1. Introduction
Oral administration is not only convenient but also offers good patient compliance; however, after the drug is taken orally and absorbed in the small intestine, it needs to overcome three barriers: the chemical barrier, the physical barrier, and the biological barrier [
1,
2]. The chemical barriers encompass the harsh gastrointestinal (GI) environment, including variable pH levels and digestive enzymes, while the physical barriers involve the intestinal mucus layer and tightly packed epithelial cells. Additionally, biological barriers arise from intestinal efflux pumps (e.g., P-glycoprotein), drug-metabolizing enzymes, gut microbiota, etc. These multilayered challenges impose stringent demands on a drug’s solubility, stability, and permeability, restricting its clinical translation [
3,
4,
5]. To address these limitations, nanocarriers have emerged as a promising strategy for oral drug delivery. They protect drugs from degradation in the GI tract, enhance mucus penetration and epithelial permeation through optimized size or surface modifications [
6], improve drug dispersion and solubility via nanoscale formulation [
7], and enable co-delivery of efflux inhibitors to counteract pump-mediated drug expulsion, thereby collectively enhancing bioavailability and therapeutic efficacy.
Researchers revealed that negatively charged hydrophilic particles can rapidly penetrate the mucosal layer, while positively charged nanoparticles show higher cellular uptake efficiency by epithelial cells [
8]. However, carriers focusing solely on overcoming a single barrier fail to simultaneously achieve both mucosal penetration and efficient epithelial absorption.
Intriguingly, viruses efficiently traverse both mucosal and epithelial barriers. Further studies attribute this capability to their unique surface architecture: a mixture of positively and negatively charged amino acid groups endows viruses with hydrophilicity and electroneutrality, enabling evasion of mucosal entrapment and clearance [
9]. Upon penetrating the mucosal layer, viruses utilize specialized fusion proteins to enhance intestinal epithelial entry.
Inspired by these findings, bioengineered materials imitating viral surface properties have been developed. When loaded with drugs, these materials effectively traverse the dual barriers of the intestinal mucosal layer and epithelial cells, significantly improving oral drug absorption and bio-availability [
10,
11,
12]. Virus-like particles are nanoscale structures that imitate the natural virus structure but lack the viral genome. They exhibit good bio-compatibility and immunogenicity, making them valuable in drug delivery applications [
13]. In recent years, researchers have exploited the characteristics of viruses to develop biomimetic nanocarriers for oral drug delivery. Yang et al. [
14] designed a surface ligand-switchable, virus-mimicking, multifunctional nanocarrier (Pep/Gal-PNP). Following oral administration, this carrier can expose trans-membrane peptide fragments via a charge reversal mechanism in the acidic gastrointestinal environment, efficiently traversing the intestinal mucosal barrier and targeting liver cells under physiological pH conditions. This design not only enhances the intestinal absorption efficiency of the drug but also achieves precise delivery to specific organs, demonstrating promising application prospects. Wu’s study [
15] introduces P-R8-Pho NPs, a biomimetic nanoparticle system that efficiently delivers insulin orally by overcoming mucus and epithelial barriers. The nanoparticles’ unique design allows them to penetrate mucus and then enter cells upon encountering IAP in the intestine, leading to improved insulin absorption and hypoglycemic effects. Zhang et al. [
16] prepared and characterized hydrophilic, electrically neutral MSN-NH2@COOH/CPP5 nanoparticles that mimic viruses. The modified nanoparticles exhibited no significant toxicity in preliminary in vitro and in vivo studies. These virus-mimicking nanoparticles can effectively overcome the barriers of the mucus layer and intestinal epithelium for the oral delivery of protein and peptide drugs.
In 1985, H.W. Kroto et al. [
17] discovered fullerenes, a class of carbon nanomaterials characterized by a hollow spherical structure, among which C60 (a spherical 32 hedron) exhibits the highest stability, with a large specific surface area and exceptional structural robustness. The molecular structure of fullerenes contains both C–C and C=C, conferring strong reducing properties. Although poorly water-soluble in their native form, fullerenes can be functionalized to yield water-soluble derivatives with enhanced bio-compatibility, making them promising candidates for drug delivery applications [
18]. Additionally, their intrinsic antioxidant capacity enables synergistic therapeutic effects when combined with pharmaceuticals [
19].
Curcumin (CUR) is a naturally occurring diketone phenolic compound extracted from the rhizome of Curcuma longa (Zingiberaceae family), demonstrating multifaceted pharmacological properties including antitumor, anti-inflammatory, antioxidant, choleretic, and hypolipidemic activities [
20]. Despite its broad therapeutic potential, CUR has not yet been approved for clinical applications due to critical pharmacokinetic limitations [
21].
Inspired by viral structural strategies to overcome the dual physiological barriers of intestinal absorption, we developed a fullerene-based nanocarrier co-modified with cell-penetrating peptide KLPVM (CPP5) [
16] and phosphoserine (Pser). This design achieved electroneutral surface properties and enhanced hydrophilicity, enabling efficient CUR encapsulation while ensuring mucus penetration. Following intestinal alkaline phosphatase (IAP)-triggered Pser cleavage at the epithelial interface, the nanocarrier underwent from neutrality reversal to positive [
22,
23], simultaneously exposing CPP5 with validated membrane-penetrating capability [
15]. This study systematically evaluates the bioavailability enhancement of curcumin through rationally designed nanocarriers and aims to develop a novel delivery platform specifically designed to address the challenges associated with orally administered drugs exhibiting poor membrane permeability, thereby enhancing their gastrointestinal absorption and therapeutic efficacy.
3. Materials and Methods
3.1. Materials
Fullerene C60(C60) was procured from Aladdin Biochemical Technology Co., Ltd. Shanghai, China, O-phospho-L-serine (Pser), N-Hydroxy succinimide (NHS), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and Diethyl bromomalonate were obtained from Yien Chemical Technology Co., Ltd. Shanghai, China, Coomassie Brilliant Blue G-250 (CBB), Curcumin, and Resveratrol were provided by Haohong Biomedical Technology Co., Ltd. Shanghai, China; 1-diphenyl-2-picrylhydrazyl (DPPH·) was provided by Thierry Chemical Industry Development Co., Ltd. Shanghai, China, Cell-penetrating pentapeptides (CPP5, KLPVM peptide) was chemically synthesized by Chu Peptide Biotechnology Co., Ltd. Beijing, China.
3.2. Synthesis of the Carrier
The carboxylated fullerene derivative (C60-COOH) was synthesized according to Semenov’s method [
30]. Specifically, 10 mg of C60 and 10 mg ethyl bromomalonate were dissolved in 5 mL toluene. After adding 20 molar equivalents of NaH, the solution was evacuated, heated to 60 °C under an inert gas (Ar) atmosphere, and stirred for 3 h. The reaction was terminated with 0.1 mL methanol, resulting in immediate solid precipitation. The product was isolated by vacuum filtration, sequentially washed twice with toluene, 2 M H
2SO
4, and deionized water, then vacuum-dried to obtain C60-COOH.
