
OPUS-BFactor: Predicting Protein B-Factor with Sequence and Structure Information



Abstract
1. Introduction
2. Results
2.1. Performance of Different B-Factor Prediction Methods
2.2. Correlation Between Protein B-Factors and the pLDDT Values from Structure Prediction Methods
2.3. Evaluation of Different Evolutionary Profiles
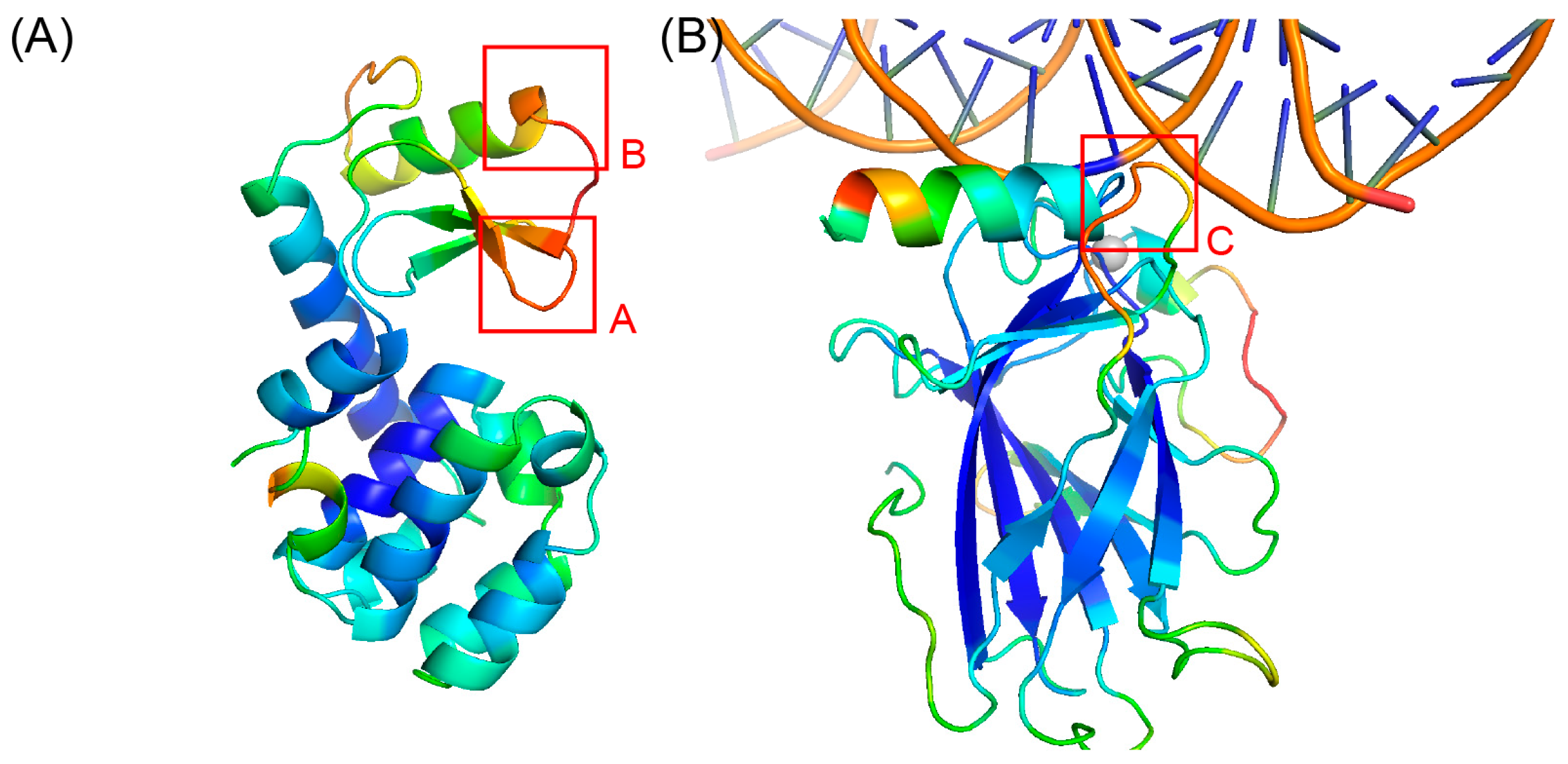
2.4. Case Study
3. Method
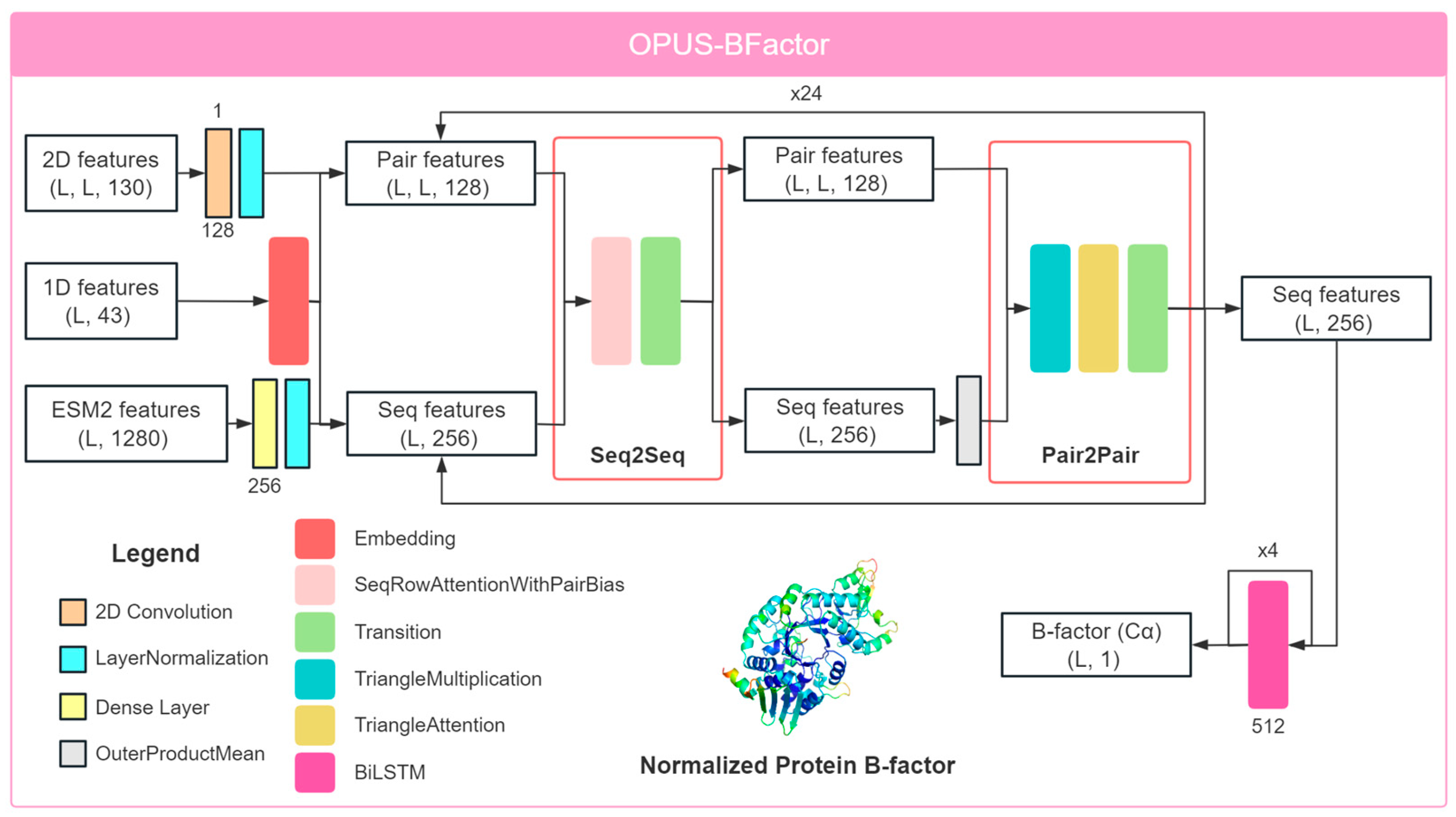
3.1. Framework of OPUS-BFactor
3.2. Datasets
3.3. Performance Metrics
3.4. Data and Software Availability
4. Concluding Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| pLDDT | predicted local distance difference test |
References
- Sun, Z.T.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef] [PubMed]
- Bramer, D.; Wei, G.W. Blind prediction of protein B-factor and flexibility. J. Chem. Phys. 2018, 149, 134107. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Argos, P. Reliability of atomic displacement parameters in protein crystal structures. Acta Crystallogr. Sect. D Struct. Biol. 1999, 55, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M.; Torkkila, E.; Riikonen, P. Accuracy of Protein Flexibility Predictions. Proteins 1994, 19, 141–149. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of Chain Flexibility in Proteins—A Tool for the Selection of Peptide Antigens. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Murthy, M.R.N. Protein thermal stability: Insights from atomic displacement parameters (B values). Protein Eng. Des. Sel. 2000, 13, 9–13. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhao, J.; Wang, Z.X. Flexibility analysis of enzyme active sites by crystallographic temperature factors. Protein Eng. Des. Sel. 2003, 16, 109–114. [Google Scholar] [CrossRef]
- Radivojac, P.; Obradovic, Z.; Smith, D.K.; Zhu, G.; Vucetic, S.; Brown, C.J.; Lawson, J.D.; Dunker, A.K. Protein flexibility and intrinsic disorder. Protein Sci. 2004, 13, 71–80. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Ma, J.P. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef]
- Mlynek, G.; Djinovic-Carugo, K.; Carugo, O. B-Factor Rescaling for Protein Crystal Structure Analyses. Crystals 2024, 14, 443. [Google Scholar] [CrossRef]
- Carugo, O. Atomic displacement parameters in structural biology. Amino Acids 2018, 50, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O. Uses and Abuses of the Atomic Displacement Parameters in Structural BiologyStructural biology. In Data Mining Techniques for the Life Sciences; Carugo, O., Eisenhaber, F., Eds.; Springer: New York, NY, USA, 2022; pp. 281–298. [Google Scholar]
- Pan, X.Y.; Shen, H.B. Prediction of Protein B-factor Profile based on Feature Selection and Kernel Learning. In Proceedings of the 2009 Chinese Conference on Pattern Recognition, Nanjing, China, 4–6 November 2009; Volume 1–2, pp. 588–592. [Google Scholar]
