2.1. DFT Studies
The pursuit of improving the therapeutic effectiveness and reducing the toxicity of tacrine derivatives led to a comprehensive investigation focusing on compounds containing an organic selenium component [
20]. This study sought to address the limitations of traditional tacrine formulations, particularly their hepatotoxicity, by leveraging selenium’s unique properties, which include antioxidative and catalytic potential akin to glutathione peroxidase [
19].
The research commenced with extensive theoretical studies to explore the mechanisms of action of these selenium-containing compounds. By employing advanced computational methods, including DFT, the study investigated molecular descriptor energies such as HOMO, LUMO, and their associated energy gaps [
21] These descriptors provided critical insights into the compounds
’ stability, reactivity, and potential for interaction with biological targets. The DFT analyses also allowed the prediction of electron donation and reception capabilities, shedding light on how these compounds might interact with active sites in acetylcholinesterase, the key enzyme targeted in Alzheimer’s disease therapy [
22].
A pivotal aspect of the study was the optimization of synthetic routes for producing these novel tacrine derivatives. This optimization involved identifying favorable reaction conditions, selecting appropriate precursors, and integrating selenium atoms into the molecular framework. The process was informed by Quantitative Structure-Activity Relationship (QSAR) analyses, molecular docking studies, and pharmacokinetic simulations. QSAR provided a quantitative link between chemical structures and their biological activities, enabling the prioritization of compounds with the highest potential efficacy [
23].
Molecular docking studies revealed how these derivatives might interact with acetylcholinesterase at the molecular level, highlighting binding affinities and the specific interactions of selenium atoms within the active site. Toxicological predictions were performed to evaluate the potential risks associated with these compounds, including hepatotoxicity and mutagenicity, ensuring that the synthesized derivatives maintained a favorable safety profile [
24].
To validate the theoretical findings, some experimental studies were carried out in parallel, such as toxicity and antioxidant tests for compound
1; the results were previously published by our group [
19]. Initial efforts focused on synthesizing compounds that exhibited the most promising profiles in silico. The synthesized derivatives will be subjected to rigorous in vitro testing to evaluate their ability to inhibit acetylcholinesterase and reduce oxidative stress, which is a hallmark of Alzheimer’s pathology [
25]. Encouraging candidates from in vitro tests advanced to in vivo studies to assess their pharmacokinetics, bioavailability, and overall therapeutic efficacy. The integration of selenium into the tacrine scaffold proved to be particularly beneficial, as selenium’s antioxidative properties appeared to mitigate oxidative damage, a significant contributor to neuronal degeneration [
26].
Another critical dimension of the research was the analysis of electronic properties, specifically focusing on the distribution of the HOMO and the LUMO. These properties are crucial for understanding the compounds’ reactivity and stability. The study revealed that the presence of selenium significantly influenced the electronic distribution, directing the HOMO electron density toward the aromatic ring containing selenium. This redistribution is believed to enhance the compound’s ability to interact with biological targets while improving stability. Interestingly, substituents on the tacrine scaffold had a relatively lesser impact on electronic distribution compared to selenium, underscoring the unique role of this element in modulating molecular properties [
27].
Moreover, pharmacokinetic profiling of the derivatives provided insights into their absorption, distribution, metabolism, and excretion (ADME) properties. The use of computational tools like PreADMET allowed for predictions regarding human intestinal absorption, blood–brain barrier penetration, and interactions with cytochrome P450 enzymes. These analyses confirmed that the incorporation of selenium not only improved the compounds’ bioavailability but also reduced the possibility of forming toxic metabolites, addressing one of the key challenges associated with tacrine use [
28].
The preliminary results reported by us [
19] form part of this broader study, which provides a detailed comparison of the electronic properties of selenium-modified tacrine derivatives with different substituents and unmodified tacrine.