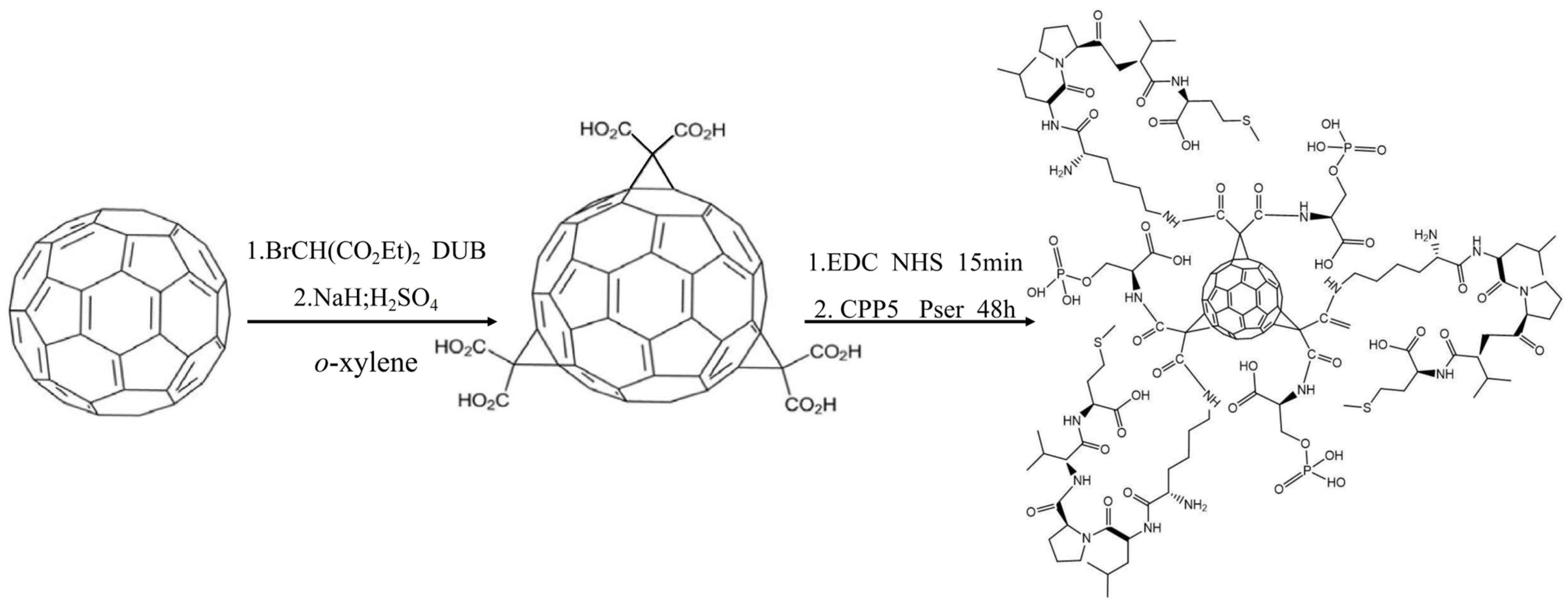
The C60-CPP5/Pser conjugate was prepared through carbodiimide-mediated coupling: 10 mg of C60-COOH was dispersed in 10 mL PBS (pH 7.4), followed by the addition of EDC and NHS at a molar ratio of 1:5:5 (C60-COOH:EDC:NHS). After 15 min activation, a three-fold molar excess of CPP5 and Pser relative to C60-COOH was introduced. The reaction proceeded under light-protected conditions with continuous stirring for 48 h, after which the product was isolated by freeze-drying. The synthetic route is depicted in
Figure 13.
3.3. Characterization of the Carrier
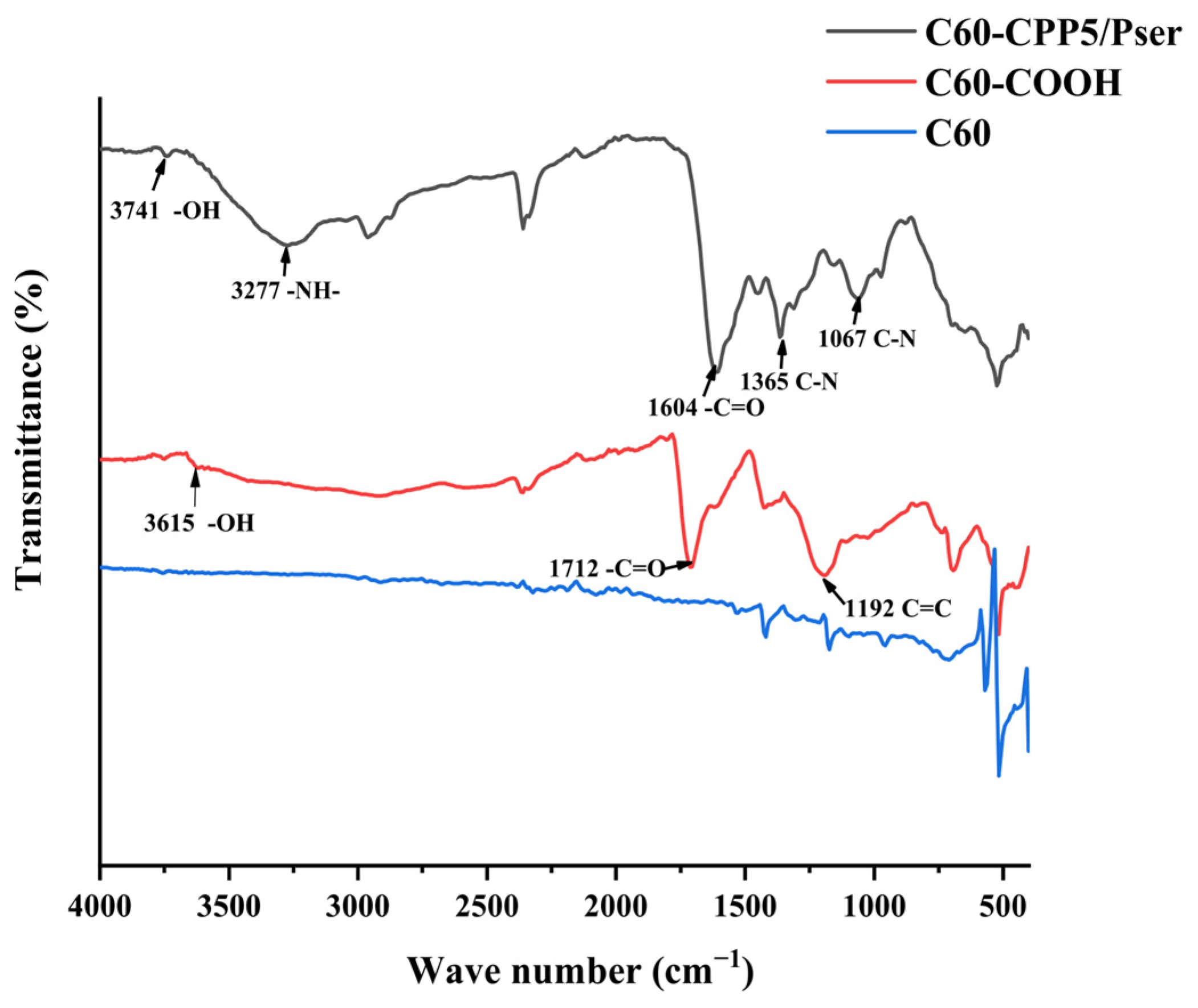
3.3.1. Physicochemical Properties of the Carrier
The dried sample powder was mounted on an SEM stub, uniformly dispersed, and subjected to high-vacuum evacuation. Prior to imaging, the specimen was sputter-coated with a gold layer to enhance conductivity. Secondary electron micrographs were acquired by Hitachi S-4800 Field Emission Scanning Electron Microscope (Hitachi, Tokyo, Japan).