- Yuan, Z.; Bailey, T.L.; Teasdale, R.D. Prediction of protein B-factor profiles. Proteins 2005, 58, 905–912. [Google Scholar] [CrossRef]
- Pandey, A.; Liu, E.; Graham, J.; Chen, W.; Keten, S. B-factor prediction in proteins using a sequence-based deep learning model. Patterns 2023, 4, 100805. [Google Scholar] [CrossRef]
- Pang, Y.-P. Use of multiple picosecond high-mass molecular dynamics simulations to predict crystallographic B-factors of folded globular proteins. Heliyon 2016, 2, e00161. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, X.; Miao, Z.; Zhang, X.; Jiang, B.; Liu, M. Prediction of Protein B-factor Profiles based on Bidirectional Long Short-Term Memory Network. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Weiss, M.S. On the interrelationship between atomic displacement parameters (ADPs) and coordinates in protein structures. Acta Crystallogr. Sect. D Struct. Biol. 2007, 63, 1235–1242. [Google Scholar] [CrossRef]
- Kidera, A.; Go, N. Refinement of Protein Dynamic Structure—Normal Mode Refinement. Proc. Natl. Acad. Sci. USA 1990, 87, 3718–3722. [Google Scholar] [CrossRef]
- Kidera, A.; Go, N. Normal Mode Refinement—Crystallographic Refinement of Protein Dynamic Structure. 1. Theory and Test by Simulated Diffraction Data. J. Mol. Biol. 1992, 225, 457–475. [Google Scholar] [CrossRef]
