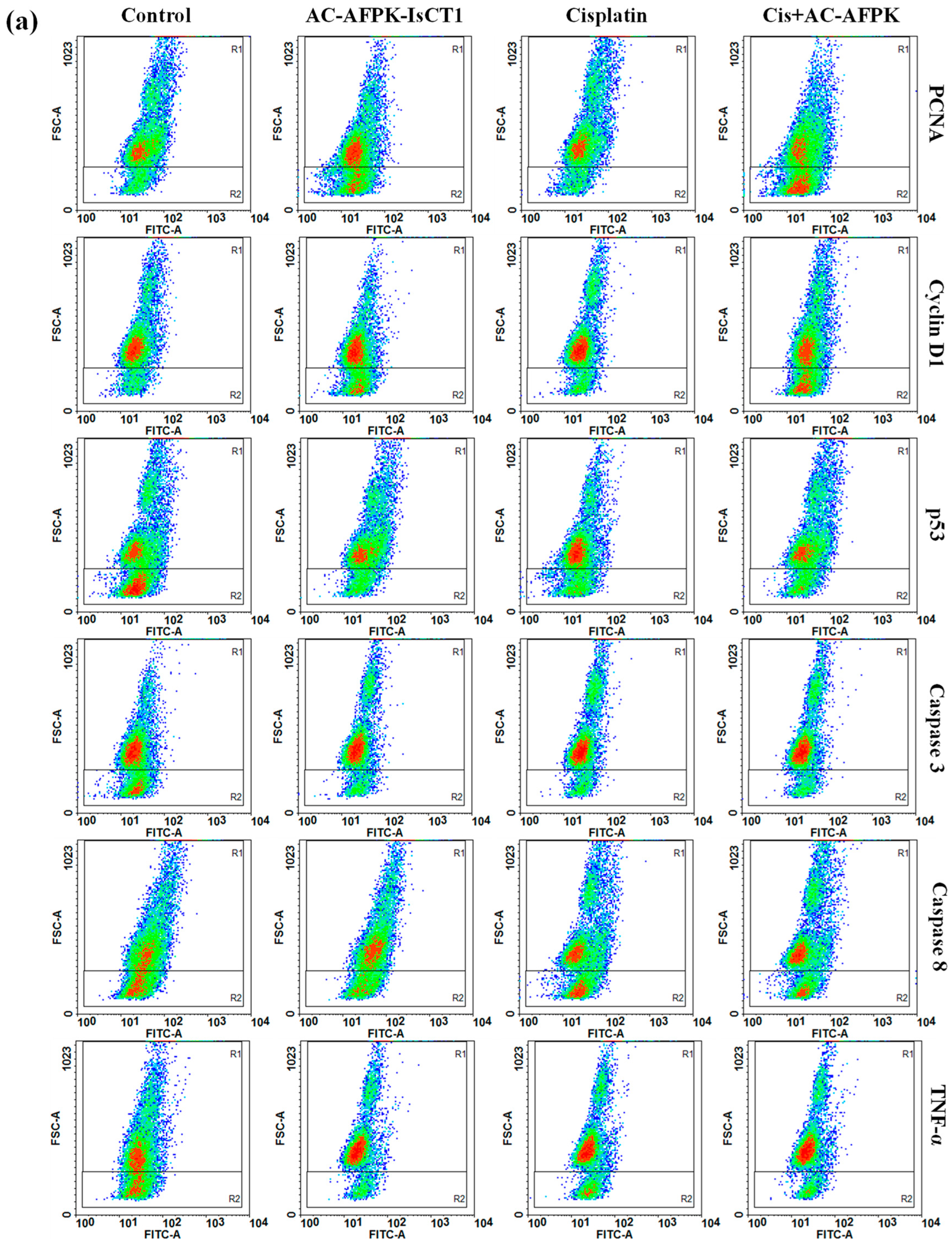
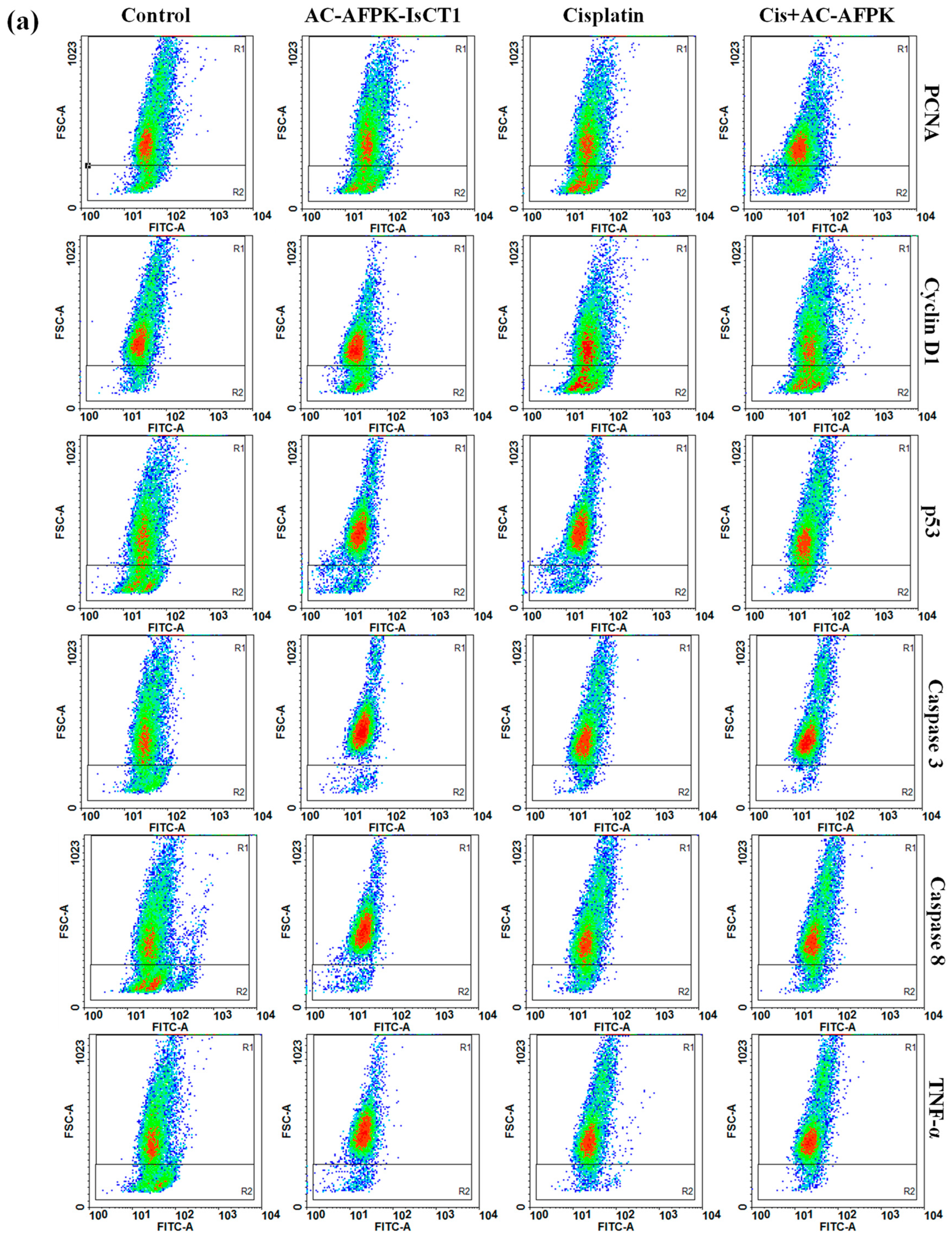
2.6. Effect of Cisplatin and AC-AFPK-IsCT1 Peptide on the Expression of Proteins Involved in Cell Death and Proliferation in Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Solid-Phase Peptide Synthesis (SPPS), Purification, and Analysis
4.2. Cell Culture
4.3. Cytotoxic Activity Determination Using the MTT Colorimetric Method
4.4. CFSE-DA Proliferation Assay
4.5. Cell Cycle Phase and DNA Fragmentation Analysis via Flow Cytometry
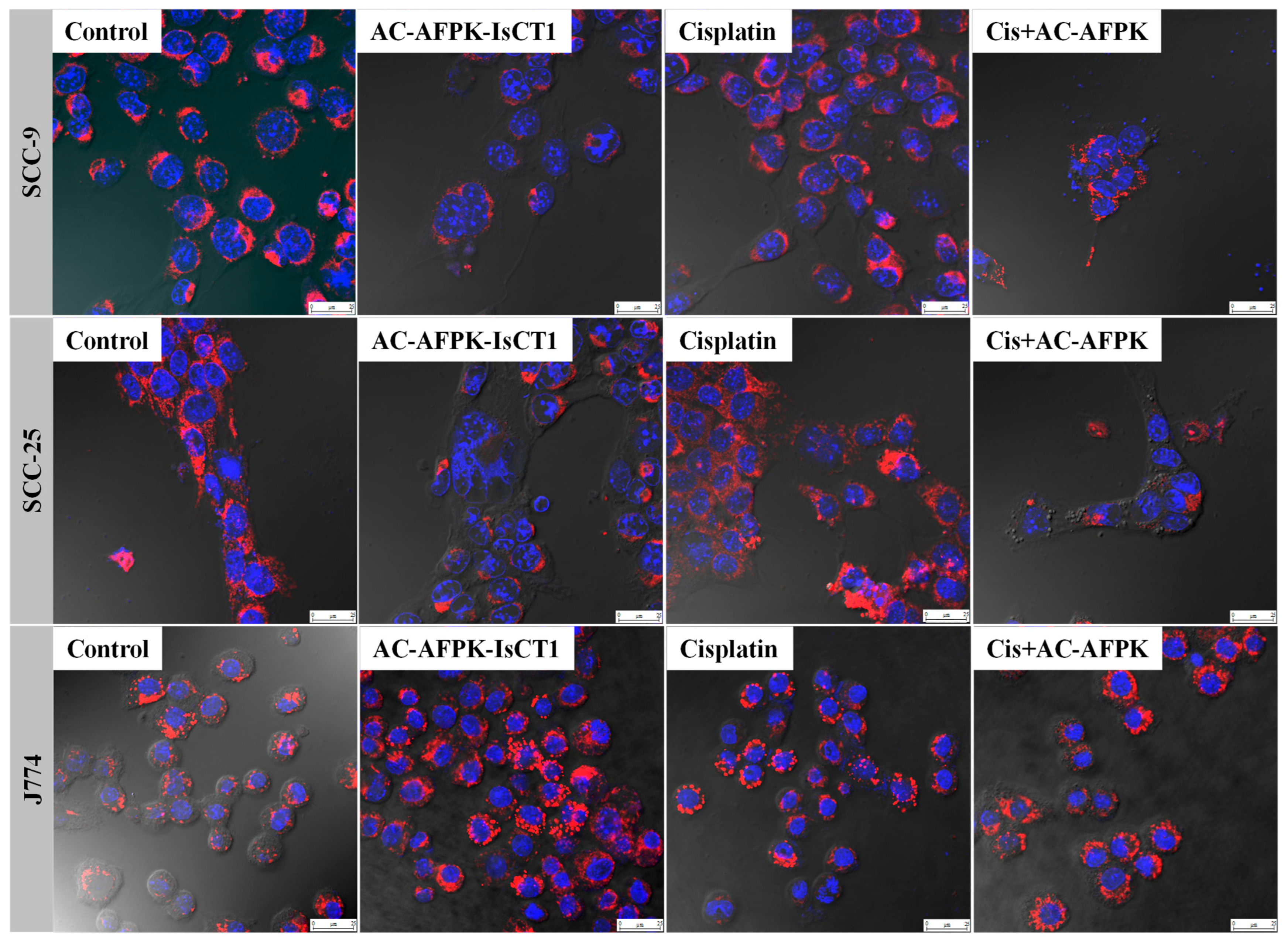
4.6. Morphological Evaluation via Laser Confocal Microscopy
4.7. Evaluation of Cellular Marker Expression via Flow Cytometry
4.8. SynergyFinder 2.0 Analysis of Multiple Drug Combinations
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouaoud, J.; Simon, C.; Martinez, P.; Grégoire, V.; Puppels, G.J.; Bertolus, C.; Breuskin, I.; Koljenović, S.; Machiels, J.-P.; Elkabets, M.; et al. Unmet Needs and Perspectives in Oral Cancer Prevention. Cancers 2022, 14, 1815. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, D.K.; Wong, R.J.; Patel, S.G.; Montero, P.H.; Migliacci, J.C.; Ganly, I.; Shah, J.P. Survival outcomes after treatment of cancer of the oral cavity (1985–2015). Oral Oncol. 2019, 90, 115–121. [Google Scholar] [CrossRef]
- Lapke, N.; Lee, L.-Y.; Liao, C.-T.; Yen, T.-C.; Wang, H.-M.; Ng, S.-H.; Lin, C.-Y.; Lu, Y.-J.; Chen, S.-J. Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 44194–44210. [Google Scholar] [CrossRef]
- Parmar, A.; Glenny, A.-M.; Worthington, H.V.; Macluskey, M.; Conway, D.I.; Clarkson, J.E.; Mc Goldrick, N.; Chan, K.K. Interventions for the treatment of oral cavity and oropharyngeal cancer: Chemotherapy. Cochrane Database Syst. Rev. 2021, 2021, CD006386. [Google Scholar] [CrossRef]
