3.3. Synthesis
((
2R,
3R,
4R,
5R)-5-(6-amino-9H-purin-9-yl)-3-((
tert-butyldimethylsilyl)oxy)-4-fluorotetrahydrofuran-2-yl)methyl sulfamate (
4). The general procedure A (Method A2) was followed with compound
2 (590 mg, 1.54 mmol), prepared as previously described [
22], in DMAc (2.4 mL), sulfamoyl chloride (426 mg, 3.69 mmol) in MeCN (1.9 mL). The crude product was purified by column chromatography (Eluent CH
2Cl
2/MeOH 94:6) to give compound
4 (570 mg, 1.23 mmol, 80%) as a white solid.
1H NMR (CD
3CN, 500 MHz): δ 0.00 (s, 3H), 0.04 (s, 3H), 0.84 (s, 9H), 3.87 (dd,
J = 12.2 Hz,
J = 3.4 Hz, 1H), 4.03 (dd,
J = 12.2 Hz,
J = 2.3 Hz, 1H), 4.30–4.33 (m, 1H), 5.51 (ddd,
JHF = 18.5 Hz,
J = 7.2 Hz,
J = 4.6 Hz, 1H), 5.76 (ddd,
JHF = 51.9 Hz,
J = 4.6 Hz,
J = 1.8 Hz, 1H), 6.05 (br s, 4H), 6.26 (dd,
JHF = 18.3 Hz,
J = 1.8 Hz, 1H), 8.11 (s, 1H), 8.24 (s, 1H).
19F NMR (CD
3CN, 470 MHz): δ −202.0 (dt,
JFH = 51.9 Hz,
JFH = 18.5 Hz).
13C NMR (CD
3CN, 150 MHz): δ −5.4, −5.3, 19.0, 26.2, 61.8, 74.8 (d,
JCF = 14.8 Hz), 82.0, 87.9 (d,
JCF = 33.6 Hz), 92.4 (d,
JCF = 189.9 Hz), 120.7, 140.5, 150.3, 154.1, 157.0. HRMS-ESI (
m/
z) calcd for C
16H
28FN
6O
5SiS [M+H]
+ 463.1595, found 463.1602. The NMR spectrum is identical to that previously reported [
13].
((
2R,
3S,
4S,
5R)-5-(6-amino-9H-purin-9-yl)-4-azido-3-((
tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methyl sulfamate (
5). The general procedure A (Method A2) was followed with compound
3 (250 mg, 0.61 mmol), prepared as previously described [
23], in DMAc (1 mL), sulfamoyl chloride (170 mg, 1.47 mmol) in MeCN (0.75 mL) to give compound
5 (265 mg, 0.54 mmol, 89%) as a white solid. The compound was used in the next step without further purification.
1H NMR (MeOD, 500 MHz): δ 0.23 (s, 3H), 0.24 (s, 3H), 1.00 (s, 9H), 4.28–4.32 (m, 2H), 4.40–4.43 (m, 1H), 4.72–4.74 (m, 1H), 4.87–4.89 (m, 1H), 6.17 (d,
J = 6.1 Hz, 1H), 8.23 (s, 1H), 8.30 (s, 1H).
13C NMR (MeOD, 125 MHz): δ −4.8, −4.6, 21.3, 26.3, 66.1, 68.8, 74.1, 84.6, 87.6, 120.6, 141.1, 150.6, 154.1, 157.5. HRMS-ESI (
m/
z) calcd for C
16H
28N
9O
5SSi [M+H]
+ 486.1703, found 486.1710.
((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-3-((tert-butyldimethylsilyl)oxy)-4-fluorotetrahydrofuran-2-yl)methyl ((tert-butoxycarbonyl)-D-alanyl)sulfamate (6). The general procedure B was followed with compound 4 (375 mg, 0.81 mmol), Boc-D-Ala-OSu (232 mg, 0.81 mmol), and DBU (0.18 mL, 1.22 mmol) in DMF (10.8 mL). The crude was purified by column chromatography (Eluent CH2Cl2/MeOH 94:6 to 9:1) to give compound 6 (392 mg, 0.61 mmol, 76%) as a white foam. 1H NMR (DMSO-d6, 600 MHz): δ −0.02 (s, 3H), 0.00 (s, 3H), 0.82 (s, 9H), 1.17 (d, J = 7.1 Hz, 3H), 1.33 (s, 9H), 3.75–3.83 (m, 2H), 3.91–3.95 (m, 1H), 4.16–4.20 (m, 1H), 4.37–4.46 (m, 1H), 5.66 (d, JHF = 52.1 Hz, 1H), 6.27 (dd, JHF = 17.3 Hz, J = 2.3 Hz, 1H), 6.46 (br s, 1H), 7.40 (br s, 2H), 8.14 (s, 1H), 8.27 (s, 1H). 19F NMR (DMSO-d6, 565 MHz): δ −201.5 (m). 13C NMR (DMSO-d6, 150 MHz): δ −5.6, −5.5, 18.1, 18.5, 25.8, 28.2, 51.3, 62.2, 74.0, 77.8, 81.5, 85.7 (d, JCF = 32.2 Hz), 91.2 (d, JCF = 190.2 Hz), 119.0, 139.1, 148.8, 152.7, 154.8, 156.1, 176.1. HRMS-ESI (m/z) calcd for C24H41FN7O8SiS [M+H]+ 634.2491, found 634.2489.
((2R,3S,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-azido-3-((tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methyl ((tert-butoxycarbonyl)-D-alanyl)sulfamate (7). The general procedure B was followed with compound 5 (438 mg, 0.90 mmol), Boc-D-Ala-OSu (258 g, 0.90 mmol), and DBU (0.20 mL, 1.35 mmol) in DMF (12 mL). The crude was purified by column chromatography (Eluent EtOAc with 1% AcOH) to give compound 7 (408 mg, 0.62 mmol, 69%) as a white foam. 1H NMR (CD3CN, 600 MHz): δ 0.18 (s, 3H), 0.19 (s, 3H), 0.96 (s, 9H), 1.27 (d, J = 7.1 Hz, 3H), 1.37 (s, 9H), 4.00–4.02 (m, 1H), 4.30–4.32 (m, 1H), 4.44 (dd, J = 11.2 Hz, J = 3.7 Hz, 1H), 4.55–4.60 (m, 2H), 4.78–4.79 (m, 1H), 5.82 (br s, 1H), 6.09 (d, J = 7.1 Hz, 1H), 8.34 (s, 1H), 8.38 (s, 1H). 13C NMR (CD3CN, 150 MHz): δ −4.7, −4.6, 18.6, 19.6, 26.1, 28.6, 53.7, 66.5, 68.1, 73.8, 79.7, 84.5, 86.5, 120.2, 141.1, 150.6, 154.1, 156.5, 156.7, 171.7. HRMS-ESI (m/z) calcd for C24H41N10O8SSi [M+H]+ 657.2599, found 657.2601.
((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methyl (D-alanyl)sulfamate salt (8). The general procedure C (Method C2) was followed with compound 6 (498 mg, 0.78 mmol), TFA (9.7 mL), and CH2Cl2/H2O (8/2 mL) to give compound 8 (450 mg, 0.69 mmol, 89%) as a white solid (2 TFA salt). 1H NMR (D2O, 500 MHz): δ 1.52 (d, J = 7.1 Hz, 3H), 3.81 (dd, J = 13.1 Hz, J = 3.7 Hz, 1H), 3.89 (q, J = 7.1 Hz, 1H), 3.98 (dd, J = 13.1 Hz, J = 2.3 Hz, 1H), 4.42–4.46 (m, 1H), 5.51 (ddd, JHF = 17.2 Hz, J = 7.6 Hz, J = 4.8 Hz, 1H), 5.76 (ddd, JHF = 51.1 Hz, J = 4.8 Hz, J = 2.0 Hz, 1H), 6.51 (dd, JHF = 18.4 Hz, J = 2.0 Hz, 1H), 8.45 (s, 1H), 8.51 (s, 1H). 19F NMR (D2O, 470 MHz): δ −200.8 (dt, JFH = 51.1 Hz, JFH = 17.6 Hz). 13C NMR (D2O, 150 MHz): δ 51.3, 59.2, 73.8 (d, JCF = 14.9 Hz), 81.5, 87.3 (d, JCF = 34.8 Hz), 91.4 (d, JCF = 191.9 Hz), 116.2 (q, JCF = 291.4 Hz), 117.2, 119.0, 143.3, 144.6, 147.9, 150.0, 162.9 (q, JCF = 35.3 Hz, TFA), 176.4. HPLC purity: (96.6%, tR = 4.48 min). HRMS-ESI (m/z) calcd for C13H18FN7O6S [M+H]+ 420.1102, found 420.1102.
((2R,3S,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-azido-3-hydroxytetrahydrofuran-2-yl)methyl (D-alanyl)sulfamate salt (9). To a solution of compound 7 (165 mg, 0.25 mmol, 1 equiv.) in H2O (2.5 mL) was added TFA (10 mL) at 20 °C. The mixture was stirred for 12 h and concentrated under reduced pressure. The residue was taken up with a small amount of MeOH. By addition of Et2O, a precipitate appeared, which was isolated by filtration to give compound 9 (91 mg, 0.13 mmol, 54%) as a white solid (2 TFA salt). 1H NMR (DMSO-d6, 500 MHz): δ 1.29 (d, J = 7.1 Hz, 3H), 3.48–3.50 (m, 1H), 4.07 (dd, J = 11.1 Hz, J = 4.3 Hz, 1H), 4.14–4.17 (m, 1H), 4.19 (dd, J = 11.1 Hz, J = 3.7 Hz, 1H), 4.57–4.59 (m, 1H), 4.67–4.69 (m, 1H), 6.06 (d, J = 6.1 Hz, 1H), 6.17 (d, J = 5.4 Hz, 1H), 7.35 (br s, 1H), 7.81 (br s, 2H), 8.16 (s, 1H), 8.39 (s, 1H). 13C NMR (DMSO-d6, 150 MHz): δ 17.2, 50.8, 64.2, 67.2, 71.5, 83.0, 84.8, 118.9, 139.3, 149.3, 152.9, 156.1, 173.4. HPLC purity: (94.9%, tR = 4.59 min). HRMS-ESI (m/z) calcd for C13H19N10O6S [M+H]+ 443.1210, found 443.1208.