- Diamond, R. On the Use of Normal-Modes in Thermal Parameter Refinement—Theory and Application to the Bovine Pancreatic Trypsin-Inhibitor. Acta Crystallogr. Sect. A Found. Crystallogr. 1990, 46, 425–435. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef]
- Yang, J.Y.; Wang, Y.; Zhang, Y. ResQ: An Approach to Unified Estimation of B-Factor and Residue-Specific Error in Protein Structure Prediction. J. Mol. Biol. 2016, 428, 693–701. [Google Scholar] [CrossRef]
- Bramer, D.; Wei, G.W. Multiscale weighted colored graphs for protein flexibility and rigidity analysis. J. Chem. Phys. 2018, 148, 054103. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Sarparast, S.; Zaimi, A.; Ebert, M.; Goldsmith, M.-R.J.A. Advanced atom-level representations for protein flexibility prediction utilizing graph neural networks. arXiv 2024, arXiv:2408.12519. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Wang, C.; Fan, H.; Quan, R.; Yang, Y.J.A. ProtChatGPT: Towards Understanding Proteins with Large Language Models. arXiv 2024, arXiv:2402.09649. [Google Scholar]
- Lv, L.; Lin, Z.; Li, H.; Liu, Y.; Cui, J.; Chen, C.Y.-C.; Yuan, L.; Tian, Y. ProLLaMA: A Protein Language Model for Multi-Task Protein Language Processing. IEEE Trans. Artif. Intell. 2024, 1–12. [Google Scholar] [CrossRef]
- Su, J.; Han, C.; Zhou, Y.; Shan, J.; Zhou, X.; Yuan, F. SaProt: Protein Language Modeling with Structure-aware Vocabulary. bioRxiv 2023. [Google Scholar] [CrossRef]