The sample was dispersed in ethanol at a 1:500 mass ratio and homogenized via ultrasonication. The resulting colloidal suspension was transferred to a quartz cuvette for simultaneous determination of hydrodynamic diameter and zeta potential by SZ-100V Fully Automated Laser Particle Size Analyzer (HORIBA, Ltd., Kyoto, Japan).
The sample suspension was deposited on a copper TEM grid and air-dried to ensure complete ethanol evaporation. Bright-field TEM imaging was performed using a FEI G2-200KV Field Emission Transmission Electron Microscope (Thermo Fisher Scientific, Waltham, MA, USA).
The sample was vacuum-dried with potassium bromide (KBr) powder, homogenized at a 1:100 mass ratio, and pressed into transparent pellets. Fourier-transform infrared (FTIR) spectra were acquired in the 400~4000 cm−1 range by IRAffinity-1 Fourier Transform Infrared Spectrophotometer (Shimadzu Corporation, Kyoto, Japan).
3.3.2. Hydrophilicity Characterization
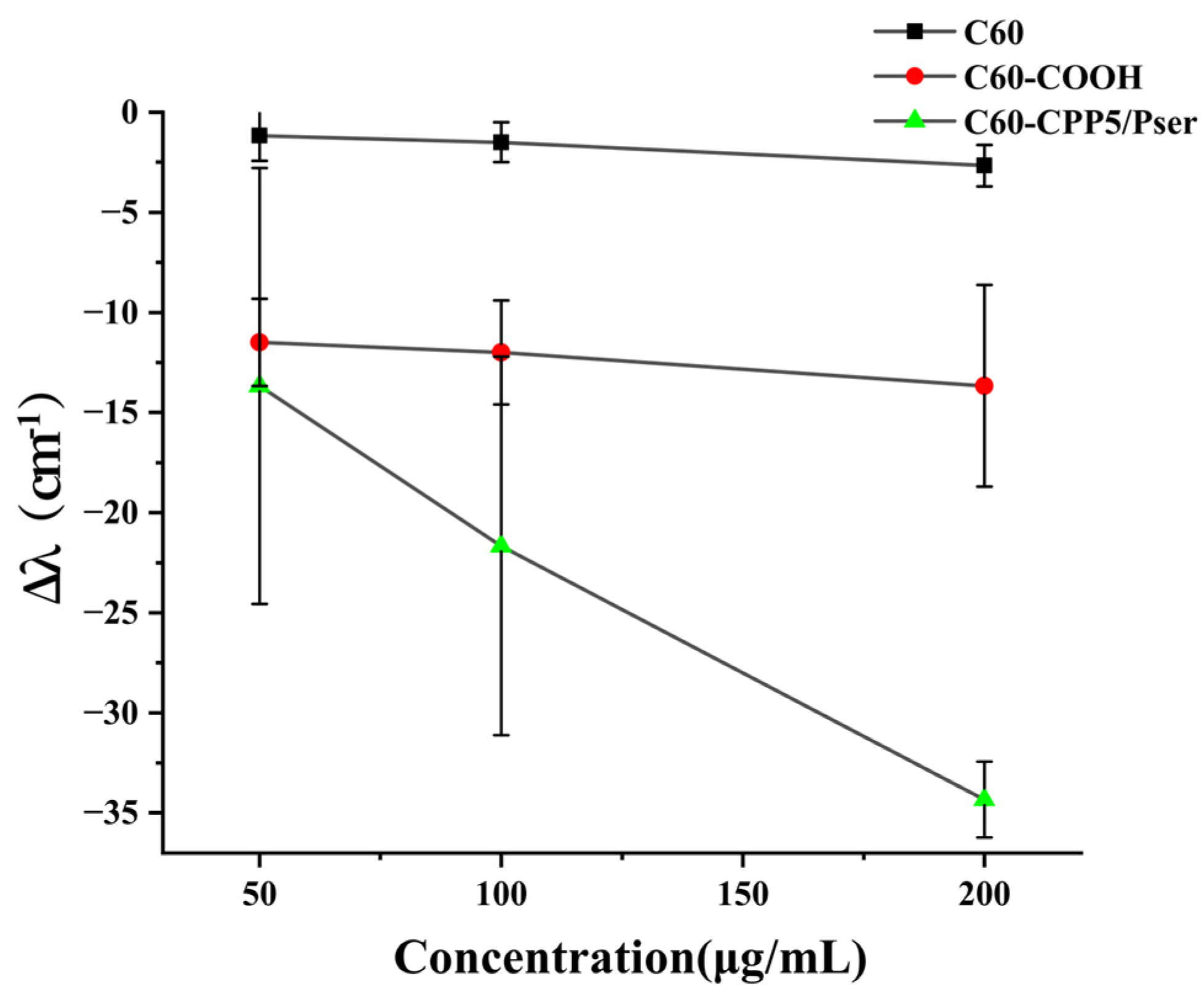
The hydrophilicity of nanocarrier surfaces is correlated with oral absorption efficacy [
31]. To evaluate the micro-environmental hydrophilicity of nanocarriers, a co-incubation assay with Coomassie Brilliant Blue G-250 (CBB G-250) dye was performed. Briefly, C60, C60-COOH, and C60-CPP5/Pser were weighed and dispersed in ultrapure water to prepare suspensions at concentrations of 50 μg·mL
−1, 100 μg·mL
−1, and 200 μg·mL
−1, respectively. These suspensions were mixed with an equal volume of 0.1 mM CBB G-250 solution and incubated for 30 min. The maximum absorption wavelength within the 400~800 nm range was measured using UQS-2110007 Double-Beam UV-Vis Spectrophotometer (Mapada Instruments Co., Ltd., Shanghai, China). The shift distance of the maximum absorption wavelength (Δλ) was calculated by comparing the results with a control group containing free CBB solution.