Figure 2 presents the results for the most promising compounds. The tacrine derivatives (compounds
12–
14) exhibit HOMO energy levels ranging from −6.66 eV to −6.74 eV, which are significantly higher than that of the original tacrine molecule (−8.89 eV). This difference indicates a lower ionization potential, suggesting that these derivatives can more readily donate electrons [
29]. As a result, they may interact more effectively with biological targets through electron donation. The similar HOMO levels observed among the selenium-containing compounds highlight the influence of selenium on these electronic properties [
30].
The LUMO energy levels remain relatively consistent across the derivatives, averaging around −5.62 eV, which is slightly higher than that of the parent tacrine molecule (−5.72 eV). This consistency suggests that the electron-accepting capability is largely preserved. Notably, selenium appears to influence the spatial distribution of LUMO density, shifting it toward regions potentially involved in interactions with acetylcholinesterase, which may enhance binding affinity [
27].
A key observation from
Figure 2 is that the HOMO–LUMO energy gap in the tacrine derivatives is significantly reduced, averaging around 1.1 eV, compared to 3.16 eV in the parent tacrine molecule. This narrower energy gap suggests increased chemical reactivity and enhanced charge-transfer capability, which are relevant features for potential enzyme inhibition mechanisms [
28].
Although not shown in the figure, the electronegativity values of the derivatives were previously reported by Sacramento et al. (2024) [
19]. These values are lower (5.93–6.18 eV) than that of the parent tacrine molecule (7.30 eV), further supporting their potential for improved interactions in biological systems. Selenium plays a key role in this decrease, contributing to the development of compounds that may act on multiple biological pathways. The incorporation of selenium into the tacrine scaffold significantly alters the electronic distribution of the derivatives [
31].
The incorporation of selenium into tacrine derivatives significantly alters the electron distribution, particularly within the HOMO orbitals. As shown in
Figure 3, selenium-containing compounds (
1,
2, and
4) exhibit a pronounced localization of HOMO electron density on the aromatic ring bearing the selenium atom, in contrast to the parent tacrine molecule, where the HOMO is more evenly distributed. This shift enhances the electron-donating capability of the selenium moiety, potentially improving interactions with biological targets such as acetylcholinesterase. While substituents like methyl groups (electron-donating) or halogens (electron-withdrawing) exert some influence, their effects are modest when compared to the substantial impact of selenium. Overall, selenium plays a pivotal role in modulating the reactivity and interaction potential of these derivatives [
32].
The comparison with tacrine underscores the significant improvements achieved through molecular modification. The original tacrine, with its high GAP and electronegativity, exhibits lower reactivity and limited interaction potential with biological targets. In contrast, the derivatives demonstrate optimized electronic properties, making them more suitable for therapeutic applications. The reduced GAP and electronegativity in the derivatives align with the goal of enhancing efficacy while minimizing toxicity [
33].
The incorporation of selenium and the structural modifications to the tacrine derivatives have significantly influenced their electronic properties. The data indicate that these changes lead to improved HOMO–LUMO energy levels and a reduced energy gap, suggesting increased molecular reactivity. This enhances their potential to interact with biological targets, as supported by molecular orbital analysis and other reactivity parameters [
9]. These findings provide a strong foundation for further experimental validation and highlight the potential of selenium-containing tacrine derivatives as promising candidates for Alzheimer’s disease therapy. By systematically analyzing these electronic parameters, the study offers valuable insights into the rational design of safer and more effective drugs.
The electronic properties of the tacrine derivatives and unmodified tacrine, as depicted in
Figure 3, reveal critical insights into their stability, reactivity, and potential therapeutic application. A detailed analysis of these compounds’ HOMO and LUMO energies provides a deeper understanding of their electronic distributions, which significantly influence their behavior in biological systems [
27].
The GAP between HOMO and LUMO is a crucial parameter for determining a compound’s chemical reactivity. In this study, the derivatives exhibit relatively lower energy gaps, suggesting increased chemical reactivity compared to tacrine [
10].