- Adkins, D.; Ley, J.; Oppelt, P.; Gay, H.A.; Daly, M.; Paniello, R.C.; Jackson, R.; Pipkorn, P.; Rich, J.; Zevallos, J.; et al. Impact on health-related quality of life of induction chemotherapy compared with concurrent cisplatin and radiation therapy in patients with head and neck cancer. Clin. Oncol. 2019, 31, e123–e131. [Google Scholar] [CrossRef]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is Cisplatin-Induced Cell Death Always Produced by Apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [CrossRef]
- Khoo, X.H.; Paterson, I.C.; Goh, B.H.; Lee, W.L. Cisplatin-resistance in oral squamous cell carcinoma: Regulation by tumor cell-derived extracellular vesicles. Cancers 2019, 11, 1166. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Castedo, M.; Martins, I.; Michels, J.; Kroemer, G.; Kepp, O.; Vitale, I.; Senovilla, L. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Shadidi, M.; Sioud, M. Selective targeting of cancer cells using synthetic peptides. Drug Resist. Updat. 2003, 6, 363–371. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Hoskin, D.W.; Power Coombs, M.R. Anticancer activities of natural and synthetic peptides. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1117, pp. 131–147. [Google Scholar] [CrossRef]
- Dai, L.; Yasuda, A.; Naoki, H.; Corzo, G.; Andriantsiferana, M.; Nakajima, T. IsCT, a novel cytotoxic linear peptide from scorpion Opisthacanthus madagascariensis. Biochem. Biophys. Res. Commun. 2001, 286, 820–825. [Google Scholar] [CrossRef]