3.3.3. Antioxidant Characterization
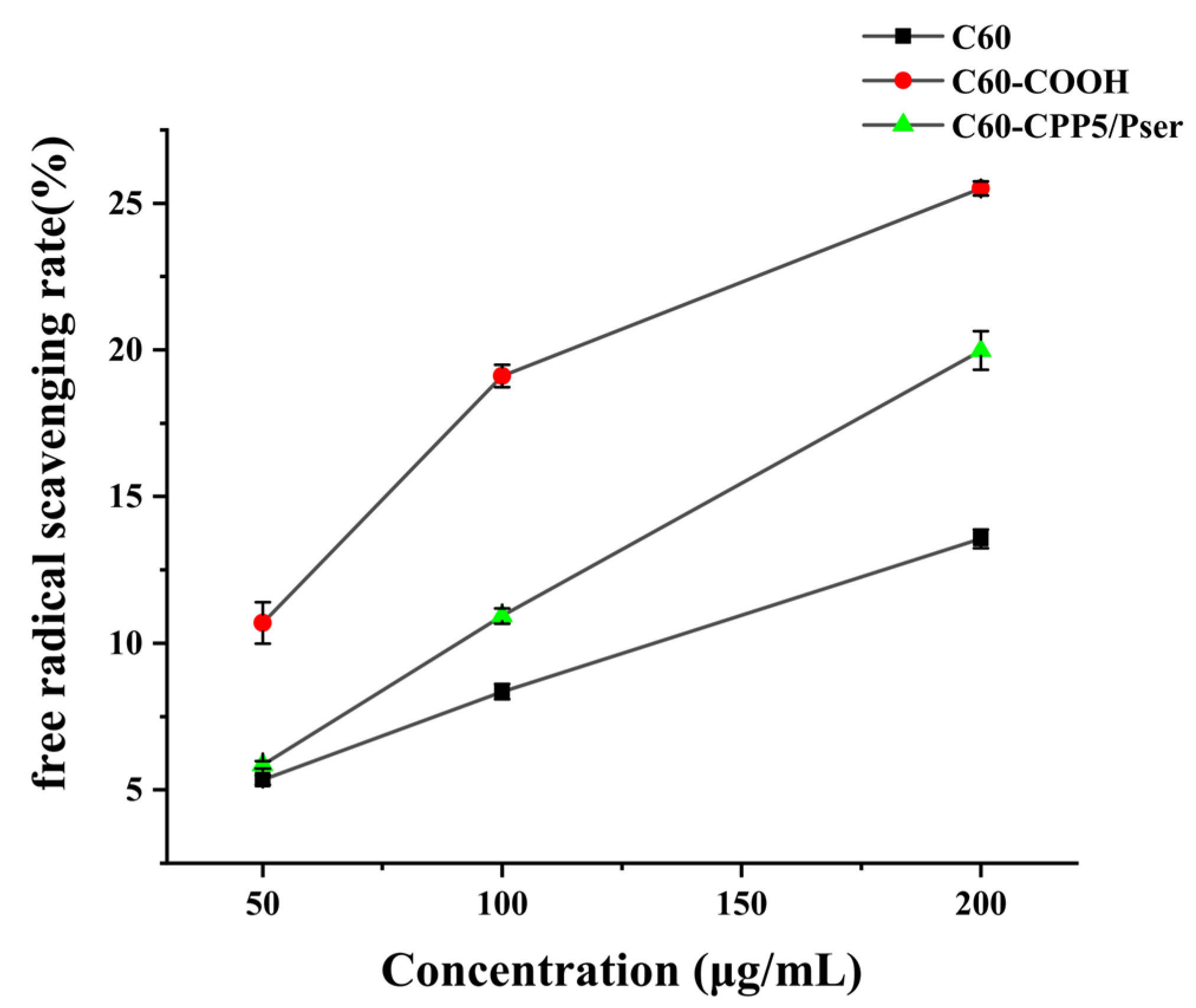
The antioxidant capacity can be evaluated through electron transfer mechanisms where antioxidants donate an electron to pair with the lone electron of the DPPH radical (DPPH·), inducing a chromatic shift from purple to yellow at 517 nm. The scavenging efficacy correlates inversely with absorbance intensity, as stronger radical-neutralizing capacity results in greater absorbance reduction [
32]. Given C60’s intrinsic electron-accepting properties and well-documented antioxidant activity through high reactivity toward free radicals [
33], we assessed the antioxidant performance of C60-CPP5/Pser to determine how structural modifications influence fullerene’s radical-scavenging functionality.
A precise amount of C60, C60-COOH, and C60-CPP5/Pser was accurately individually weighed and dispersed in ultrapure water to prepare nanoparticle suspensions with concentrations of 50 μg·mL−1, 100 μg·mL−1, and 200 μg·mL−1. These suspensions were then mixed with an equal volume of 0.1 mM DPPH· ethanol solution and incubated for 30 min. UV absorption at 517 nm was measured to evaluate the antioxidant activity.
3.4. Drug Loading and Releasing
3.4.1. Drug Loading
Analytical measurements were conducted using an Ultimate 3000 high-performance liquid chromatography (HPLC) system (Thermo Fisher Scientific, Waltham, MA, USA). A HPLC method for quantifying CUR was established under optimized chromatographic conditions: a Hypersil GOLDTM C18 column (200 mm × 4.6 mm, 5 μm) was employed with a mobile phase comprising acetonitrile and 0.1% phosphoric acid aqueous solution (48:52, v/v). The separation protocol operated at a flow rate of 1 mL·min−1, column temperature of 25 °C, and detection wavelength of 430 nm. Sample injections (10 μL) were performed using an autosampler, ensuring retention time reproducibility across triplicate runs.
Dried C60-CPP5/Pser was weighed and dispersed in deionized water and absolute ethanol via ultrasonication. Curcumin (CUR) was separately dissolved in ethanol, with the curcumin-to-carrier (mole-to-mole ratio) feeding ratio of 1:15, the ultrasonic time of 120 min, and the ethanol-to-water solvent ratio of 10:5. The CUR solution was then combined with the C60-CPP5/Pser suspension, followed by further ultrasonication. The mixture was subjected to rotary evaporation to form a thin film, which was subsequently reconstituted with 20 mL of deionized water under ultrasonication. The resulting suspension was centrifuged, and the supernatant was collected and lyophilized to obtain C60-CPP5/Pser@CUR. The drug loading methodology was optimized through orthogonal experimental design, with the experimental results analyzed with IBM SPSS Statistics 25.
The drug loading (DL) and entrapment efficiency (EE) were calculated according to Equations (1) and (2) based on the drug content determined using the established CUR method.