The energy gap also influences the compounds’ electronic excitation and charge transfer potential. For instance, selenium-containing compounds appear to direct the HOMO and LUMO electron densities toward specific regions of the molecule, optimizing their interactions with the biological target [
34]. This targeted electronic distribution enhances the therapeutic potential of these derivatives while minimizing off-target effects [
12].
The electronegativity values of the tacrine derivatives are relatively consistent, indicating comparable tendencies to attract electrons. This uniformity suggests that the introduction of selenium and substituents does not drastically alter the electron-attracting capabilities of the molecules, preserving their chemical behavior [
35].
Interestingly, tacrine exhibits a significantly higher electronegativity value compared to the derivatives. This observation suggests that the derivatives are more stable than tacrine, as lower electronegativity values generally indicate a reduced likelihood of engaging in rapid, potentially harmful electron transfer reactions. The improved stability of the derivatives supports their potential as safer alternatives to tacrine, especially in mitigating the hepatotoxic effects associated with tacrine’s metabolism [
36].
Figure 3 highlights how selenium and various substituents influence the electronic distribution within the tacrine scaffold. Selenium, known for its unique electron-donating and antioxidative properties, significantly impacts the HOMO electron density, directing it toward the aromatic ring containing selenium. This redistribution enhances the molecule’s ability to engage in specific interactions with biological targets, such as acetylcholinesterase, while maintaining overall molecular stability. Substituents, categorized as electron-donating or electron-withdrawing groups, further modulate the electronic properties [
27].
Neutral substituents (Compound 1): Exhibit balanced HOMO–LUMO distributions, indicating moderate stability and reactivity.
Electron-donating substituents (Compound 2): Slightly reduce the energy gap, enhancing reactivity while maintaining sufficient stability for biological applications.
Electron-withdrawing substituents (Compound 4): Increase electronegativity and stabilize the LUMO, potentially reducing off-target interactions.
These variations in electronic behavior highlight the flexibility of the tacrine scaffold in accommodating diverse chemical modifications to optimize therapeutic properties [
37].
The comparison between tacrine and its selenium-containing derivatives reveals clear improvements in electronic properties resulting from structural modifications. Tacrine exhibits the highest electronegativity (7.30 eV) and the widest HOMO–LUMO gap (3.16 eV), suggesting lower chemical reactivity and a potential for toxicity. Notably, this may be linked to a specific point of electronic instability at carbon 7, which is effectively stabilized in the proposed derivatives through the incorporation of selenium at that position [
29].
On the other hand, the derivatives—particularly those containing selenium—exhibit significantly lower HOMO–LUMO gap values (0.81–1.15 eV) and reduced electronegativity (5.93–6.18 eV). These features suggest increased chemical reactivity and an enhanced ability to interact with biological targets. Such electronic modifications highlight the potential of these molecules as more effective candidates for the treatment of Alzheimer’s disease [
38].
Specifically, tacrine derivatives exhibited a distinctive distribution pattern, with HOMO electron density situated between the selenium atom and the substituent ring. Meanwhile, the LUMO was predominantly located on the amino-acridine ring. In summary, while aniline derivatives tended to draw energy from the HOMO orbital, selenium-containing compounds directed HOMO density towards the selenium atom and the adjacent ring. These findings provide valuable insights into the design and development of novel therapeutic agents with improved efficacy and safety profiles [
10].
In order to verify whether it was really the influence of selenium, we analyzed a derivative replacing selenium with the oxygen atom, which has similar characteristics. We observed that it contains different HOMO profiles than derivatives containing selenium (
Figure S1; Supplementary Materials).
MEPs offer a valuable visual representation of electron density distribution within a molecule. Regions of red and blue in these maps correspond to areas of high (negative) and low (positive) electron density, respectively (
Figure 4). By examining MEPs of compounds in their ground states, we can effectively compare the influence of various substituents on molecular charge distribution [
18].