- Acevedo, I.C.C.; Silva, P.I., Jr.; Silva, F.D.; Araujo, I.; Alves, F.L.; Oliveira, C.S.; Oliveira, V.X., Jr. IsCT-based analogs intending better biological activity. J. Pept. Sci. 2019, 25, e3219. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Orozco, R.M.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.D.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Njiki, S.; Mbemi, A.; Yedjou, C.G.; Tchounwou, P.B. Pharmacological Effects of Cisplatin Combination with Natural Products in Cancer Chemotherapy. Int. J. Mol. Sci. 2022, 23, 1532. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatina e além: Mecanismos moleculares de ação e desenvolvimento de resistência a medicamentos na quimioterapia do câncer. Radiol. Oncol. 2019, 53, 148. [Google Scholar] [CrossRef]
- Su, X.; Dong, C.; Zhang, J.; Su, L.; Wang, X.; Cui, H.; Chen, Z. Combination therapy of anti-cancer bioactive peptide with Cisplatin decreases chemotherapy dosing and toxicity to improve the quality of life in xenograft nude mice bearing human gastric cancer. Cell Biosci. 2014, 4, 7. [Google Scholar] [CrossRef]
- Toribio, M.; Henriques, J.C.G.; Gonzaga, W.J.C.; Miranda, C.S.S.; Campos, E.L.; Lorenzon, E.N.; Monteiro, G.J. The combination of peptides and cisplatin for the treatment of oral cancer. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2024, 137, e308. [Google Scholar] [CrossRef]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Pérez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Aldossary, S.A. Review on pharmacology of cisplatin: Clinical use, toxicity and mechanism of resistance of cisplatin. Biomed. Pharmacol. J. 2019, 12, 7–15. [Google Scholar] [CrossRef]
- Lin, Y.-X.; Wang, J.; An, H.-W.; Qi, B.; Wang, Y.; Wang, L.; Shi, J.; Mei, L.; Wang, H. Peptide-Based Autophagic Gene and Cisplatin Co-delivery Systems Enable Improved Chemotherapy Resistance. Nano Lett. 2019, 19, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.G.d.S.; de Oliveira, C.S.; Oliveira, V.X., Jr.; Alves, R.C.B.; Poyet, J.