(
2R,
3R,
4S,
5R,
6R)-2-(acetoxymethyl)-6-((1-((
2R,
3R,
4S,
5R)-2-(6-amino-9H-purin-9-yl)-5-(((N-((
tert-butoxycarbonyl)-D-alanyl)sulfamoyl)oxy)methyl)-4-((
tert-butyldimethylsilyl)oxy)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (
10). The general procedure E was followed with compound
7 (269 mg, 0.41 mmol), glucosyl-
O-propargyl ether (175 mg, 0.45 mmol), prepared as previously described [
50], sodium ascorbate (8 mg, 0.04 mmol) and CuSO
4.5H
2O (5 mg, 0.02 mmol) in
t-BuOH/H
2O (4 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 9:1) to give compound
10 (276 mg, 0.26 mmol, 64%) as a white solid.
1H NMR (CD
3CN, 600 MHz): δ −0.18 (s, 3H), 0.01 (s, 3H), 0.73 (s, 9H), 1.23 (d,
J = 7.4 Hz, 3H), 1.33 (s, 9H), 1.85 (s, 3H), 1.92 (s, 3H), 1.97 (s, 3H), 2.02 (s, 3H), 3.82 (ddd,
J = 10.1 Hz,
J = 4.7 Hz,
J = 2.5 Hz, 1H), 3.98 (br s, 1H), 4.10 (dd,
J = 12.4 Hz,
J = 2.5 Hz, 1H), 4.25 (dd,
J = 12.4 Hz,
J = 4.7 Hz, 1H), 4.28 (dd,
J = 10.4 Hz,
J = 3.6 Hz, 1H), 4.38–4.39 (m, 1H), 4.44–4.46 (m, 1H), 4.68 (d,
J = 12.4 Hz, 1H), 4.71 (d,
J = 8.1 Hz, 1H), 4.81 (d,
J = 12.4 Hz, 1H), 4.85–4.88 (m, 2H), 5.02 (t,
J = 9.7 Hz, 1H), 5.18 (t,
J = 9.7 Hz, 1H), 5.78 (br s, 1H), 5.96 (br s, 1H), 6.28 (br s, 2H), 6.80 (d,
J = 5.6 Hz, 1H), 7.94 (s, 1H), 8.19 (s, 1H), 8.39 (br s, 1H).
13C NMR (CD
3CN, 150 MHz): δ −4.9, −4.8, 18.3, 19.7, 20.8, 20.9 (2C), 21.0, 25.9, 28.6, 53.7, 62.7, 62.9, 66.2, 68.2, 69.2, 71.9, 72.5, 72.8, 73.4, 79.7, 85.1, 86.3, 100.3, 120.3, 126.0, 141.0, 144.3, 150.6, 154.1, 156.4, 156.8, 170.2, 170.5, 170.9, 171.4, 182.0. HRMS-ESI (
m/
z) calcd for C
41H
63N
10O
18SSi [M+H]
+ 1043.3812, found 1043.3835.
((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3-hydroxy-4-(4-((((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)methyl (D-alanyl)sulfamate salt (11). A solution of compound 10 (70 mg, 0.06 mmol, 1 equiv.) in H2O (2 mL) and TFA (8 mL) was stirred for 12 h at 20 °C. Volatiles were removed under reduced pressure, and the residue was dissolved in MeOH (1 mL), and a solution of MeONa (5.4 M) in MeOH (0.05 mL, 0.3 mmol, 5 equiv.) was added at 20 °C. The mixture was stirred for 1 h and quenched with an aqueous solution of TFA (10%), then concentrated under reduced pressure. The residue was dissolved in THF (1 mL), and a solution of 37% triethylamine trihydrofluoride (0.1 mL, 0.24 mmol, 4 equiv.) was added at 20 °C. The mixture was stirred for 12 h, and volatiles were removed under reduced pressure. The crude was purified by HPLC RP-18 (MeCN/H2O, linear gradient) to give compound 11 (17 mg, 0.26 mmol, 32%) as a white solid (2 TFA salt). 1H NMR (D2O, 600 MHz): δ 1.48 (d, J = 7.2 Hz, 3H), 3.24–3.26 (m, 1H), 3.33–3.37 (m, 1H), 3.41–3.45 (m, 2H), 3.66 (dd, J = 12.7 Hz, J = 6.3 Hz, 1H), 3.85–3.87 (m, 1H), 3.89–3.93 (m, 1H), 4.46–4.48 (m, 1H), 4.51–4.57 (m, 2H), 4.62–4.64 (m, 1H), 4.88 (d, J = 12.7 Hz, 1H), 4.99 (d, J = 12.7 Hz, 1H), 5.05 (t, J = 5.6 Hz, 1H), 5.99 (t, J = 5.6 Hz, 1H), 6.99 (d, J = 4.9 Hz, 1H), 8.26 (s, 1H), 8.39 (s, 1H), 8.60 (s, 1H). 13C NMR (D2O, 150 MHz): δ 16.3, 51.0, 60.6, 61.7, 65.8, 68.3, 69.5, 69.8, 72.9, 75.6, 75.9, 83.2, 86.3, 101.4, 116.2 (q, JCF = 291.1 Hz), 119.2, 126.6, 142.7, 143.7, 144.6, 148.1, 149.9, 162.9 (q, JCF = 35.8 Hz), 175.4. HPLC purity: (88.5%, tR = 2.42 min). HRMS-ESI (m/z) calcd for C22H33N10O12S [M+H]+ 661.2000, found 661.2016.
diisopropyl ((
E)-4-(((
2R,
3R,
4R,
5R)-2-(6-(
N-benzoylbenzamido)-9H-purin-9-yl)-4-((
tert-butyldimethylsilyl)oxy)-5-(((
tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-3-yl)oxy)-1,1-difluorobut-2-en-1-yl)phosphonate (
13). The general procedure F was followed with compound
12 (320 mg, 0.44 mmol), prepared as previously described [
33], TBDMSCl (218 mg, 1.45 mmol), imidazole (196 mg, 2.89 mmol), and DMAP (4.9 mg, 0.04 mmol) in DMF (5 mL). The crude product was purified by column chromatography (Eluent Pentane/EtOAc 7:3) to give compound
13 (333 mg, 0.35 mmol, 79%) as a white foam.
1H NMR (CDCl
3, 500 MHz): δ 0.06 (s, 3H), 0.08 (s, 3H), 0.11 (s, 6H), 0.90 (s, 9H), 0.92 (s, 9H), 1.33 (t,
J = 6.2 Hz, 6H), 1.35 (dd,
J = 6.2 Hz,
JHP = 1.7 Hz, 6H), 3.78 (dd,
J = 11.7 Hz,
J = 3.1 Hz, 1H), 3.98 (dd,
J = 11.7 Hz,
J = 3.4 Hz, 1H), 4.13 (dt,
J = 5.7 Hz,
J = 3.1 Hz, 1H), 4.22–4.31 (m, 2H), 4.36 (dd,
J = 5.7 Hz,
J = 3.6 Hz, 1H), 4.53–4.55 (m, 1H), 4.79–4.86 (m, 2H), 5.95–6.03 (m, 1H), 6.18 (d,
J = 3.6 Hz, 1H), 6.26–6.33 (m, 1H), 7.33–7.36 (m, 4H), 7.46–7.49 (m, 2H), 7.84–7.86 (m, 4H), 8.37 (s, 1H), 8.65 (s, 1H).
19F NMR (CDCl
3, 565 MHz): δ −109.5 (ddd,
JFF = 298.3 Hz,
JFP = 113.2 Hz,
JFH = 12.4 Hz), −110.3 (ddd,
JFF = 298.3 Hz,
JFP = 113.2 Hz,
JFH = 12.4 Hz).
31P NMR (CDCl
3, 243 MHz): δ 4.3 (t,
JPF = 113.2 Hz).