- Li, M.; Tan, P.; Ma, X.; Zhong, B.; Yu, H.; Zhou, Z.; Ouyang, W.; Zhou, B.; Hong, L.; Tan, Y. ProSST: Protein Language Modeling with Quantized Structure and Disentangled Attention. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lin, Z.M.; Akin, H.; Rao, R.S.; Hie, B.; Zhu, Z.K.; Lu, W.T.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Ding, X.; Hsu, W.; Munir, A.; Xu, Y.; Li, Y. pLM4ACE: A protein language model based predictor for antihypertensive peptide screening. Food Chem. 2024, 431, 137162. [Google Scholar] [CrossRef]
- Xu, X.; Bonvin, A.M.J.J. DeepRank-GNN-esm: A graph neural network for scoring protein–protein models using protein language model. Bioinform. Adv. 2024, 4, vbad191. [Google Scholar] [CrossRef]
- Zeng, S.; Wang, D.; Jiang, L.; Xu, D. Parameter-efficient fine-tuning on large protein language models improves signal peptide prediction. Genome Res. 2024, 34, 1445–1454. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Carugo, O. pLDDT Values in AlphaFold2 Protein Models Are Unrelated to Globular Protein Local Flexibility. Crystals 2023, 13, 1560. [Google Scholar] [CrossRef]
- Weaver, L.H.; Matthews, B.W. Structure of Bacteriophage-T4 Lysozyme Refined at 1.7 a Resolution. J. Mol. Biol. 1987, 193, 189–199. [Google Scholar] [CrossRef]