-L.; Maria, D.A. Antitumoral and Antiproliferative Potential of Synthetic Derivatives of Scorpion Peptide IsCT1 in an Oral Cavity Squamous Carcinoma. Molecules 2024, 29, 4533. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.X.; Silva, P.I.; Torres, M.D.T.; Pedron, C.N.; Silva, F.D.; Andrade, V.B.; Oliveira, C.S.; de la Fuente-Nunez, C. Synthetic Peptide Derived from Scorpion Venom Displays Minimal Toxicity and Anti-infective Activity in an Animal Model. ACS Infect. Dis. 2021, 7, 2736–2745. [Google Scholar] [CrossRef]
- Yang, S.K.; Han, Y.C.; He, J.R.; Yang, M.; Zhang, W.; Zhan, M. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed. Pharmacother. 2020, 130, 110521. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Ito, H.; Matsui, H. The cisplatin-derived increase of mitochondrial reactive oxygen species enhances the effectiveness of photodynamic therapy via transporter regulation. Cells 2019, 8, 918. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Fischer, U.; Schulze-Osthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005, 12, 942–961. [Google Scholar] [CrossRef]
- Kaufmann, T.; Strasser, A.; Jost, P.J. Fas death receptor signalling: Roles of Bid and XIAP. Cell Death Differ. 2011, 19, 42–50. [Google Scholar] [CrossRef]
- Zhao, C. Exploring cell death pathways in oral cancer: Mechanisms, therapeutic strategies, and future perspectives. Discov. Oncol. 2025, 16, 395. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Cabral, L.G.d.S.; de Oliveira, C.S.; Alves, R.C.B.; Oliveira, V.X., Jr.; Poyet, J.-L.; Mari, D.A. Synergistic Effect of the Association of BR2 Peptide with 2-Aminoethyl Dihydrogen Phosphate in Triple-Negative Breast Cancer Cells. Arch. Breast Cancer 2023, 10, 148–158. [Google Scholar] [CrossRef]