13C NMR (CDCl
3, 150 MHz): δ −5.3, −5.2, −4.7, −4.5, 18.2, 18.6, 23.8 and 23.9 (d,
JCP = 4.1 Hz), 24.3 (d,
JCP = 3.4 Hz, 2C), 25.8, 26.2, 61.8, 69.3, 69.8, 73.9 (d,
JCP = 7.1 Hz, 2C), 82.1, 85.1, 87.3, 116.8 (dt,
JCF = 258.9 Hz,
JCP = 220.7 Hz), 122.7 (dt,
JCF = 22.3 Hz,
JCP = 13.4 Hz), 128.3, 128.8 (4C), 129.6 (4C), 133.1 (2C), 134.2 (2C), 134.4 (dt,
JCF = 9.8 Hz,
JCP = 5.6 Hz), 143.7, 151.9, 152.3, 152.7, 172.4. HRMS-ESI (
m/
z) calcd for C
46H
67F
2N
5O
9PSi
2 [M+H]
+ 958.4183, found 958.4179.
tert-butyl (tert-butoxycarbonyl)(9-((2R,3R,4R,5R)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-(((E)-4-(diisopropoxyphosphoryl)-4,4-difluorobut-2-en-1-yl)oxy)tetrahydrofuran-2-yl)-9H-purin-6-yl)carbamate (14). To a solution of compound 13 (300 mg, 0.44 mmol, 1 equiv.) in EtOH (3.5 mL), CH3NH2 (2.1 mL, 33% in EtOH) was added at 20 °C. The mixture was stirred for 1 h and followed by TLC. When the conversion was quantitative, the solvent was removed under reduced pressure. It was dissolved in DMF (4 mL), and triethylamine (0.17 mL, 1.20 mmol, 3 equiv.) and DMAP (5 mg, 0.04 mmol, 0.5 equiv.) were added at 20 °C. The solution was cooled, and (Boc)2O (0.27 mL, 1.20 mmol, 3 equiv.) was added dropwise at 0 °C. The reaction mixture was stirred for 12 h and quenched with an aqueous solution of HCl (1 M). The aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (Eluent Pentane/EtOAc 7:3) to give compound 14 (326 mg, 0.34 mmol, 78%) as a waxy oil. 1H NMR (CDCl3, 600 MHz): δ 0.07 (s, 3H), 0.08 (s, 3H), 0.12 (s, 3H), 0.13 (s, 3H), 0.91 (s, 9H), 0.94 (s, 9H), 1.33 (dd, J = 6.4 Hz, JHP = 4.3 Hz, 6H), 1.36 (dd, J = 6.4 Hz, JHP = 1.9 Hz, 6H), 1.45 (s, 18H), 3.81 (dd, J = 11.6 Hz, J = 2.6 Hz, 1H), 4.05 (dd, J = 11.6 Hz, J = 3.4 Hz, 1H), 4.16 (dt, J = 6.3 Hz, J = 2.6 Hz, 1H), 4.24 (dd, J = 4.7 Hz, J = 3.1 Hz, 1H), 4.26–4.30 (m, 1H), 4.36–4.40 (m, 1H), 4.52 (dd, J = 6.3 Hz, J = 4.7 Hz, 1H), 4.80–4.86 (m, 2H), 5.98–6.06 (m, 1H), 6.22 (d, J = 3.1 Hz, 1H), 6.29–6.33 (m, 1H), 8.54 (s, 1H), 8.84 (s, 1H). 19F NMR (CDCl3, 565 MHz): δ −109.6 (ddd, JFF = 298.4 Hz, JFP = 112.4 Hz, JFH = 12.2 Hz), −110.2 (ddd, JFF = 298.4 Hz, JFP = 112.4 Hz, JFH = 12.2 Hz). 31P NMR (CDCl3, 243 MHz): δ 4.3 (t, JPF = 112.4 Hz). 13C NMR (CDCl3, 150 MHz): δ −5.3, −5.2, −4.8, −4.5, 18.2, 18.6, 23.8 and 23.9 (d, JCP = 4.3 Hz), 24.3 (d, JCP = 3.4 Hz, 2C), 25.8, 26.2, 27.9, 61.5, 69.3, 69.4, 73.9 (d, JCP = 7.1 Hz, 2C), 82.5, 84.0, 84.7, 87.3, 116.7 (dt, JCF = 259.1 Hz, JCP = 220.8 Hz), 122.7 (dt, JCF = 21.3 Hz, JCP = 13.4 Hz), 129.4, 134.4 (dt, JCF = 9.8 Hz, JCP = 5.3 Hz), 143.4, 150.4, 150.6, 152.2, 152.6. HRMS-ESI (m/z) calcd for C42H75F2N5O11PSi2 [M+H]+ 950.4707, found 950.4697.
tert-butyl (tert-butoxycarbonyl)(9-((2R,3R,4R,5R)-4-((tert-butyldimethylsilyl)oxy)-3-(((E)-4-(diisopropoxyphosphoryl)-4,4-difluorobut-2-en-1-yl)oxy)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)carbamate (15). The general procedure G (method G1) was followed with compound 14 (666 mg, 0.70 mmol) in H2O (1.2 mL), THF (1.2 mL), and AcOH (3.75 mL). The crude product was purified by column chromatography (Eluent Cyclohexane/EtOAc 6:4 to 4:6) to give compound 15 (315 mg, 0.38 mmol, 54%) as a colorless oil. 1H NMR (CDCl3, 600 MHz): δ 0.12 (s, 6H), 0.93 (s, 9H), 1.31 (dd, J = 6.2 Hz, JHP = 2.6 Hz, 6H), 1.33 (d, J = 6.2 Hz, 6H), 1.46 (s, 18H), 3.73 (dd, J = 13.1 Hz, J = 1.4 Hz, 1H), 3.86–3.89 (m, 1H), 3.97 (dd, J = 13.1 Hz, J = 1.8 Hz, 1H), 4.08–4.11 (m, 1H), 4.21–4.23 (m, 1H), 4.57–4.58 (m, 1H), 4.72 (dd, J = 7.6 Hz, J = 4.5 Hz, 1H), 4.77–4.83 (m, 2H), 5.83–5.90 (m, 1H), 6.01 (d, J = 7.6 Hz, 1H), 6.10–6.15 (m, 1H), 8.22 (s, 1H), 8.82 (s, 1H). 19F NMR (CDCl3, 470 MHz): δ −109.7 (ddd, JFF = 315.6 Hz, JFP = 113.2 Hz, JFH = 12.3 Hz), −110.6 (ddd, JFF = 315.6 Hz, JFP = 113.2 Hz, JFH = 12.3 Hz). 31P NMR (CDCl3, 202 MHz): δ 4.2 (t, JPF = 113.2 Hz). 13C NMR (CDCl3, 150 MHz): δ −4.6, −4.5, 18.2, 23.8 and 23.9 (d, JCP = 2.3 Hz), 24.2 and 24.3 (d, JCP = 1.4 Hz), 25.8, 27.9, 62.9, 69.1, 71.6, 73.9 and 74.0 (d, JCP = 3.7 Hz), 80.9, 84.3, 89.5, 89.6, 116.6 (dt, JCF = 258.6 Hz, JCP = 220.6 Hz), 122.7 (dt, JCF = 21.2 Hz, JCP = 13.4 Hz), 130.4, 134.0 (dt, JCF = 10.1 Hz, JCP = 5.8 Hz), 144.9, 150.5, 151.4, 151.6, 152.0. HRMS-ESI (m/z) calcd for C36H61F2N5O11PSi [M+H]+ 836.3843, found 836.3840.
((2R,3R,4R,5R)-5-(6-(bis(tert-butoxycarbonyl)amino)-9H-purin-9-yl)-3-((tert-butyldimethylsilyl)oxy)-4-(((E)-4-(diisopropoxyphosphoryl)-4,4-difluorobut-2-en-1-yl)oxy)tetrahydrofuran-2-yl)methyl sulfamate (16). The general procedure A (Method A2) was followed with compound 15 (100 mg, 0.12 mmol) in DMAc (0.6 mL), sulfamoyl chloride (33 mg, 0.29 mmol) in MeCN (0.5 mL) to give compound 16 (105 mg, 0.10 mmol, 84%) with DMAc (13% w/w) as a colorless oil. This compound was used in the next step without further purification to avoid its degradation. 1H NMR (CDCl3, 600 MHz): δ 0.12 (s, 6H), 0.91 (s, 9H), 1.33 (dd, J = 6.2 Hz, JHP = 2.4 Hz, 6H), 1.36 (d, J = 6.2 Hz, 6H), 1.45 (s, 18H), 4.20–4.27 (m, 2H), 4.30–4.33 (m, 2H), 4.47–4.49 (m, 1H), 4.53–4.55 (m, 1H), 4.58–4.60 (m, 1H), 4.80–4.87 (m, 2H), 5.88 (br, s, 2H), 5.93–5.96 (m, 1H), 6.20 (d, J = 4.2 Hz, 1H), 6.20–6.24 (m, 1H), 8.44 (s, 1H), 8.85 (s, 1H). 19F NMR (CDCl3, 565 MHz): δ −109.7 (ddd, JFF = 334.1 Hz, JFP = 114.2 Hz, JFH = 12.8 Hz, 1F), −110.5 (ddd, JFF = 334.1 Hz, JFP = 114.2 Hz, JFH = 12.8 Hz, 1F). 31P NMR (CDCl3, 243 MHz): δ 4.0 (t, JPF = 114.2 Hz). 13C NMR (CDCl3, 150 MHz): δ −4.8, −4.6, 18.1, 23.8 and 23.9 (d, JCP = 4.7 Hz), 24.2 and 24.3 (d, JCP = 2.3 Hz), 25.8, 27.9, 67.9, 69.3, 70.5, 74.3 and 74.4 (d, JCP = 7.2 Hz), 80.6, 82.7, 84.3, 87.5, 116.5 (dt, JCF = 258.4 Hz, JCP = 220.7 Hz), 123.3 (dt, JCF = 21.3 Hz, JCP = 13.7 Hz), 128.9, 134.4 (dt, JCF = 9.8 Hz, JCP = 5.7 Hz), 143.8, 150.4, 150.6, 152.4, 152.7. HRMS-ESI (m/z) calcd for C36H62F2N6O13SiPS [M+H]+ 915.3571, found 915.3564.