3.4.2. In Vitro Drug Release
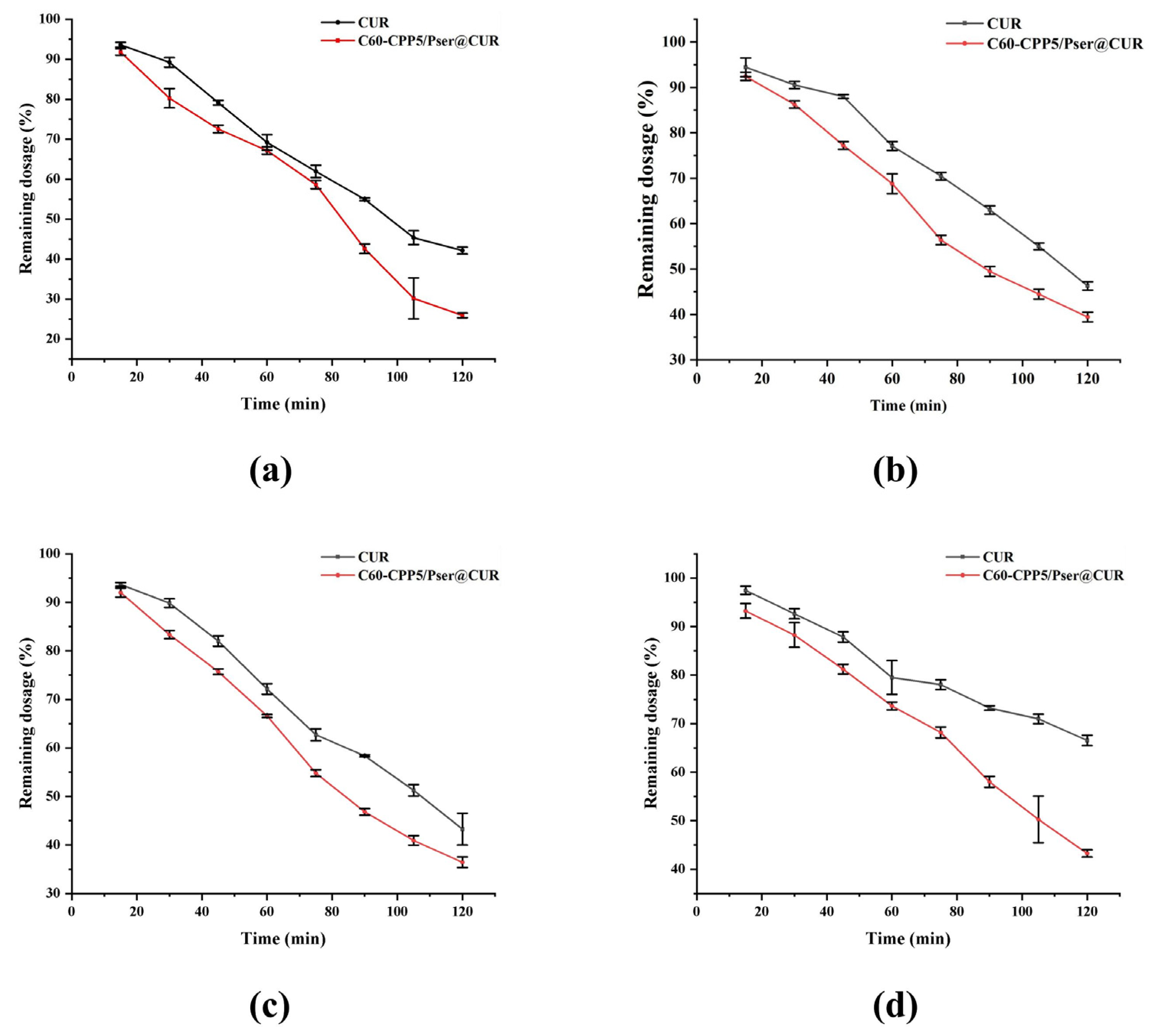
CUR and C60-CPP5/Pser@CUR were weighed, respectively, and dispersed in PBS buffer solution, then packed into dialysis bags (MD38.1 kDa). The release media were designed to simulate physiological conditions of the human gastrointestinal tract: pH 2.0 (simulated gastric fluid), pH 6.8 (simulated intestinal fluid), and pH 7.4 (to mimic the alkaline colonic terminus). Release experiments were conducted in PBS buffers (pH 2.0, 6.8, and 7.4) containing 1% Tween-80 and 10% ethanol, with continuous agitation at 120 rpm and 37 °C. Aliquots (0.5 mL) were collected at predetermined intervals (0.5, 1, 2, 4, 6, 8, 12, and 24 h), with the release medium replenished immediately after each sampling to maintain sink conditions. The experiment was repeated three times.
The drug release was quantified using a validated HPLC method. Cumulative release percentages were plotted against time, and release kinetics were analyzed using four models: zero-order kinetics, first-order kinetics, Higuchi equation, and Weibull distribution function. Key parameters, including T50 (time required for 50% drug dissolution) and Td (time required for 63.2% drug dissolution), were calculated to evaluate pH-dependent release profiles.
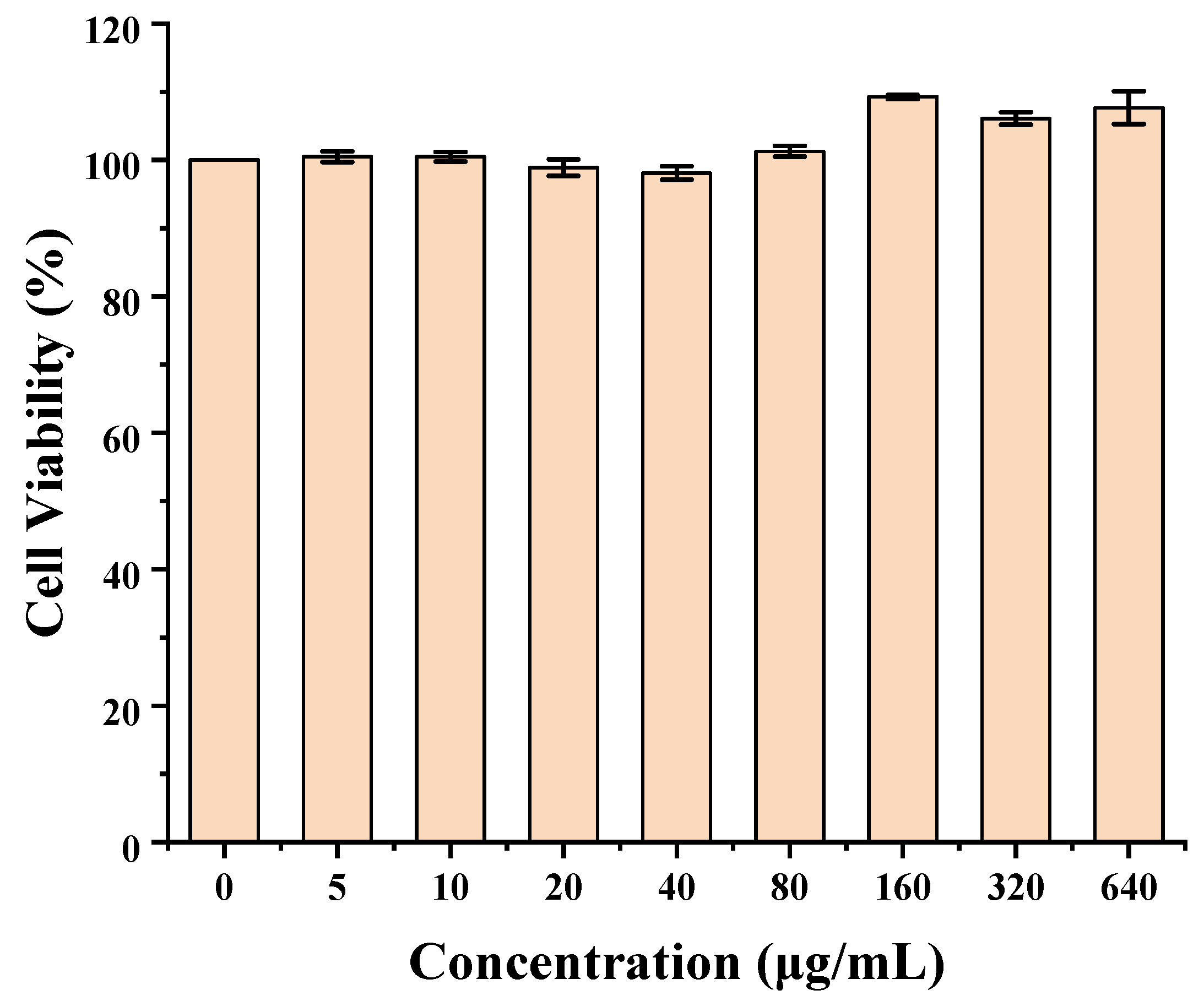
3.5. Cytotoxicity
The cytotoxicity of C60-CPP5/Pser was evaluated by the Cell Counting Kit-8 (CCK-8) assay on Caco-2 cells. Briefly, Caco-2 cells were seeded in 96-well plates at a density of 1 × 10
5 cells·mL
−1 (100 μL per well) and cultured for 24 h. After removing the medium, various concentrations of C60-CPP5/Pser were added to the wells (six replicates per concentration) and incubated for an additional 24 h. Subsequently, the supernatant was aspirated, and the cell monolayer was gently washed with D-PBS (pH 7.4), then supplemented with 10 μL of CCK-8 reagent and incubated for 3.5 h. The optical density (OD) at 450 nm was measured by a fully automated microplate reader. Cell viability was calculated with Formula (3):
(
Aexperimental group represents OD values of cells treated with varying concentrations of the formulation,
Ablank group represents OD values of cell-free wells,
Anegative control group represents OD values of wells containing culture medium alone).