- Timur, S.S.; Gürsoy, R.N. The role of peptide-based therapeutics in oncotherapy. J. Drug Target. 2021, 29, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, C.M.; Barry, M.A.; Eastman, A.; Barry, M.A.; Eastman, A. Analysis of Events Associated With Cell Cycle Arrest at G 2 Phase and Cell Death Induced by Cisplatin. JNCI J. Natl. Cancer Inst. 1990, 82, 749–755. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Zhao, H.; Zhang, S.; Lee, M.Y.W.T.; Lee, E.Y.C.; Zhang, Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21 WAF1, Cdt1 and the p12 subunit of DNA polymerase δ revealed in individual cells by cytometry. Oncotarget 2015, 6, 11735–11750. [Google Scholar] [CrossRef] [PubMed]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef]
- Zhou, Q.; Greene, L.A. Dpep Inhibits Cancer Cell Growth and Survival via Shared and Context-Dependent Transcriptome Perturbations. Cancers 2023, 15, 5318. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Cell | IC50 | Cell | IC50 | Cell | IC50 | Cell | IC50 |
|---|---|---|---|---|---|---|---|---|
| Cisplatin 24 h | SCC-9 | 94.2 | SCC-25 | 47.6 | FN1 | 73.11 | J-774 | 68.3 |
| Cisplatin 48 h | 5.2 | 15.2 | 3.06 | 8.7 | ||||
| Cis + AC-AFPK 50 µM 24 h | 0.4 | 24.6 | - | - | ||||
| Cis + AC-AFPK 30 µM 24 h | 19.9 | 31.7 | - | - | ||||
| Cis + AC-AFPK 10 µM 24 h | 44.6 | 38.8 | - | - | ||||
| Cis + AC-AFPK 50 µM 48 h | 0.8 | 4.7 | 36.21 | 48.57 | ||||
| Cis + AC-AFPK 30 µM 48 h | 1.1 | 5.1 | 34 | 42.1 | ||||
| Cis + AC-AFPK 10 µM 48 h | 1.3 | 6.7 | 31.8 | 37.5 |
| Treatment for SCC-9, FN1 and J-774 Cells | |
|---|---|
| Group 1 | Cisplatin |
| Group 2 | Cisplatin (50 μM)/AC-AFPK-IsCT1 10 µM |
| Group 3 | Cisplatin (50 μM)/AC-AFPK-IsCT1 30 µM |
| Group 4 | Cisplatin (50 μM)/AC-AFPK-IsCT1 50 µM |