Compound
1 exhibits negative regions near the hydrogen atoms, suggesting a slightly acidic character at these sites. Conversely, the most positive regions are likely associated with heteroatoms or the π system of the aromatic ring, implying basic or nucleophilic properties. Compound
2 demonstrates a more polarized charge distribution, with distinct regions of positive and negative charge. The selenium atom, for instance, is likely associated with a region of partial positive charge [
39]. Similarly, compound
4 presents a polarized charge distribution, with the halogen atom bearing a partial negative charge. This observation highlights that, unlike the HOMO and LUMO maps, substituents significantly influence the distribution of electrostatic potential.
2.2. Pharmacokinetic Properties
The data presented in
Table 1 provide a comprehensive overview of the pharmacokinetic properties of tacrine and its selenium-modified derivatives, highlighting key parameters such as HIA (%), BBBP (Log
CC), permeability in the central nervous system (CNS) (Log
PS), permeability across membranes (Log
P 8 × 10
−6 ca
−2 nm/s), and the Log
P values. These parameters are critical in understanding the absorption, distribution, metabolism, and excretion (ADME) characteristics of the compounds, ultimately influencing their therapeutic potential [
16].
The HIA (%), which measures the intestinal absorption, demonstrates that most of the selenium-modified derivatives exhibit improved absorption compared to tacrine, which has an HIA of 94.17%. Notably, compounds such as 3, 9, and 15 achieve near-perfect absorption, with values of 99.65%, 99.94%, and 100%, respectively. This enhanced absorption indicates the potential for these derivatives to exhibit better bioavailability than tacrine, making them promising candidates for oral administration [
18].
The BBBP (Log
CC) values, which reflect the plasma protein binding capacity, vary across the derivatives. Tacrine exhibits a relatively high BBBP value of 0.21, suggesting significant plasma protein interaction, which can influence the drug’s free concentration in circulation. Several derivatives, such as compounds
3 and
15, display negative BBBP values, indicating reduced protein binding. This characteristic could enhance the free drug fraction, potentially improving therapeutic efficacy. However, derivatives with higher PBH values, such as compound
17 (0.12), may exhibit prolonged circulation times due to stronger protein binding [
36].
The CNS permeability (Log
PS) values offer insights into the compounds’ ability to cross the blood–brain barrier (BBB), a crucial factor for neuroactive drugs targeting central nervous system disorders like Alzheimer’s disease. Tacrine shows a Log
PS of −1.70, indicating limited BBBP penetration. In comparison, most derivatives exhibit similar or slightly better permeability values, with compound
15 demonstrating the least restriction at −1.13. This improvement suggests that the selenium-modified derivatives may have enhanced potential for effective central nervous system delivery [
40].
The membrane permeability (Log
P 8 × 10
−6 ca
−2 nm/s) values further emphasize the enhanced transport properties of the derivatives. Tacrine demonstrates a permeability value of 1.58, while the derivatives show comparable or slightly lower values, indicating similar abilities to permeate cellular membranes. Compound
1, with a permeability of 1.54, closely matches tacrine’s performance, whereas compound
12 exhibits a lower value of 0.98, which might suggest reduced membrane-crossing efficiency for this specific derivative [
23].
The Log
P values, a measure of lipophilicity, are critical for understanding the compounds’ solubility and membrane permeability. Tacrine has a Log
P of 2.83, indicating moderate lipophilicity. In contrast, most derivatives exhibit higher Log
P values, ranging from 3.51 to 5.75. Compounds like
13 (5.75) and
12 (5.39) are particularly lipophilic, which could enhance their ability to penetrate lipid-rich environments such as the BBB. However, excessively high lipophilicity might also pose challenges related to reduced solubility in aqueous environments and potential toxicity [
34].
Overall, the pharmacokinetic analysis underscores the improvements achieved in the selenium-modified derivatives compared to tacrine. Enhanced AIH values indicate better absorption, while variations in PBH and permeability values suggest that the derivatives have been optimized for better bioavailability and central nervous system targeting. The increased Log
P values highlight the improved lipophilicity of the derivatives, which could contribute to their therapeutic efficacy. These findings provide valuable insights for selecting the most promising candidates for further in vitro and in vivo evaluations, paving the way for the development of safer and more effective treatments for neurodegenerative diseases like Alzheimer’s [
35].