((2R,3R,4R,5R)-5-(6-(bis(tert-butoxycarbonyl)amino)-9H-purin-9-yl)-3-((tert-butyldimethylsilyl)oxy)-4-(((E)-4-(diisopropoxyphosphoryl)-4,4-difluorobut-2-en-1-yl)oxy)tetrahydrofuran-2-yl)methyl ((tert-butoxycarbonyl)-D-alanyl)sulfamate (17). The general procedure B was followed with compound 16 (285 mg, 0.31 mmol), Boc-D-Ala-OSu (89 mg, 0.31 mmol), and DBU (0.07 mL, 0.46 mmol) in DMF (4.4 mL). The crude was purified by column chromatography (Eluent EtOAc/CycloHex 9:1) to give compound 17 (195 mg, 0.18 mmol, 58%) as a waxy oil. 1H NMR (Acetone-d6, 500 MHz): δ 0.19 (s, 3H), 0.21 (s, 3H), 0.97 (s, 9H), 1.29 (dd, J = 6.3 Hz, JHP = 1.8 Hz, 6H), 1.31 (d, J = 6.4 Hz, 3H), 1.33 (d, J = 6.2 Hz, 6H), 1.39 (s, 9H), 1.44 (s, 18H), 4.01–4.06 (m, 1H), 4.28–4.38 (m, 4H), 4.41–4.46 (m, 1H), 4.75–4.83 (m, 4H), 5.96–6.04 (m, 2H), 6.26–6.31 (m, 1H), 6.39–6.41 (m, 1H), 8.77 (s, 1H), 8.83 (s, 1H). 19F NMR (Acetone-d6, 470 MHz): δ −109.8 (ddd, JFF = 291.6 Hz, JFP = 113.4 Hz, JFH = 12.9 Hz), −110.5 (ddd, JFF = 291.6 Hz, JFP = 113.4 Hz, JFH = 12.9 Hz). 31P NMR (Acetone-d6, 202 MHz): δ 3.7 (t, JPF = 111.6 Hz). 13C NMR (Acetone-d6, 150 MHz): δ −4.5, −4.4, 18.6, 19.9, 23.9 and 24.0 (d, JCP = 3.1 Hz), 24.3 (d, JCP = 3.3 Hz, 2C), 26.2, 27.9, 28.7, 53.2, 68.9, 69.6, 71.9, 74.3 and 74.4 (d, JCP = 6.4 Hz), 79.0, 82.2, 84.0, 84.3, 87.4, 117.7 (dt, JCF = 257.6 Hz, JCP = 222.3 Hz), 122.7 (dt, JCF = 22.1 Hz, JCP = 13.5 Hz), 129.7, 135.8 (dt, JCF = 10.3 Hz, JCP = 5.6 Hz), 145.1, 151.0, 151.2, 152.7, 153.9, 155.9, 181.0. HRMS-ESI (m/z) calcd for C44H75F2N7O16PSSi [M+H]+ 1086.4466, found 1086.4462.
((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-(((E)-4-(diisopropoxyphosphoryl)-4,4-difluorobut-2-en-1-yl)oxy)-3-hydroxytetrahydrofuran-2-yl)methyl (D-alanyl)sulfamate salt (18). The general procedure C (Method C2) was followed with compound 17 (317 mg, 0.29 mmol), TFA (3.6 mL), and CH2Cl2/H2O (3:0.7 mL). The crude was purified by HPLC RP-18 (MeCN/H2O, linear gradient) to give compound 18 (112 mg, 0.12 mmol, 43%) as a white solid (2 TFA salt). 1H NMR (MeOD, 600 MHz): δ 1.31 (d, J = 6.1 Hz, 6H), 1.34 (dd, J = 6.1 Hz, JHP = 1.8 Hz, 6H), 1.52 (d, J = 7.1 Hz, 3H), 3.87 (q, J = 7.1 Hz, 1H), 4.32–4.36 (m, 2H), 4.43–4.47 (m, 2H), 4.48–4.53 (m, 2H), 4.75–4.80 (m, 2H), 5.91–5.99 (m, 1H), 6.27 (d, J = 4.1 Hz, 1H), 6.29–6.33 (m, 1H), 8.42 (s, 1H), 8.62 (s, 1H). 19F NMR (MeOD, 470 MHz): δ −111.5 (m), −111.2 (m). 31P NMR (MeOD, 202 MHz): δ 4.2 (t, JPF = 114.6 Hz). 13C NMR (MeOD, 150 MHz): δ 17.3, 24.0 (d, JCP = 4.9 Hz, 2C), 24.3 and 24.4 (d, JCP = 1.2 Hz), 52.0, 70.2, 70.5, 72.2, 75.9 (d, JCP = 7.3 Hz, 2C), 83.7, 85.0, 88.8, 117.8 (dt, JCF = 252.8 Hz, JCP = 225.1 Hz), 120.4, 122.6 (dt, JCF = 21.9 Hz, JCP = 13.8 Hz), 136.9 (dt, JCF = 9.8 Hz, JCP = 5.6 Hz), 143.4, 146.3, 149.8, 152.4, 174.1. HPLC purity: (77.6%, tR = 10.53 min). HRMS-ESI (m/z) calcd for C23H37F2N7O10PS [M+H]+ 672.2028, found 672.2036.
2′,3′-
O-isopropylidene-5′-
N-(3-amino-cyclobut-3-ene-1,2-dione)aminodeoxyadenosine (
21). To a solution of
20 (0.92 g, 3 mmol, 1 equiv.), prepared as previously described [
36,
37], in MeOH (20 mL), dimethyl squarate (0.89 g, 6.26 mmol, 2 equiv.) was added at 20 °C. The mixture was stirred for 4 h and concentrated under reduced pressure. The residue was dissolved in MeOH (70 mL), and a solution of NH
3 (7 N) in MeOH (4.87 mL) was added at 20 °C. The mixture was stirred for 16 h, and the reaction mixture was filtered. The solid was washed with pentane to give compound
21 (0.58 g, 1.45 mmol, 48%) as a white foam. The product was used in the next step without further purification.
1H NMR (DMSO-d
6, 500 MHz): δ 1.32 (s, 3H), 1.54 (s, 3H), 3.70–3.76 (m, 1H), 3.94 (br s, 1H), 4.24–4.27 (m, 1H), 5.00–5.02 (m, 1H), 5.43 (s, 1H), 6.20 (s, 1H), 7.37 (br s, 2H), 7.44 (br s, 1H), 8.19 (s, 1H), 8.33 (s, 1H).
13C NMR (DMSO-d
6, 150 MHz): δ 25.7, 27.5, 27.3, 45.6, 81.6, 83.6, 89.2, 114.2, 119.6, 140.4, 149.2, 153.4, 156.6, 170.1, 183.8.
2′,3′-O-isopropylidene-5′-N-(N-tert-butoxycarbonyl-D-alanyl]-(3amino-cyclobut-3-ene-1,2-dione))aminodeoxy adenosine (22). The general procedure B was followed with compound 21 (200 mg, 0.5 mmol), Boc-D-Ala-OSu (180 mg, 0.63 mmol), and DBU (0.19 mL, 1.2 mmol) in DMF (5 mL) at 60 °C for 4h. The crude mixture was purified by column chromatography (Eluent EtOAc/MeOH 9:1) to give compound 22 (140 mg, 0.244 mmol, 49%) as a white solid. 1H NMR (DMSO-d6, 500 MHz): δ 1.19–1.20 (m, 5H), 1.31 (s, 3H), 1.36 (s, 18H), 1.53 (s, 3H), 3.69–3.74 (m, 1H), 3.81–3.87 (m, 2H), 4.23–4.27 (m, 1H), 5.01–5.03 (m, 1H), 5.42 (s, 1H), 6.18 (s, 1H), 6.87 (br s, 1H), 7.36 (s, 1H), 7.85 (br s, 1H), 8.17 (s, 1H), 8.33 (s, 1H). 13C NMR (DMSO-d6, 150 MHz): δ 18.1, 25.7, 27.5, 28.6, 45.5, 49.7, 78.2, 81.7, 83.2, 83.6, 89.2, 113.8, 114.2, 119.6, 140.3, 149.3, 153.4, 155.5, 156.6, 170.2, 175.5, 183.7. HRMS-ESI (m/z) calcd for C25H33N8O8 [M+H]+ 573.2421 found 573.2428.
5′-N-(N-D-alanyl]-(3-amino-cyclobut-3-ene-1,2-dione))aminodeoxy adenosine salt (23). The general procedure C (Method C2) was followed with compound 22 (210 mg, 0.37 mmol), TFA (5 mL), and CH2Cl2/H2O (4/1 mL). The crude mixture was purified by HPLC RP-C18 (MeCN/H2O, linear gradient) to give compound 23 (100.8 mg, 0.153 mmol, 64%) as a white solid (2 TFA salt). 1H NMR (DMSO-d6, 500 MHz): δ 1.38–1.43 (m, 3H), 3.86–3.94 (m, 1H), 4.05–4.13 (m, 3H),4.18–4.23 (m, 1H), 4.61–4.65 (m, 1H), 5.93 (d, J = 5.5 Hz, 1H), 7.80 (br s, 1H), 8.10 (br s, 2H), 8.26 (br s, 2H), 8.28 (s, 1H), 8.43 (s, 1H), 11.98 (s, 1H). 13C NMR (DMSO-d6, 150 MHz): δ 16.6, 45.8, 48.5, 70.8, 73.1, 83.2,87.7, 119.1, 140.7, 148.9, 150.1, 154.1, 169.3, 172.1, 172.8, 181.4, 189.3. HPLC purity: (97.1%, tR = 3.10 min). HRMS-ESI (m/z) calcd for C17H21N8O6 [M+H]+ 433.1584, found 433.1585.
tert-butyl (
tert-butoxycarbonyl)(9-((
3aR,
4R,
6R,
6aR)-6-(((
tert-butoxycarbonyl)(
N-((
tert-butoxycarbonyl)-D-alanyl)sulfamoyl)amino)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)carbamate (
28). The general procedure B was followed with compound
26 (1.2 g, 1.76 mmol), prepared as previously described [
20], Boc-D-Ala-OSu (640 mg, 2.23 mmol), and DBU (0.6 mL, 4.22 mmol) in DMF (25 mL). The crude was purified by flash chromatography (Eluent CH
2Cl
2/MeOH 9:1) to give compound
28 (1.1 g, 1.33 mmol, 76%) as a white foam.