3.6. In Vitro Absorption
3.6.1. Cellular Uptake
A method for HPLC quantification of CUR in cellular uptake studies was established, which involved validation of specificity, precision, stability, and recovery rates, along with the preparation of a calibration curve (see
Supplementary Materials). Cellular samples containing CUR demonstrated excellent linearity within the concentration range of 1~100 μg·mL
−1, with a regression equation of
y = 0.972
x + 0.022 (R² = 0.9995).
Caco-2 cells were seeded into the donor chamber of Transwell inserts at a density of 1 × 10
5 cell·mL
−1 and cultured in complete medium. Transepithelial electrical resistance (TEER) was measured on the 0th, 3rd, 5th, 7th, 10th, and 15th days to monitor monolayer integrity. A TEER value exceeding 700 Ω·cm² was established as the threshold for successful formation of the human intestinal epithelial barrier model. Following model validation, the cell surface was equilibrated with PBS for 30 min and rinsed to remove debris. CUR solutions (50, 100, and 200 μg·mL
−1) were prepared in PBS containing 0.1% DMSO. Subsequently, 500 μL of each CUR solution was added to the apical compartment and incubated for 10, 20, or 60 min. Cellular uptake was terminated by rapid washing with ice-cold PBS to remove surface-bound drug and nonviable cells. Cells were harvested using a cell scraper, lysed, and divided into two aliquots: one for quantifying CUR content via HPLC and the other for determining total cellular protein concentration using a BCA protein assay kit. Cellular uptake was calculated according to Equation (4). Single-factor analysis of uptake time and drug concentration was performed to optimize parameters for subsequent experiments.
(
CCUR: Intracellular CUR Concentration,
Cprotein: Intracellular Protein Concentration)
PBS solution containing 200 μg·mL−1 CUR (with 0.1% DMSO) was prepared. The established Caco-2 cell monolayer model imitating human intestinal epithelium was equilibrated in pH 7.4 PBS at 37 °C within a CO2 incubator for 30 min. After removal of PBS, the cell surface was rinsed three times. Subsequently, 500 μL of CUR solution or nanocarrier dispersion was added to the AP side of the Transwell plate, followed by 20 min of incubation. The uptake process was terminated by ice-cold PBS, and residual solutions were removed by washing. Cells were then scraped into centrifuge tubes, sonicated, and quantified for uptake measurements.
3.6.2. Mucus Penetration Assay
HPLC methodology was developed and validated for the determination of CUR in intestinal mucus. The method demonstrated good linearity within the concentration range of 1~20 μg·mL−1, with a calibration curve of y = 0.5981x − 0.0933 (R² = 0.9998).
For mucus preparation, intestinal segments from the distal duodenum to the proximal cecum were excised from rats. Luminal contents were flushed with ice-cold PBS (pH 7.4), and the mucosal layer was mechanically scraped. The harvested mucus was homogenized, centrifuged, and subjected to ethanol precipitation for impurity removal. The resulting pellet was dried and reconstituted in 50 mM sodium carbonate buffer (pH 7.4). After centrifugation at 5000 r·min−1 for 15 min at 4 °C, supernatants were collected and pooled for subsequent analysis.