2.3. Toxicological Properties
Table 2 presents toxicological predictions for tacrine and its selenium-modified derivatives, focusing on three critical parameters: carcinogenicity in humans, hepatotoxicity in humans, and skin sensitization. These parameters are pivotal in assessing the safety profiles of potential drug candidates and ensuring that modifications do not introduce unacceptable risks [
36].
For carcinogenicity, the majority of the derivatives are classified as “plausible” for their potential to induce carcinogenic effects in humans. This classification suggests that while the risk exists, it is not definitive and warrants further investigation. Compounds
8,
13,
14, and
15 stand out as exceptions, with no evidence or indication of carcinogenic potential. This distinction highlights these derivatives as potentially safer options in terms of long-term use, particularly when compared to tacrine, which is also categorized as “plausible” for carcinogenicity. The widespread plausible classification across the derivatives emphasizes the need for additional preclinical and clinical studies to confirm the safety of these compounds [
41].
For hepatotoxicity, tacrine is distinctly marked as “certain”, reflecting its well-documented hepatotoxic effects, which have been a significant limitation in its therapeutic use. In contrast, none of the selenium-modified derivatives exhibit indications of hepatotoxicity. This stark difference underscores the success of the structural modifications, particularly the incorporation of selenium, in mitigating the hepatotoxic effects traditionally associated with tacrine. This improvement aligns with the broader goal of developing safer acetylcholinesterase inhibitors for Alzheimer’s disease [
23].
Regarding skin sensitization, most compounds show no signs of inducing skin sensitization, with only compounds
3 and
9 marked as “plausible”. Tacrine, on the other hand, is categorized as “probable”, indicating a higher probability of causing adverse skin reactions compared to the derivatives. The limited instances of plausible skin sensitization among the derivatives suggest that selenium incorporation does not significantly increase the risk of allergic reactions through skin contact, further supporting their safety profiles [
34].
Overall, the toxicological predictions indicate that the selenium-modified derivatives offer significant improvements over tacrine in terms of reduced hepatotoxicity and skin sensitization risks. However, the potential carcinogenicity of several derivatives remains a concern that must be addressed through further testing. Compounds such as
8,
13,
14, and
15, which show no indication of carcinogenicity, hepatotoxicity, or skin sensitization, emerge as particularly promising candidates for further development. These findings highlight the potential of selenium-modified tacrine derivatives to serve as safer alternatives for the treatment of neurodegenerative diseases, paving the way for further experimental validation and optimization [
35].
2.4. Metabolic Prediction
Employing the ADMET Predictor
®, a metabolic prediction for tacrine was conducted. The prediction identified potential metabolic pathways and primary metabolites (M1a, M2a, M3a, and M4a) derived from the parent molecule. Tacrine was predicted to be extensively metabolized by CYP1A2 and CYP2D6 enzymes [
34].
These enzymes catalyze structural modifications, resulting in the formation of metabolites M1a, M2a, M3a, and M4a. As aligned with previous literature, M1a, the predominant metabolite, is recognized as the primary reactive intermediate responsible for toxicity. Subsequent metabolites include M2a, M3a, and M4a (
Figure 5).
We performed a metabolic prediction for our compounds that showed better theoretical results. Metabolic prediction studies revealed that our compounds exhibited favorable theoretical outcomes in comparison to tacrine. Notably, the likelihood of forming reactive intermediates was significantly reduced. The primary metabolic transformation observed was the substitution of one methyl group with a hydroxyl group (-OH) [
30].
We conducted a comprehensive analysis of the metabolic profiles of all proposed derivatives and compared them with the metabolic profile of tacrine in the context of the CYP enzyme complex. Our evaluation revealed a significant formation of the probable metabolites M1b and M2b in compound
12 (
Figure 6), accounting for 35% and 30% of the total metabolic products, respectively [
40].