1H NMR (CDCl
3, 500 MHz): δ 1.32 (d,
J = 7.4 Hz, 3H), 1.39 (s, 3H), 1.43 (s, 18H), 1.44 (s, 9H), 1.62 (s, 3H), 4.04–4.15 (m, 3H), 4.20 (dd,
J = 15.4 Hz,
J = 5.9 Hz, 1H), 4.53–4.58 (m, 1H), 4.83–4.95 (m, 1H), 5.13–5.17 (m, 1H), 5.38–5.41 (m, 1H), 6.18 (d,
J = 2.1 Hz, 1H), 8.21 (s, 1H), 8.87 (s, 1H).
13C NMR (CDCl
3, 125 MHz): δ 16.4, 25.6, 27.4, 27.9, 28.0, 28.4, 50.3, 82.4, 83.9, 84.5, 85.3, 85.4, 90.7, 114.9, 129.6, 144.0, 150.5, 150.7, 152.4, 152.7, 171.7. HRMS-ESI (
m/
z) calcd for C
31H
48N
8O
12S [M+H]
+ 757.3191, found 757.3193.
tert-butyl (
tert-butoxycarbonyl)(9-((
2R,
3R,
4R,
5R)-5-(((
tert-butoxycarbonyl)(
N-((
tert-butoxycarbonyl)-D-alanyl)sulfamoyl)amino)methyl)-4-((
tert-butyldimethylsilyl)oxy)-3-fluorotetrahydrofuran-2-yl)-9H-purin-6-yl)carbamate (
29). The general procedure B was followed with compound
27 (185 mg, 0.24 mmol), prepared as previously described [
20], Boc-D-Ala-OSu (70 mg, 0.24 mmol), and DBU (55 µL, 0.36 mmol) in DMF (3.3 mL). The crude was purified by column chromatography (Eluent Pentane/EtOAc 1:1) to give compound
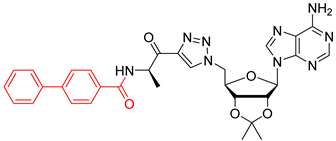
29 (173 mg, 0.18 mmol, 77%) as a white foam.
1H NMR (CD
3CN, 600 MHz): δ 0.16 (s, 3H), 0.19 (s, 3H), 0.95 (s, 9H), 1.19 (d,
J = 7.2 Hz, 3H), 1.25 (s, 9H), 1.40 (s, 18H), 1.41 (s, 9H), 3.97–4.03 (m, 2H), 4.14–4.17 (m, 1H), 4.34–4.35 (m, 1H), 4.80–4.83 (m, 1H), 5.53–5.63 (m, 1H), 5.77 (br s, 1H), 6.30 (dd,
JFH = 17.5 Hz,
J = 2.4 Hz, 1H), 8.44 (s, 1H), 8.81 (s, 1H).
19F NMR (CD
3CN, 470 MHz): δ −205.6 (m).
13C NMR (CD
3CN, 150 MHz): δ −4.7, −4.5, 18.2, 18.7, 26.1, 27.9, 28.1, 28.7, 50.4, 53.3, 73.0 (d,
JCF = 15.9 Hz), 80.9, 83.1, 85.0, 85.9, 88.3 (d,
JCF = 32.9 Hz), 93.3 (d,
JCF = 190.8 Hz), 130.1, 146.0, 151.0, 151.5, 152.8, 153.6, 155.6, 157.2, 179.5. HRMS-ESI (
m/
z) calcd for C
39H
66FN
8O
13SiS [M+H]
+ 933.4223, found 933.4211.
(R)-2-amino-N-(N-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)sulfamoyl)propenamide salt (30). The general procedure C (Method C2) was followed with compound 28 (528 mg, 0.62 mmol), TFA (7.5 mL), and CH2Cl2/H2O (6/1.5 mL). The crude was purified by HPLC RP-C18 (MeCN/H2O, linear gradient) to give compound 30 (219 mg, 0.34 mmol, 55%) as a white solid (2 TFA salt). 1H NMR (D2O, 500 MHz): δ 1.52 (d, J = 7.2 Hz, 3H), 3.37–3.38 (m, 2H), 3.97 (q, J = 7.2 Hz, 1H), 4.31–4.34 (m, 1H), 4.41–4.44 (m, 1H), 4.80–4.83 (m, 1H), 6.04 (d, J = 5.8 Hz, 1H), 8.37 (s, 1H), 8.39 (s, 1H). 13C NMR (D2O, 125 MHz): δ 16.4, 44.3, 50.4, 71.0, 73.5, 83.7, 89.1, 119.3, 142.8, 147.1, 148.2, 151.7, 173.0. HRMS-ESI (m/z) calcd for C13H20N8O6S [M+H]+ 417.1305, found 417.1304.
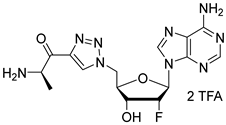
(R)-2-amino-N-(N-(((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methyl)sulfamoyl)propenamide salt (31). The general procedure C (Method C2) was followed with compound 29 (173 mg, 0.18 mmol), TFA (2.3 mL), and CH2Cl2/H2O (2/0.5 mL) to give compound 31 (105 mg, 0.16 mmol, 88%) as a white solid (2 TFA salt). 1H NMR (D2O, 500 MHz): δ 1.47 (d, J = 7.2 Hz, 3H), 3.33 (dd, J = 14.3 Hz, J = 4.5 Hz, 1H), 3.42 (dd, J = 14.3 Hz, J = 2.8 Hz, 1H), 3.85 (q, J = 7.2 Hz, 1H), 4.28–4.32 (m, 1H), 4.70 (ddd, JHF = 18.3 Hz, J = 7.2 Hz, J = 4.7 Hz, 1H), 5.50 (ddd, JHF = 52.1 Hz, J = 4.7 Hz, J = 2.4 Hz, 1H), 6.34 (dd, JHF = 17.3 Hz, J = 2.4 Hz, 1H), 8.28 (s, 1H), 8.32 (s, 1H). 19F NMR (D2O, 470 MHz): δ −203.9 (dt, JFH = 52.1 Hz, JFH = 17.3 Hz, 1F). 13C NMR (D2O, 125 MHz): 16.6, 43.4, 51.0, 69.5 (d, JCF = 17.1 Hz), 81.6, 87.1 (d, JCF = 33.9 Hz), 93.0 (d, JCF = 187.9 Hz), 119.1, 141.3, 148.3, 150.6, 154.1, 175.3. HPLC purity: (98.3%, tR = 2.08 min). HRMS-ESI (m/z) calcd for C13H19FN8O5S [M+H]+ 419.1261, found 419.1264.
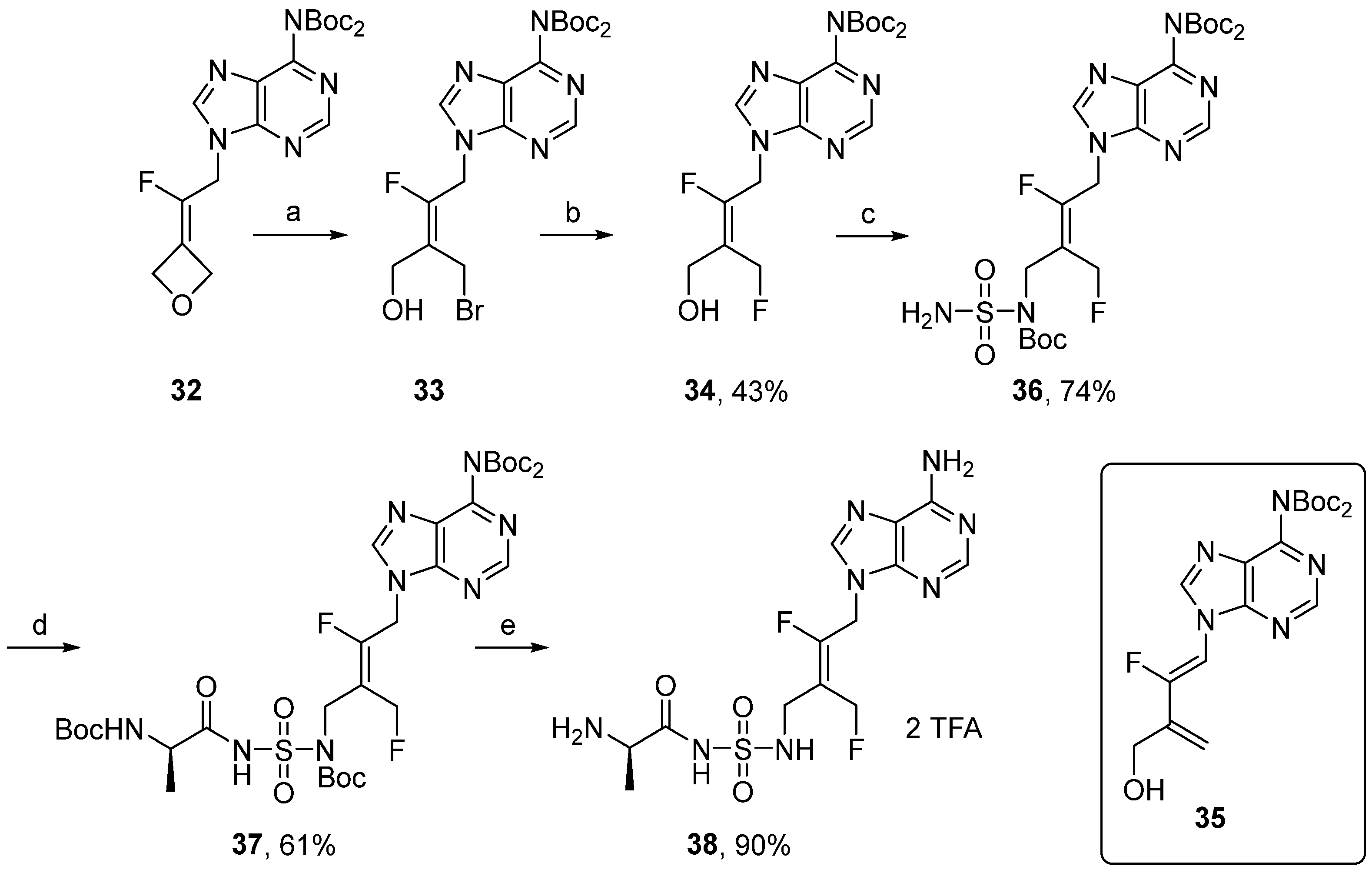
tert-butyl (
E)-(
tert-butoxycarbonyl)(9-(2,4-difluoro-3-(hydroxymethyl)but-2-en-1-yl)-9H-purin-6-yl)carbamate (
34). A solution of compound
33 (247 mg, 0.48 mmol, 1 equiv.), prepared as previously described [
40], in
t-BuOH (5 mL) was added CsF (111 mg, 0.96 mmol, 2.0 equiv.). The reaction mixture was stirred at 70 °C for 4 h, and the solvent was removed under reduced pressure. The residue was taken up with EtOAc/H
2O, and the aqueous layers were extracted three times with EtOAc. The combined organic layers were washed with brine, dried over MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (Eluent Pentane/EtOAc 1:1 to 6:4) to give compound
34 (93 mg, 0.21 mmol, 43%) as a colorless oil.