| Treatment for SCC-25 cells | |
| Group 5 | Cisplatin |
| Group 6 | Cisplatin (25 μM)/AC-AFPK-IsCT1 10 µM |
| Group 7 | Cisplatin (25 μM)/AC-AFPK-IsCT1 20 µM |
| Group 8 | Cisplatin (25 μM)/AC-AFPK-IsCT1 40 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, L.G.d.S.; de Oliveira, C.S.; Oliveira, V.X., Jr.; de Abreu Paulo, E.P.; Poyet, J.-L.; Maria, D.A. Evaluation of the Antitumor and Antiproliferative Potential of Synthetic Peptides Derived from IsCT1, Associated with Cisplatin, in Squamous Cell Carcinoma of the Oral Cavity. Molecules 2025, 30, 2594. https://doi.org/10.3390/molecules30122594
Cabral LGdS, de Oliveira CS, Oliveira VX Jr., de Abreu Paulo EP, Poyet J-L, Maria DA. Evaluation of the Antitumor and Antiproliferative Potential of Synthetic Peptides Derived from IsCT1, Associated with Cisplatin, in Squamous Cell Carcinoma of the Oral Cavity. Molecules. 2025; 30(12):2594. https://doi.org/10.3390/molecules30122594
Chicago/Turabian StyleCabral, Laertty Garcia de Sousa, Cyntia Silva de Oliveira, Vani Xavier Oliveira, Jr., Ellen Paim de Abreu Paulo, Jean-Luc Poyet, and Durvanei Augusto Maria. 2025. "Evaluation of the Antitumor and Antiproliferative Potential of Synthetic Peptides Derived from IsCT1, Associated with Cisplatin, in Squamous Cell Carcinoma of the Oral Cavity" Molecules 30, no. 12: 2594. https://doi.org/10.3390/molecules30122594
APA StyleCabral, L. G. d. S., de Oliveira, C. S., Oliveira, V. X., Jr., de Abreu Paulo, E. P., Poyet, J.-L., & Maria, D. A. (2025). Evaluation of the Antitumor and Antiproliferative Potential of Synthetic Peptides Derived from IsCT1, Associated with Cisplatin, in Squamous Cell Carcinoma of the Oral Cavity. Molecules, 30(12), 2594. https://doi.org/10.3390/molecules30122594