- Kaur, H.; Sasidhar, Y.U. Molecular dynamics study of an insertion/duplication mutant of bacteriophage T4 lysozyme reveals the nature of α → β transition in full protein context. Phys. Chem. Chem. Phys. 2013, 15, 7819–7830. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Allen, M.D.; Fersht, A.R. Crystal structure of a superstable mutant of human p53 core domain—Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Luo, Z.; Yan, Y.; Wang, Q.; Ma, J. OPUS-Rota5: A highly accurate protein side-chain modeling method with 3D-Unet and RotaFormer. Structure 2024, 32, 1001–1010.e1002. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.D.; Zhou, Y.Q. Improving prediction of protein secondary structure, backbone angles, solvent accessibility and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks. Bioinformatics 2019, 35, 2403–2410. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Q.H.; Ma, J.P. OPUS-TASS: A protein backbone torsion angles and secondary structure predictor based on ensemble neural networks. Bioinformatics 2020, 36, 5021–5026. [Google Scholar] [CrossRef]
- Lu, M.Y.; Dousis, A.D.; Ma, J.P. OPUS-PSP: An orientation-dependent statistical all-atom potential derived from side-chain packing. J. Mol. Biol. 2008, 376, 288–301. [Google Scholar] [CrossRef]
- Xu, G.; Ma, T.Q.; Zang, T.W.; Sun, W.T.; Wang, Q.H.; Ma, J.P. OPUS-DOSP: A Distance- and Orientation-Dependent All-Atom Potential Derived from Side-Chain Packing. J. Mol. Biol. 2017, 429, 3113–3120. [Google Scholar] [CrossRef]