1H NMR (CDCl
3, 500 MHz): δ 1.44 (s, 18H), 2.33 (br s, 1H), 4.34 (s, 2H), 5.14 (dd,
JHF = 21.7 Hz,
JHF = 1.8 Hz, 2H), 5.35 (dd,
JHF = 47.8 Hz,
JHF = 2.2 Hz, 2H), 8.14 (s, 1H), 8.85 (s, 1H).
19F NMR (CDCl
3, 470 MHz): δ −107.4 (t,
JFH = 21.7 Hz), −212.1 (t,
JFH = 47.8 Hz).
13C NMR (CDCl
3, 125 MHz): δ 27.9, 41.0 (d,
JCF = 29.6 Hz), 56.3 (dd,
JCF = 8.7 Hz,
JCF = 1.2 Hz), 78.6 (dd,
JCF = 166.7 Hz,
JCF = 10.1 Hz), 84.1, 118.8 (dd,
JCF = 15.5 Hz,
JCF = 13.4 Hz), 128.5, 144.5, 150.5, 150.6, 152.5, 153.2, 153.7 (dd,
JCF = 269.6 Hz,
JCF = 10.8 Hz). HRMS-ESI (
m/
z) calcd for C
20H
28F
2N
5O
5 [M+H]
+ 456.2059, found 456.2054.
tert-butyl (E)-(tert-butoxycarbonyl)(9-(4-((tert-butoxycarbonyl)(sulfamoyl)amino)-2-fluoro-3-(fluoromethyl)but-2-en-1-yl)-9H-purin-6-yl)carbamate (36). The general procedure D was followed with compound 34 (120 mg, 0.26 mmol), tert-butyl sulfamoylcarbamate (155 mg, 0.78 mmol), triphenylphosphine (102 mg, 0.39 mmol), and diisopropyl azodicarboxylate (0.05 mL, 0.27 mmol) in dry THF (5.2 mL). The crude product was purified by column chromatography (EtOAc/Pentane 1:2) to give compound 36 (123 mg, 0.19 mmol, 74%) as a white foam. 1H NMR (CDCl3, 600 MHz): δ 1.44 (s, 9H), 1.46 (s, 18H), 4.57 (br s, 2H), 5.16 (d, JHF = 21.1 Hz, 2H), 5.33 (d, JHF = 47.8 Hz, 2H), 5.40 (br s, 2H), 8.15 (s, 1H), 8.85 (s, 1H). 19F NMR (CDCl3, 565 MHz): δ −103.1 (t, JFH = 21.1 Hz), −208.1 (t, JFH = 47.8 Hz). 13C NMR (CDCl3, 150 MHz): δ 27.9, 28.1, 41.1 (d, JCF = 30.1 Hz), 43.1 (d, JCF = 8.4 Hz), 79.6 (dd, JCF = 165.9 Hz, JCF = 10.4 Hz), 84.1, 85.3, 114.9 (dd, JCF = 13.9 Hz, JCF = 12.1 Hz), 128.6, 144.4, 150.6, 150.7, 151.9, 152.6, 153.1, 155.6 (dd, JCF = 263.6 Hz, JCF = 11.7 Hz). HRMS-ESI (m/z) calcd for C25H38F2N7O8S [M+H]+ 634,2471 found 634.2474.
tert-butyl (E)-(tert-butoxycarbonyl)(9-(4-((N-((tert-butoxycarbonyl)-D-alanyl)sulfamoyl)amino)-2-fluoro-3-(fluoromethyl)but-2-en-1-yl)-9H-purin-6-yl)carbamate (37). The general procedure B was followed with compound 36 (130 mg, 0.20 mmol), Boc-D-Ala-OSu (59 mg, 0.20 mmol), and DBU (46 µL, 0.31 mmol) in DMF (2.6 mL). The crude was purified by column chromatography (Eluent EtOAc/Pentane 1:1 to 1:0) to give compound 37 (100 mg, 0.12 mmol, 61%) as a white foam. 1H NMR (CDCl3, 600 MHz): δ 1.31 (d, JHF = 6.9 Hz, 3H), 1.40 (s, 9H), 1.44 (s, 18H), 1.45 (s, 18H), 4.69 (br s, 2H), 5.17 (d, JHF = 21.3 Hz, 2H), 5.26 (d, JHF = 47.8 Hz, 2H), 8.18 (s, 1H), 8.86 (s, 1H). 19F NMR (CDCl3, 565 MHz): δ −103.9–103.6 (m), −209.6 (t, JFH = 47.8 Hz). 13C NMR (CDCl3, 150 MHz): δ 18.1, 27.8, 27.9, 28.4, 41.1 (d, JCF = 29.4 Hz), 44.1, 52.1, 78.4 (dd, JCF = 166.4 Hz, JCF = 9.6 Hz), 80.1, 84.1, 84.4, 116.2 (m), 128.4, 144.8, 150.2, 150.6, 152.3, 152.7, 153.3, 154.6 (dd, JCF = 262.3 Hz, JCF = 10.9 Hz), 156.0, 177.7. HRMS-ESI (m/z) calcd for C33H51F2N8O11S [M+H]+ 805.3366, found 805.3391.
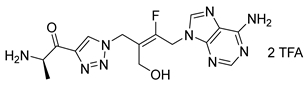
(R,E)-2-amino-N-(N-(4-(6-amino-9H-purin-9-yl)-3-fluoro-2-(fluoromethyl)but-2-en-1-yl)sulfamoyl)propenamide salt (38). The general procedure C (Method C2) was followed with compound 37 (100 mg, 0.12 mmol), TFA (1.4 mL) and CH2Cl2/H2O (1.2/0.3 mL) to give compound 38 (76 mg, 0.11 mmol, 90%) as a white solid (2 TFA salt). 1H NMR (MeOD, 600 MHz): δ 1.52 (d, J = 7.1 Hz, 3H), 3.88 (br s, 2H), 3.92 (q, J = 7.1 Hz), 5.28 (dd, JHF = 20.6 Hz, JHF = 1.6 Hz, 2H), 5.33 (dd, JHF = 47.6 Hz, JHF = 1.8 Hz, 2H), 8.24 (s, 1H), 8.32 (s, 1H). 19F NMR (CDCl3, 565 MHz): δ −105.5 (m), −211.3 (m).13C NMR (MeOD, 150 MHz): δ 17.1, 39.0 (d, JCF = 9.1 Hz), 41.8 (d, JCF = 28.8 Hz), 50.5, 79.3 (dd, JCF = 163.1 Hz, JCF = 9.2 Hz), 116.6 (dd, JCF = 15.1 Hz, JCF = 12.6 Hz), 118.1 (q, JCF = 292.1 Hz), 119.7, 143.8, 149.8, 150.6, 154.6, 156.5 (dd, JCF = 261.7 Hz, JCF = 10.8 Hz), 163.4 (q, JCF = 32.4 Hz), 170.1. HPLC purity: (95.8%, tR = 3.23 min). HRMS-ESI (m/z) calcd for C13H19N8O3F2S [M+H]+ 405.1269, found 405.1271.