Primarily catalyzed by CYP2C19, CYP2C9, and CYP3A4 isoforms, the biotransformation of compound
13 resulted in multiple oxygenated metabolites, of which M1c and M2c, each with 20%, emerged as the major metabolites
Figure 7.
This finding suggests that the metabolic pathways catalyzed by CYP2D6, CYP2D9, and CYP3A4 are predominantly utilized during the biotransformation of these compounds. Notably, despite the high metabolic activity observed, the metabolites generated from these derivatives do not exhibit structural characteristics typically associated with hepatotoxicity. This observation highlights the potential safety of these compounds, as the absence of hepatotoxic metabolites could reduce the possibility of adverse liver-related effects, a common concern in the development of compounds targeting the CYP complex [
23].
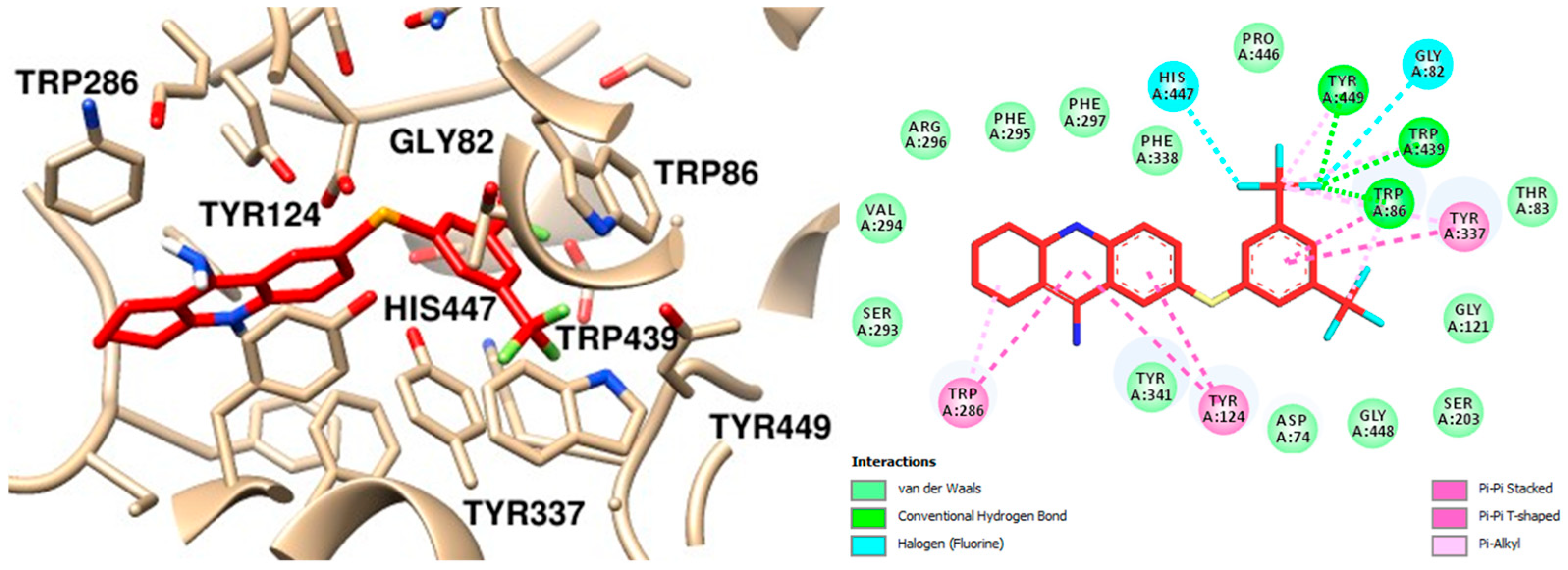
2.6. Molecular Dynamics
To ensure the reliability of our results and validate the system’s stability, we combined molecular docking with molecular dynamics (MD) simulations. The docking results for the complexes AChE–tacrine, AChE–compound
12, and AChE–compound
13, as well as the AChE apoprotein, were subjected to 100 ns MD simulations to analyze their atomic behavior in detail. Root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and the number of hydrogen bonds were calculated for the three complexes. Simulations of the AChE apoprotein were also conducted to better understand its intrinsic atomic behavior. The RMSD results from triplicate simulations of the protein backbone for each system revealed that the AChE apoprotein tends to stabilize around 40 ns, with RMSD values ranging between 0.4 and 0.6 nm. This indicates overall structural stability in the absence of ligands (
Figure S3A, Supplementary Materials). A similar stabilization pattern was observed in the RMSD for the protein backbones for systems with ligands (
Figure S3B,D,F; Supplementary Materials).