- Yang, J.Y.; Anishchenko, I.; Park, H.; Peng, Z.L.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Q.H.; Ma, J.P. OPUS-X: An open-source toolkit for protein torsion angles, secondary structure, solvent accessibility, contact map predictions and 3D folding. Bioinformatics 2022, 38, 108–114. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the 3rd International Conference on Learning Representations, San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.M.; Chen, Z.F.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation, Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Xu, G.; Wang, Q.H.; Ma, J.P. OPUS-Mut: Studying the Effect of Protein Mutation through Side-Chain Modeling. J. Chem. Theory Comput. 2023, 19, 1629–1640. [Google Scholar] [CrossRef]
- Haas, J.; Barbato, A.; Behringer, D.; Studer, G.; Roth, S.; Bertoni, M.; Mostaguir, K.; Gumienny, R.; Schwede, T. Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins 2018, 86, 387–398. [Google Scholar] [CrossRef] [PubMed]
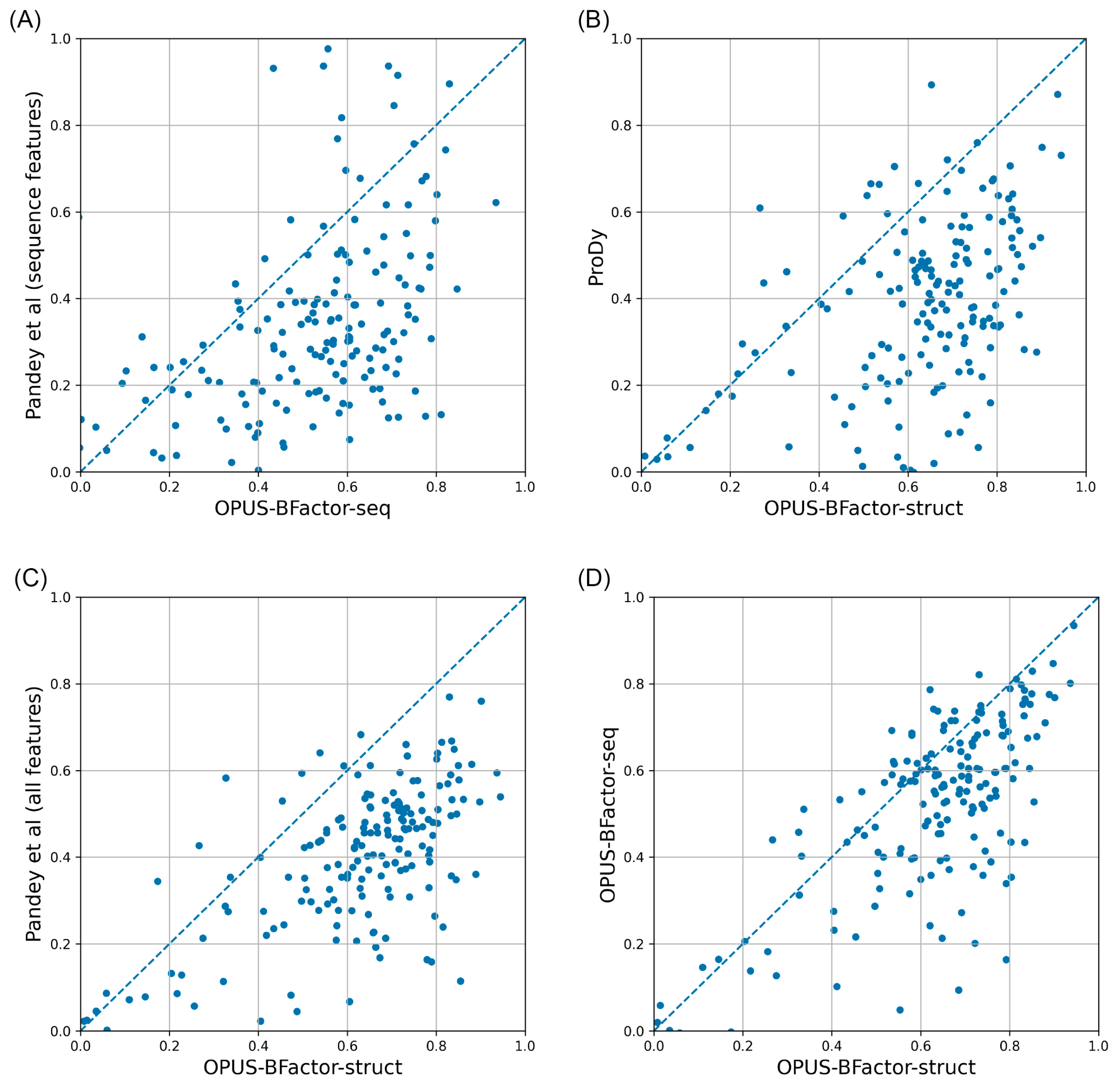

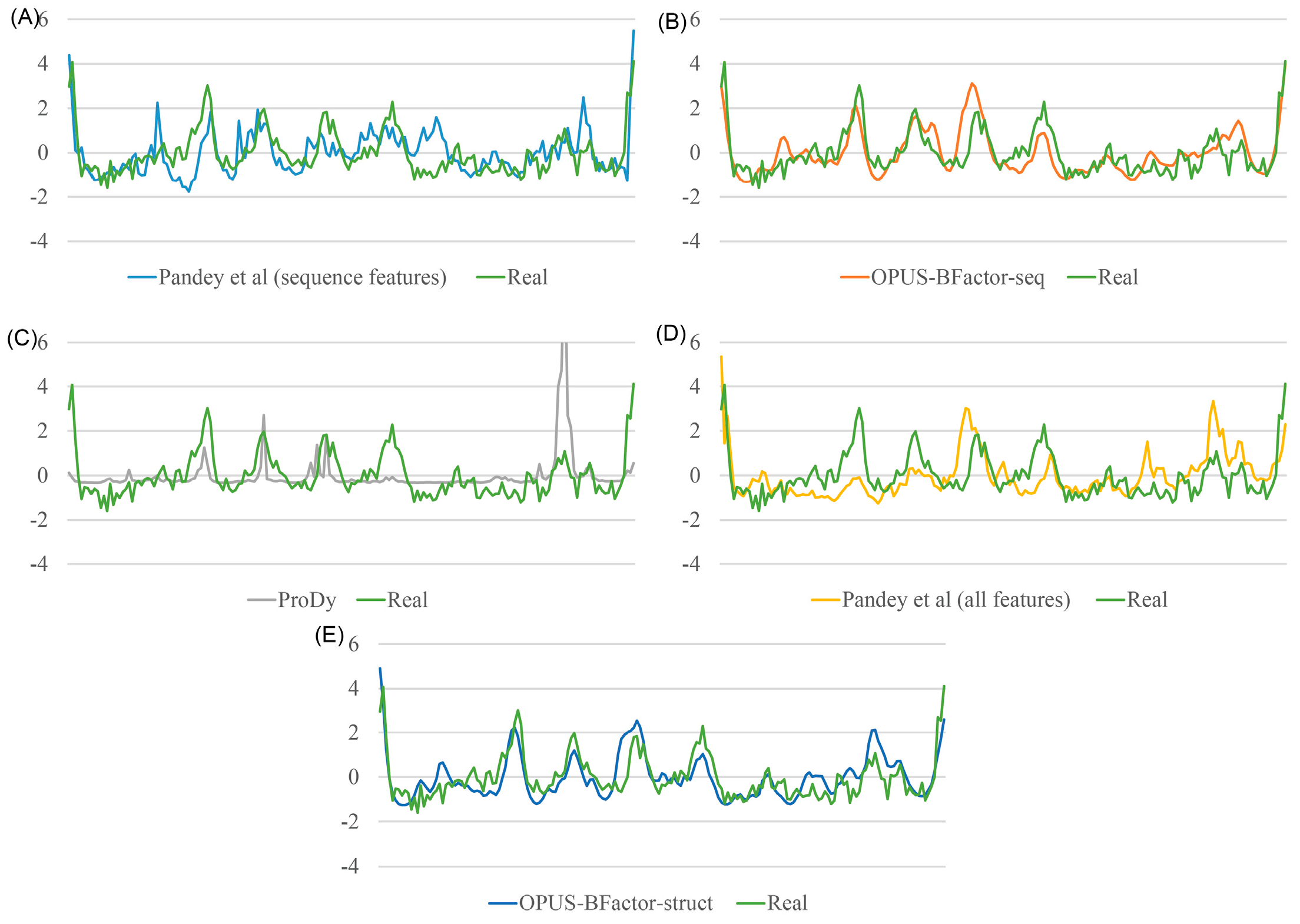
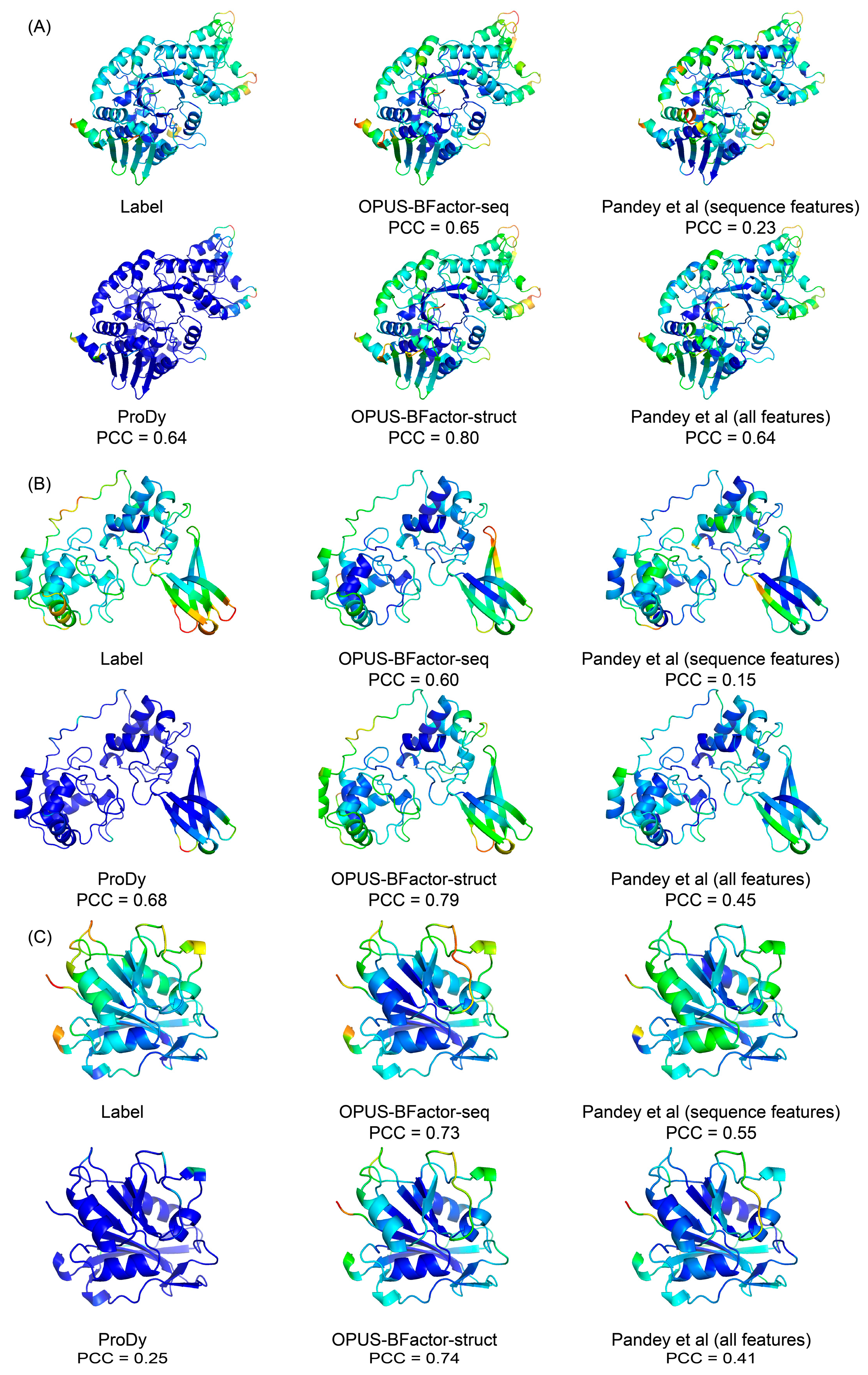
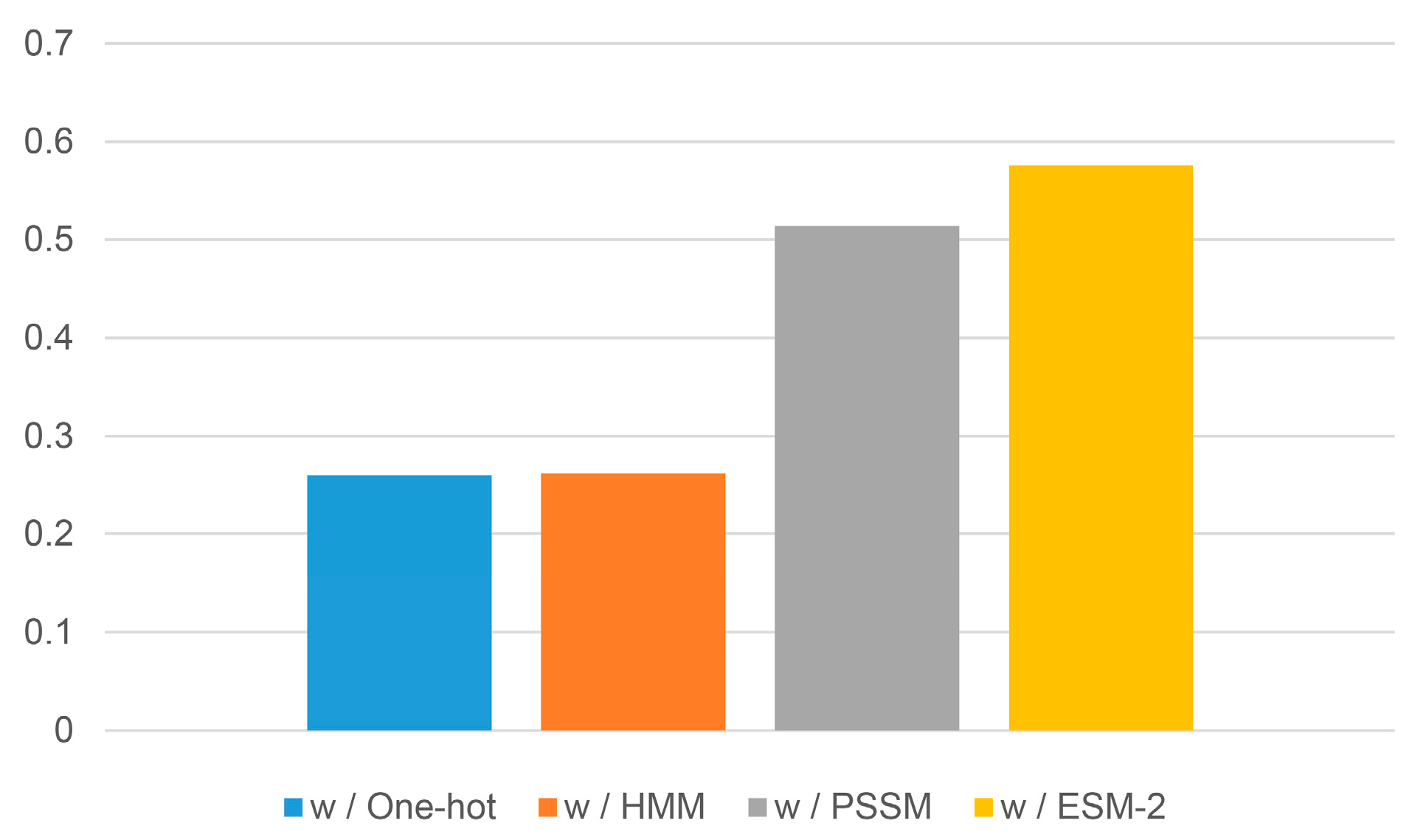

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAMEO65 | CASP15 | CAMEO82 | |
|---|---|---|---|
| Sequence-based methods | |||
| Pandey et al. [16] | 0.37 (0.30) | 0.20 (0.24) | 0.33 (0.30) |
| OPUS-BFactor-seq | 0.50 (0.56) | 0.34 (0.41) | 0.58 (0.60) |
| Structure-based methods | |||
| ProDy [39] | 0.31 (0.33) | 0.25 (0.28) | 0.43 (0.44) |