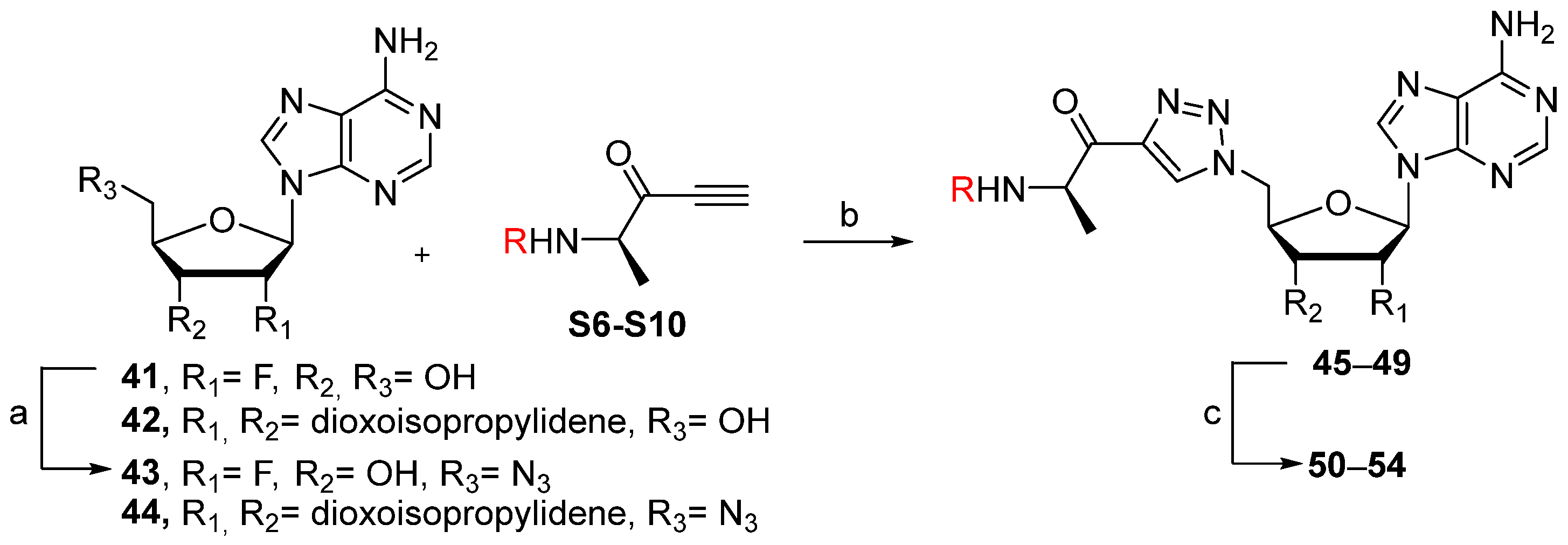
tert-butyl ((
R)-1-(1-(((
2R,
3R,
4R,
5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)carbamate (
45). The general procedure E was followed with compound
43 (410 mg, 1.39 mmol), prepared as previously described [
43], alkyne
S6 (295 mg, 1.53 mmol), sodium ascorbate (28 mg, 0.14 mmol), and CuSO
4.5H
2O (17 mg, 0.07 mmol) in
t-BuOH/H
2O (9 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 94:6) to give compound
45 (470 g, 0.94 mmol, 68%) as a white solid.
1H NMR (DMSO-d
6, 500 MHz): δ 1.26 (d,
J = 7.4 Hz, 3H), 1.32 (s, 9H), 4.34 (dt,
J = 7.4 Hz,
J = 3.2 Hz, 1H), 4.69–4.77 (m, 1H), 4.82 (dd,
J = 14.8 Hz,
J = 7.4 Hz, 1H), 4.88 (dd,
J = 14.8 Hz,
J = 2.8 Hz, 1H), 4.89–4.95 (m, 1H), 5.50 (ddd,
JHF = 52.6 Hz,
J = 4.5 Hz,
J = 1.3 Hz, 1H), 6.08 (d,
J = 6.4 Hz, 1H), 6.24 (dd,
JHF = 20.6 Hz,
J = 1.3 Hz, 1H), 7.24 (d,
J = 6.8 Hz, 1H), 7.33 (br s, 2H), 8.10 (s, 1H), 8.23 (s, 1H), 8.60 (s, 1H).
19F NMR (DMSO-d
6, 470 MHz): δ −201.2 (dt,
JFH = 52.6 Hz,
JFH = 20.6 Hz).
13C NMR (DMSO-d
6, 125 MHz): δ 16.6, 28.1, 51.0, 52.3, 69.7 (d,
JCF = 16.3 Hz), 78.0, 80.0, 86.5 (d,
JCF = 34.8 Hz), 93.0 (d,
JCF = 186.5 Hz), 119.2, 128.7, 140.0, 144.6, 148.6, 152.7, 155.2, 156.1, 193.4. HRMS-ESI (
m/
z) calcd for C
20H
27FN
9O
5 [M+H]
+ 492.2119, found 492.2123.
N-((
R)-1-(1-(((
3aR,
4R,
6R,
6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)heptanamide (
46). The general procedure E was followed with compound
44 (400 mg, 1.20 mmol), prepared as previously described [
44], alkyne
S7 (277 mg, 1.32 mmol), sodium ascorbate (24 mg, 0.12 mmol), and CuSO
4.5H
2O (15 mg, 0.06 mmol) in
t-BuOH/H
2O (8 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 1:0 to 98:2) to give compound
46 (511 mg, 0.95 mmol, 79%) as a brown foam.
1H NMR (CDCl
3, 500 MHz): δ 0.80 (t,
J = 7.0 Hz, 3H), 1.19–1.25 (m, 6H), 1.35 (s, 3H), 1.44 (d,
J = 7.6 Hz, 3H), 1.56 (s, 3H), 2.18 (t,
J = 7.0 Hz, 2H), 4.56–4.61 (m, 1H), 4.84–4.86 (m, 2H), 5.18–5.20 (m, 1H), 5.38–5.39 (m, 1H), 5.44–5.52 (m, 1H), 6.07 (s, 1H), 6.61 (s, 1H), 6.67–6.72 (m, 1H), 7.86 (s, 1H), 8.03 (s, 1H), 8.25 (s, 1H).
13C NMR (CDCl
3, 125 MHz): δ 14.1, 18.8, 22.5, 25.4, 25.6, 27.2, 28.9, 31.6, 36.6, 52.0, 81.8, 84.1, 84.2, 85.3, 90.7, 115.1, 120.4, 128.3, 140.4, 145.3, 148.8, 152.9, 155.9, 172.9, 193.4. HRMS-ESI (
m/
z) calcd for C
25H
36N
9O
5 [M+H]
+ 542.2839, found 542.2841.
N-((
R)-1-(1-(((
3aR,
4R,
6R,
6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)-[1,1′-biphenyl]-4-carboxamide (
47). The general procedure E was followed with compound
44 (400 mg, 1.20 mmol), prepared as previously described [
44], alkyne
S8 (367 mg, 1.32 mmol), sodium ascorbate (24 mg, 0.12 mmol), and CuSO
4.5H
2O (15 mg, 0.06 mmol) in
t-BuOH/H
2O (8 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 1:0 to 99:1) to give compound
47 (561 mg, 0.92 mmol, 77%) as a brown foam.
1H NMR (CDCl
3, 500 MHz): δ 1.33 (s, 3H), 1.55 (s, 3H), 1.59–1.62 (m, 3H), 4.56–4.61 (m, 1H), 4.80–4.88 (m, 1H), 5.17–5.19 (m, 1H), 5.36–5.37 (m, 1H), 5.66–5.73 (m, 1H), 6.06 (br s, 1H), 6.62 (br s, 2H), 7.31–7.34 (m, 1H), 7.38–7.41 (m, 2H), 7.50–7.58 (m, 5H), 7.85–7.88 (m, 3H), 8.04–8.05 (m, 1H), 8.27 (br s, 1H).
13C NMR (CDCl
3, 125 MHz): δ 18.9, 25.4, 27.1, 29.7, 51.9, 52.7, 81.7, 84.1, 85.3, 90.6, 115.1, 120.4, 127.1 (2C), 127.2 (2C), 127.7 (2C), 128.4, 128.9 (2C), 132.8, 140.0, 140.2, 144.2, 145.2, 148.8, 153.3, 156.1, 166.6, 193.2. HRMS-ESI (
m/
z) calcd for C
31H
32N
9O
5 [M+H]
+ 610.2526, found 610.2531.
N-((
R)-1-(1-(((
3aR,
4R,
6R,
6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)-4-(hexyloxy)benzamide (
48). The general procedure E was followed with compound
44 (400 mg, 1.20 mmol), prepared as previously described [
44], alkyne
S9 (398 mg, 1.32 mmol), sodium ascorbate (24 mg, 0.12 mmol), and CuSO
4.5H
2O (15 mg, 0.06 mmol) in
tBuOH/H
2O (8 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 99:1) to give compound
48 (617 mg, 0.97 mmol, 81%) as a brown foam.
1H NMR (CDCl
3, 500 MHz): δ 0.87–0.90 (m, 3H), 1.30–1.36 (m, 4H), 1.40 (s, 3H), 1.41–1.45 (m, 2H), 1.58–1.61 (m, 6H), 1.72–1.79 (m, 2H), 3.92–3.97 (m, 2H), 4.59–4.61 (m, 1H), 4.84–4.91 (m, 2H), 5.13–5.20 (m, 1H), 5.33–5.39 (m, 1H), 5.64–5.70 (m, 1H), 6.06 (br s, 1H), 6.82–6.88 (m, 2H), 7.75–7.77 (m, 2H), 7.88 (s, 1H), 8.04 (s, 1H), 8.28 (s, 1H).
13C NMR (CDCl
3, 125 MHz): δ 14.1, 19.1, 22.7, 25.4, 25.8, 27.2, 29.2, 31.6, 52.1, 52.7, 68.3, 81.8, 84.2, 85.4, 90.7, 114.2 (2C), 115.2, 120.5, 126.2, 128.4, 129.0 (2C), 129.7, 140.6, 145.4, 149.2, 152.6, 162.0, 166.4, 193.4. HRMS-ESI (
m/
z) calcd for C
31H
40N
9O
6 [M+H]
+ 634.3102, found 634.3104.