Analysis of the RMSD for ligand heavy atoms fitted to the protein backbone showed distinct behaviors among the three ligands (
Figure S3C,E,G; Supplementary Materials). The results were consistent among triplicates for compound
12, while slightly more pronounced fluctuations were observed for tacrine and compound
13. These findings suggest that all three ligands exhibit some degree of movement within the binding site, with compound
13 demonstrating the highest stability. Overall, the RMSD data highlight the stability and conformational dynamics of AChE when bound to different ligands. While each ligand induces some level of fluctuation, the protein structure remains stable, indicating effective binding without major disruptions.
In summary, considering the AChE–tacrine complex, the protein backbone stabilizes around 0.4 nm after 20 ns, and the heavy atoms from the ligand stabilize at 0.4 nm after 40 ns, indicating initial instability followed by stability (
Figure 10A). Regarding the selenylated compound
12, both AChE and the ligand quickly stabilize around 0.4 nm, indicating a stable complex throughout the simulation (
Figure 10B), and a similar behavior was observed in the AChE–compound
13 complex (
Figure 10C). Comprehensively, the RMSD data indicate that all three ligands show initial fluctuations, which stabilize over time. Nonetheless, both compounds
12 and
13 show similar stability profiles, suggesting that they form more stable complexes with AChE compared to tacrine. The stabilization of RMSD values for the ligands implies that they remained within the binding site throughout the simulation period, without significant dissociation.
The root-mean-square fluctuation (RMSF) values were calculated to assess the flexibility of the protein chain residues during the MD simulations. RMSF quantifies the average deviation of each atom’s position from its mean position over the course of a simulation or within an ensemble of structures [
31]. The RMSF values obtained across all triplicate runs for the AChE apoprotein (
Figure S4A, Supplementary Materials) and its complexes with tacrine (
Figure S4B, Supplementary Materials), compound
12 (
Figure S4C, Supplementary Materials), and compound
13 (
Figure S4D, Supplementary Materials) indicate that most residues exhibit low fluctuation, reflecting structural stability. However, increased flexibility was observed in the region around residue 600 across all systems, consistent with the dynamic nature of the protein terminal domains. The alignment and consistency of the RMSF values across the triplicates, represented by the median line, confirm the reliability and reproducibility of the results. This consistency underscores the common area of flexibility or disorder in the AChE structure (
Figure 11) and reinforces the stability of the analyzed complexes.
In the AChE–tacrine complex, the number of hydrogen bonds fluctuates more significantly across the triplicates, with a median of approximately 1. This suggests fewer and less stable hydrogen bonds compared to involving the selenylated compounds. For the AChE–compound 12 complex, the number of hydrogen bonds shows moderate variation, with a median of around 2, indicating that compound 12 forms more stable hydrogen bonds with AChE compared to tacrine. Finally, the AChE–compound 13 complex demonstrates the highest number of hydrogen bonds, with a median of approximately 3. Additionally, the fluctuations are more moderate across the triplicates, suggesting that compound 13 forms the most stable hydrogen bonds with AChE among the tested ligands.
The medians across the triplicates for each complex are summarized in
Figure 12, reinforcing the conclusion that AChE–compound
13 exhibits the most stable and numerous hydrogen bonds, followed by compound
12 and tacrine. These findings highlight the potential of compound
13 to form stable interactions with AChE, underscoring its efficacy as a promising ligand.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}