| Pandey et al. [16] | 0.38 (0.42) | 0.33 (0.35) | 0.41 (0.43) |
| OPUS-BFactor-struct | 0.61 (0.69) | 0.48 (0.56) | 0.67 (0.69) |
| CAMEO65 | CASP15 | CAMEO82 | |
|---|---|---|---|
| pLDDT (ESMFold) | 0.28 | 0.24 | 0.38 |
| OPUS-BFactor-seq | 0.50 | 0.34 | 0.58 |
| TM Score > 0.9 (89) | 0.8 < TM Score < 0.9 (32) | Others (60) | |
|---|---|---|---|
| pLDDT (ESMFold) | 0.42 | 0.34 | 0.14 |
| OPUS-BFactor-seq | 0.61 | 0.48 | 0.33 |
| OPUS-BFactor-struct (ESMFold) | 0.63 | 0.55 | 0.34 |
| OPUS-BFactor-struct (PDB) | 0.67 | 0.64 | 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Lv, Y.; Luo, Z.; Wang, Q.; Xu, G.; Ma, J. OPUS-BFactor: Predicting Protein B-Factor with Sequence and Structure Information. Molecules 2025, 30, 2570. https://doi.org/10.3390/molecules30122570
Yang Y, Lv Y, Luo Z, Wang Q, Xu G, Ma J. OPUS-BFactor: Predicting Protein B-Factor with Sequence and Structure Information. Molecules. 2025; 30(12):2570. https://doi.org/10.3390/molecules30122570
Chicago/Turabian StyleYang, Yulu, Ying Lv, Zhenwei Luo, Qinghua Wang, Gang Xu, and Jianpeng Ma. 2025. "OPUS-BFactor: Predicting Protein B-Factor with Sequence and Structure Information" Molecules 30, no. 12: 2570. https://doi.org/10.3390/molecules30122570
APA StyleYang, Y., Lv, Y., Luo, Z., Wang, Q., Xu, G., & Ma, J. (2025). OPUS-BFactor: Predicting Protein B-Factor with Sequence and Structure Information. Molecules, 30(12), 2570. https://doi.org/10.3390/molecules30122570