4-nitrobenzyl ((
R)-1-(1-(((
3aR,
4R,
6R,
6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)carbamate (
49). The general procedure E was followed with compound
44 (400 mg, 1.20 mmol), prepared as previously described [
44], compound
S10 (365 mg, 1.32 mmol), sodium ascorbate (24 mg, 0.12 mmol), and CuSO
4.5H
2O (15 mg, 0.06 mmol) in
t-BuOH/H
2O (8 mL). The crude was purified by column chromatography (Eluent EtOAc/MeOH 1:0 to 99:1) to give compound
49 (470 mg, 0.77 mmol, 64%) as a brown foam.
1H NMR (CDCl
3, 500 MHz): δ 1.37 (s, 3H), 1.53 (t,
J = 7.6 Hz, 3H), 1.58 (s, 3H), 4.58–4.61 (m, 1H), 4.87–4.88 (m, 2H), 5.13–5.22 (m, 3H), 5.32–5.35 (m, 1H), 5.40–5.42 (m, 1H), 6.04–6.07 (m, 1H), 6.35 (br s, 2H), 7.46–7.47 (m, 2H), 7.88 (s, 1H), 7.97 (s, 1H), 8.11–8.16 (m, 1H), 8.29 (s, 1H).
13C NMR (CDCl
3, 125 MHz): δ 19.1, 25.4, 27.2, 29.8, 52.1, 53.7, 65.3, 81.9, 84.2, 85.4, 90.8, 115.2, 123.8 (2C), 128.1 (2C), 128.2, 140.6, 144.1, 145.2, 147.6, 152.9, 155.3, 155.8, 193.1. HRMS-ESI (
m/
z) calcd for C
26H
29N
10O
8 [M+H]
+ 609.2170, found 609.2173.
(R)-2-amino-1-(1-(((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)propan-1-one salt (50). The general procedure C (Method C2) with compound 45 (380 mg, 0.77 mmol), TFA (9.5 mL), and CH2Cl2/H2O (8.7/1.8 mL) to give compound 50 (470 mg, 0.68 mmol, 88%) as a white solid (2 TFA). 1H NMR (D2O, 500 MHz): δ 1.65 (d, J = 7.2 Hz, 3H), 4.58–4.61 (m, 1H), 4.73 (ddd, JHF = 22.7 Hz, J = 9.2 Hz, J = 4.4 Hz, 1H), 4.93 (q, J = 7.2 Hz, 1H), 5.00 (d, J = 3.9 Hz, 2H), 5.58 (dd, JHF = 51.6 Hz, J = 4.8 Hz, 1H), 6.42 (d, JHF = 20.3 Hz, 1H), 8.23 (s, 1H), 8.30 (s, 1H), 8.55 (s, 1H). 19F NMR (D2O, 470 MHz): δ −201.5 (ddd, JFH = 51.6 Hz, JFH = 22.7 Hz, JFH = 20.3 Hz, 1F). 13C NMR (D2O, 150 MHz): δ 16.2, 50.1, 52.6, 69.3 (d, JCF = 16.9 Hz), 79.6, 87.3 (d, JCF = 36.1 Hz), 92.8 (d, JCF = 186.1 Hz), 115.3, 118.7, 130.5, 142.8, 145.6, 147.7, 150.7, 189.7. HPLC purity: (98.5%, tR = 10.24 min). HRMS-ESI (m/z) calcd for C15H19FN9O3 [M+H]+ 392.1595, found 392.1594.
N-((R)-1-(1-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)heptanamide salt (51). The general procedure C (Method C1) was followed with compound 46 (250 mg, 0.46 mmol), HCl aq (1 mL, 3 M), and MeOH (2.5 mL). The crude was precipitated in MeOH/Et2O to give compound 51 (219 mg, 0.41 mmol, 89%) as a white solid (HCl salt). 1H NMR (DMSO-d6, 500 MHz): δ 0.83 (t, J = 7.1 Hz, 3H), 1.18–1.24 (m, 6H), 1.27–1.29 (m, 3H), 1.41–1.44 (m, 2H), 2.06–2.10 (m, 2H), 4.27–4.29 (m, 1H), 4.37–4.40 (m, 1H), 4.62–4.64 (m, 1H), 4.85–4.91 (m, 2H), 5.11–5.16 (m, 1H), 5.98 (d, J = 5.2 Hz, 1H), 8.27 (br s, 1H), 8.54 (s, 1H), 8.74 (s, 1H), 8.77 (s, 1H), 9.07 (br s, 1H), 9.78 (br s, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 14.0, 16.7, 22.0, 25.2, 28.3, 31.1, 34.8, 50.9, 51.7, 70.8, 73.4, 82.5, 88.3, 118.9, 128.9, 142.8, 144.7, 144.9, 148.2, 150.3, 172.1, 193.0. HPLC purity: (93.6%, tR = 10.83 min). HRMS-ESI (m/z) calcd for C22H32N9O5 [M+H]+ 502.2526, found 502.2527.
N-((R)-1-(1-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)-[1,1′-biphenyl]-4-carboxamide salt (52). The general procedure C (Method C1) was followed with compound 47 (250 mg, 0.41 mmol), HCl aq (1 mL, 3 M), and MeOH (2.5 mL). The crude was precipitated in MeOH/Et2O to give compound 52 (205 mg, 0.34 mmol, 83%) as a white solid (HCl salt). 1H NMR (DMSO-d6, 500 MHz): δ 1.46–1.48 (m, 3H), 4.29–4.31 (m, 1H), 4.39–4.43 (m, 1H), 4.63 (dt, J = 7.8 Hz, J = 5.1 Hz, 1H), 5.87–5.94 (m, 2H), 5.36–5.42 (m, 1H), 5.99–6.01 (m, 1H), 7.39–7.42 (m, 1H), 7.47–7.51 (m, 2H), 7.72–7.78 (m, 4H), 7.97–8.00 (m, 2H), 8.53 (d, J = 1.6 Hz, 1H), 8.75 (d, J = 4.8 Hz, 1H), 8.79 (d, J = 9.8 Hz, 1H), 8.90 (d, J = 6.2 Hz, 1H), 8.97 (br s, 1H), 9.67 (br s, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 16.4, 51.7, 52.0, 70.8, 73.4, 82.5, 88.3, 118.9, 126.4, (2C), 126.9 (2C), 128.1, 128.3 (2C), 128.8, 129.1 (2C), 132.5, 139.1, 142.7, 142.9, 144.8, 145.3, 148.3, 150.5, 165.9, 192.7. HPLC purity: (98.8%, tR = 12.32 min). HRMS-ESI (m/z) calcd for C28H28N9O5 [M+H]+ 570.2213, found 570.2214.
N-((R)-1-(1-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)-4-(hexyloxy)benzamide salt (53). The general procedure C (Method C1) was followed with compound 48 (491 mg, 0.77 mmol), HCl aq (1.9 mL, 3 M), and MeOH (4.7 mL). The crude was precipitated in MeOH/Et2O to give compound 53 (370 mg, 0.58 mmol, 76%) as a white solid (HCl salt). 1H NMR (DMSO-d6, 500 MHz): δ 0.87 (t, J = 7.0 Hz, 3H), 1.28–1.31 (m, 4H), 1.39–1.44 (m, 5H), 1.68–1.73 (m, 2H), 4.01 (t, J = 6.6 Hz, 2H), 4.28–4.30 (m, 1H), 4.38–4.42 (m, 1H), 4.61–4.64 (m, 1H), 4.88–4.91 (m, 1H), 5.31–5.38 (m, 1H), 5.98–6.00 (m, 1H), 6.95–6.98 (m, 2H), 7.84–7.86 (m, 2H), 8.53 (s, 1H), 8.65–8.66 (m, 1H), 8.74–8.75 (m, 1H), 8.76–8.78 (m, 1H), 8.96 (br s, 1H), 9.66 (br s, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 13.9, 16.4, 22.1, 25.2, 28.6, 31.0, 51.6, 51.8, 67.7, 70.8, 73.3, 82.4, 88.3, 113.8 (2C), 118.9, 125.7, 128.8, 129.4 (2C), 142.7, 144.8, 145.3, 148.3, 150.1, 161.2, 165.7, 193.1. HPLC purity: (96.5%, tR = 14.41 min). HRMS-ESI (m/z) calcd for C28H36N9O6 [M+H]+ 594.2789, found 594.2797.
4-nitrobenzyl ((R)-1-(1-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)-1-oxopropan-2-yl)carbamate salt (54). The general procedure C (Method C1) was followed with compound 49 (250 mg, 0.41 mmol), HCl aq (1 mL, 3 M), and MeOH (2.5 mL). The crude was precipitated in MeOH/Et2O to give compound 54 (195 mg, 0.32 mmol, 79%) as a white solid (HCl salt). 1H NMR (DMSO-d6, 500 MHz): δ 1.34 (t, J = 7.4 Hz, 3H), 4.27–4.29 (m, 1H), 4.38–4.41 (m, 1H), 4.62–4.64 (m, 1H), 4.89–4.91 (m, 2H), 5.02–5.07 (m, 1H), 5.15 (s, 2H), 5.98 (d, J = 4.9 Hz, 1H), 7.59 (d, J = 8.1 Hz, 2H), 7.94 (d, J = 7.4 Hz, 1H), 8.22 (d, J = 8.1 Hz, 2H), 8.52 (s, 1H), 8.75 (s, 1H), 8.77 (s, 1H), 9.01 (br s, 1H), 9.69 (br s, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 16.8, 51.6, 52.8, 64.2, 70.8, 73.4, 82.4, 88.3, 118.8, 123.5 (2C), 128.1 (2C), 128.9, 142.7, 144.5, 145.0, 145.1, 146.9, 148.2, 150.4, 155.5, 193.0. HPLC purity: (98.3%, tR = 10.74 min). HRMS-ESI (m/z) calcd for C23H25N10O8 [M+H]+ 569.1857, found 569.1860.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}