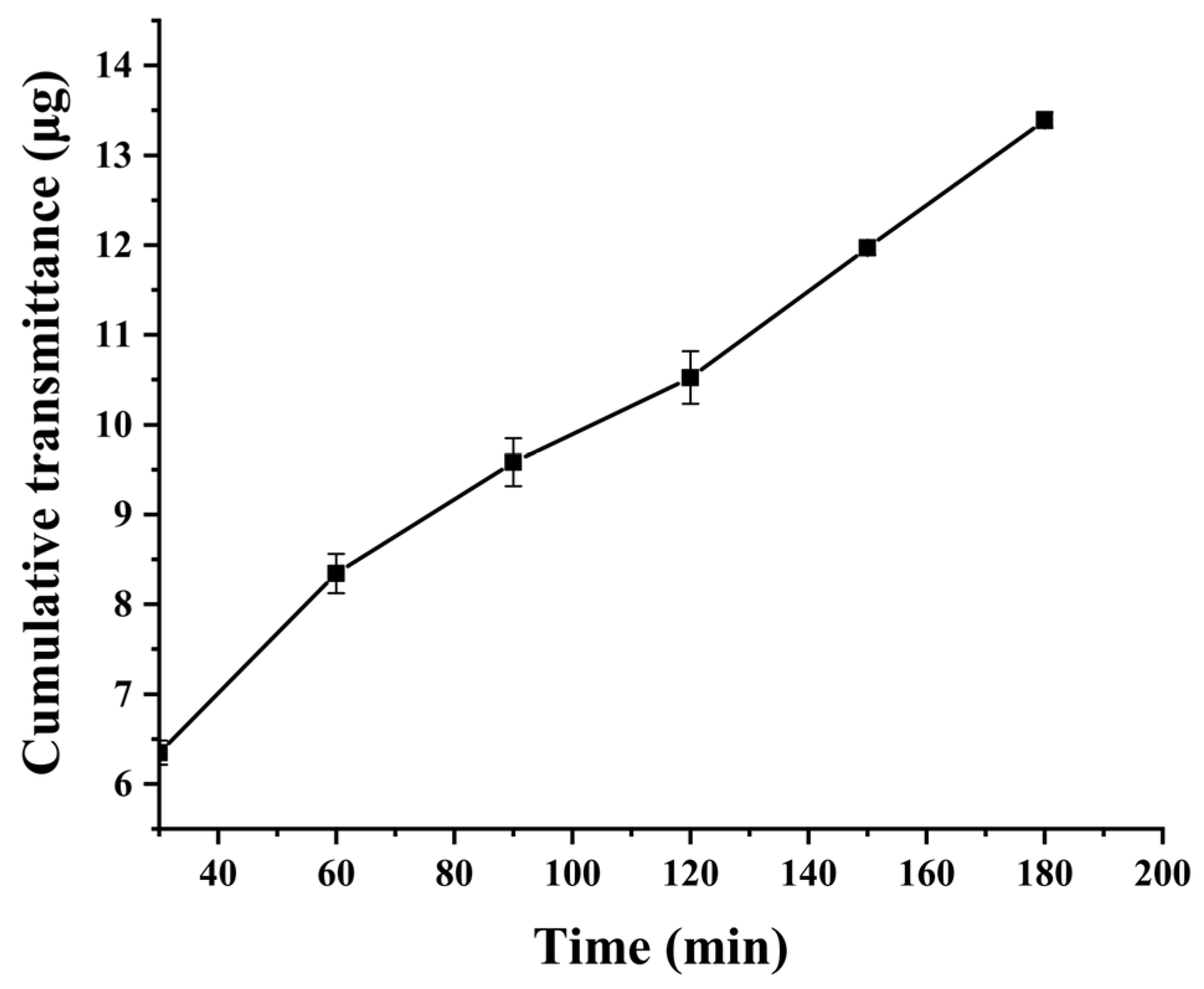
The mucus was uniformly coated into the donor compartment of the Transwell chamber, while PBS (pH 7.4) was added to the receiver compartment. After equilibration at 37 °C for 15 min, 100 μL of 20 mg·L
−1 CUR suspension and C60-CPP5/Pser@CUR dispersion (containing equivalent drug content) were separately introduced into the donor compartment. The Transwell system was then incubated at 37 °C at 100 rpm for 3 h. At predetermined time intervals, samples were collected and centrifuged, and the CUR concentration was quantified by HPLC. The cumulative mucus permeated amount (
Q, μg·cm
−2) and apparent permeability coefficient (
Papp, cm·s
−1) were calculated using Equations (5) and (6):
(
represents the drug concentration in the receiver compartment at time t;
Ct(n−1) represents the drug concentration in the receiver compartment at time t − 1)
(dQ/dT is the cumulative permeation amount per unit time; A is the effective area of the chamber membrane;
C0 is the initial concentration of CUR in the donor compartment)
3.7. In Vivo Absorption Studies
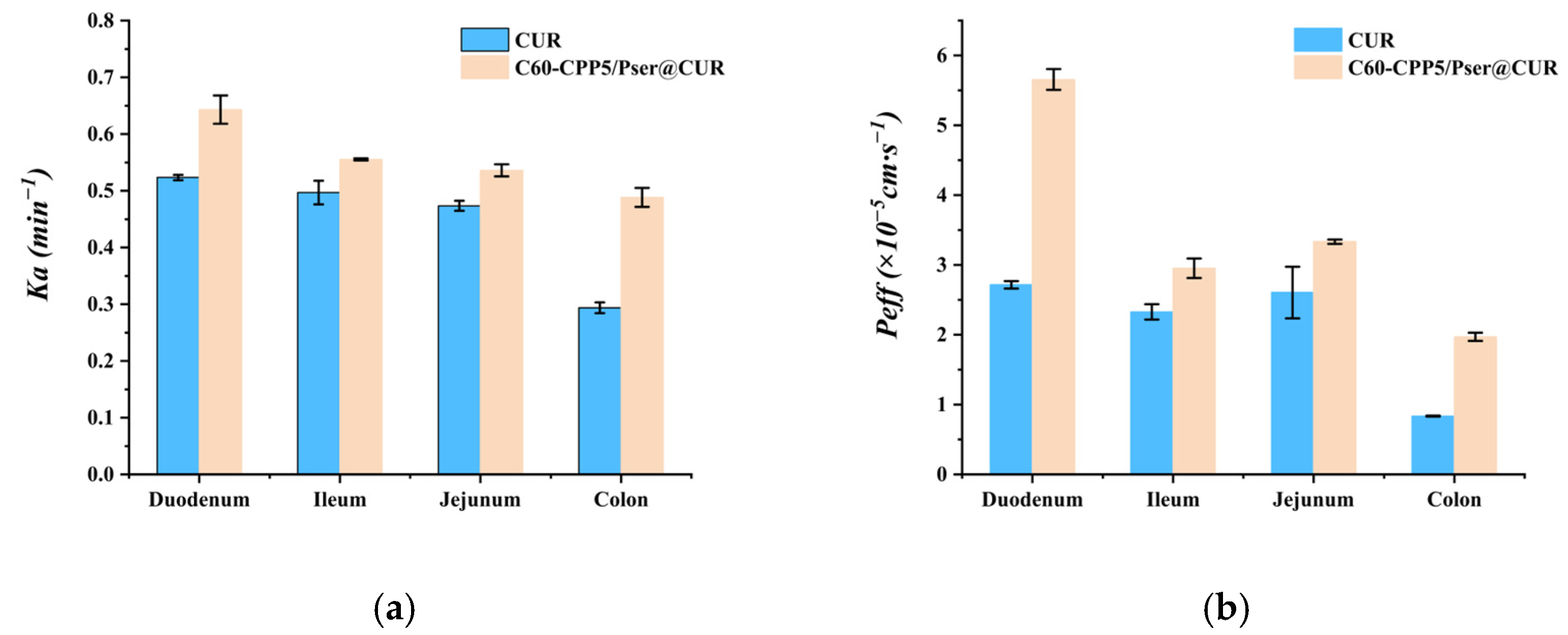
3.7.1. Single-Pass Intestinal Perfusion
The experimental animals were purchased from SPF Biotechnology Co., Ltd. (Beijing, China, Animal Production License No. SCXK (Jing) 2024-0001) and housed at the Animal Experiment Center of Inner Mongolia Medical University. All animal procedures were conducted in accordance with the guidelines approved by the Ethics Committee of Inner Mongolia Medical University (approval number: YKD202402228).
The methodology for HPLC quantification of CUR in single-pass intestinal perfusion experiments was established to ensure analytical accuracy and reproducibility, then six Sprague–Dawley (SD) rats (body weight: 220 ± 20 g; equal numbers of males and females) were fasted for 24 h with free access to water prior to the experiment. The rats were anesthetized and secured in a supine position on a surgical platform maintained at 37 °C. After abdominal hair removal and disinfection with 75% ethanol, a midline laparotomy was performed to expose the target intestinal segment. A 10 cm intestinal segment was isolated, and incisions were made at both ends. Silicone tubes were inserted into the incisions and ligated. The intestinal lumen was flushed with 0.9% NaCl solution for 5 min to remove luminal contents, followed by equilibration with blank Krebs–Henseleit solution for 15 min (flow rate, 1.5 mL·min−1). Subsequently, the test sample was perfused through the intestinal loop. Then, 1 mL intestinal circulation samples were collected at predetermined time points, with an equivalent volume of Krebs–Henseleit solution replenished after each sampling. The collected samples were centrifuged, and the supernatant was mixed with methanol to precipitate proteins. After a second centrifugation, the resulting supernatant was analyzed by HPLC.
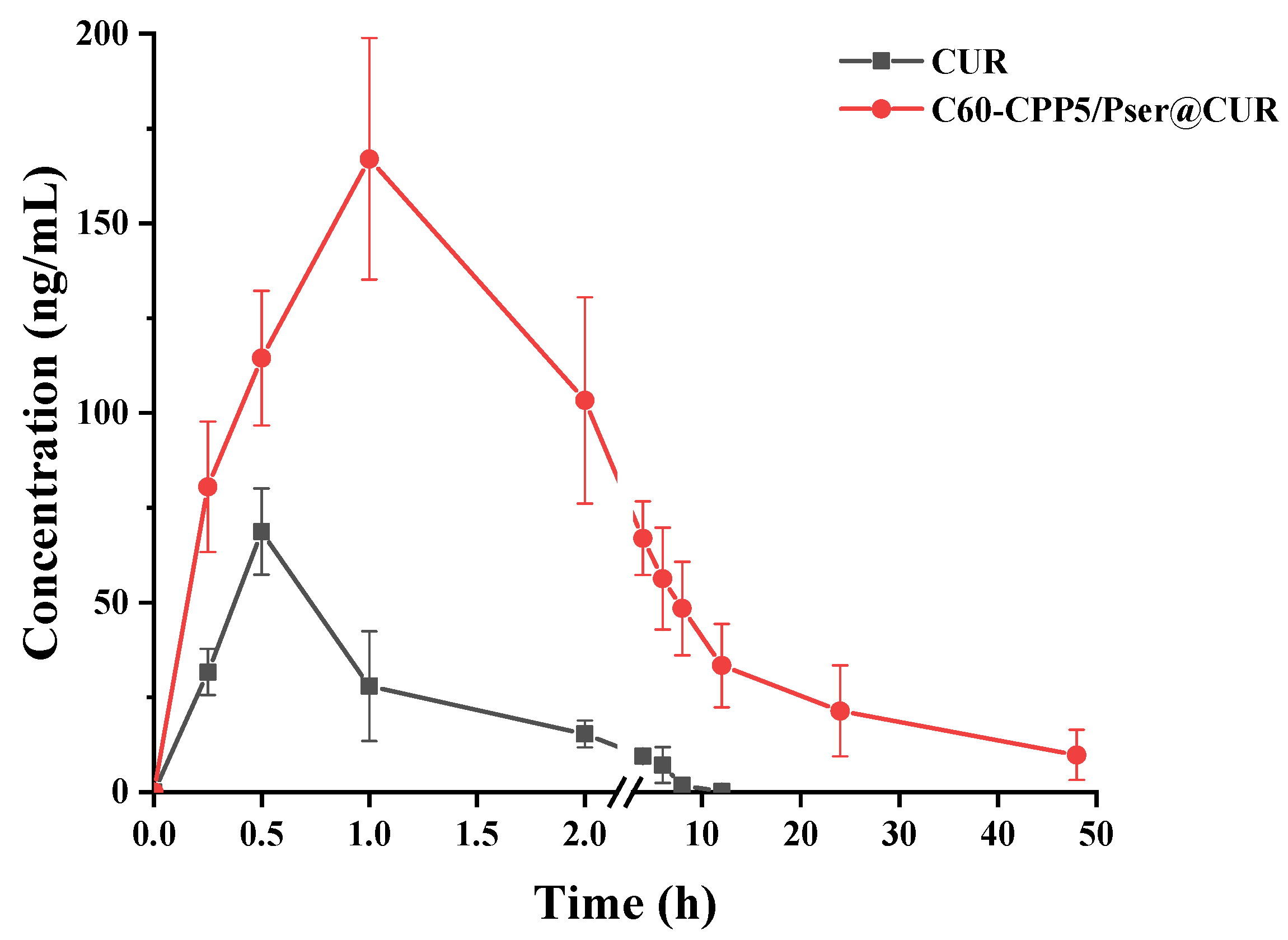
3.7.2. Pharmacokinetics Study in Rats
A HPLC-MS method for determining CUR in rat plasma was established with Resveratrol as the internal standard. The analysis was performed on a Q Exactive™ hybrid quadrupole-Orbitrap™ mass spectrometer (Thermo Fisher Scientific, USA) with the following parameters: Chromatographic separation was achieved by Agilent ZORBAX SB-C18 column (150 mm × 4.6 mm, 5 μm) with a mobile phase of acetonitrile and 0.1% formic acid aqueous solution (48:52, v/v) at a flow rate of 1 mL·min−1 while maintaining the column temperature at 30 °C. The mass spectrometer operated in positive ion mode with an electrospray ionization (ESI) source, employing multiple reaction monitoring (MRM) for detection. Key ionization parameters included a capillary voltage of 3.0 kV and nitrogen as the collision gas.
Twelve SD rats (220 ± 20 g, equal numbers of males and females) were fasted for 24 h with free access to water and randomly allocated into two groups (n = 6 per group). Both groups received CUR at a dose of 50 mg·kg−1 via oral gavage, administered as either a CUR suspension or C60-CPP5/Pser@CUR dispersion. Blood samples (~0.5 mL) were collected from the jugular vein into anticoagulant tubes at 0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h after administration. After centrifugation to collect the supernatant, samples were spiked with 20 μL of a 100 μg·mL−1 internal standard solution, extracted twice with ethyl acetate for protein precipitation, and the combined organic phases were evaporated using a refrigerated vacuum concentrator. The residue was reconstituted in 100 μL methanol through vortex mixing (4 min) and centrifugation prior to HPLC-MS analysis to quantify the CUR concentration.
3.8. Statistical Analysis
The experimental data were expressed as mean ± sd. Comparisons were performed with one-way analysis of variance (ANOVA) with SPSS 25.0 statistical software (IBM Corp., Armonk, NY, USA), and statistical significance was defined as p < 0.05.
4. Conclusions
Inspired by the viral structural features of viruses to overcome the dual barriers of intestinal mucus and epithelial membranes for oral drug delivery, we developed a charge-reversal nanocarrier (C60-CPP5/Pser). This system takes advantage of its unique surface properties: the zwitterionic coating helps to rapid mucus penetration, while subsequent hydrolysis by mucosal alkaline phosphatase triggers charge reversal to a cationic state. The exposed CPP5 further enhances transmembrane transport via caveolae-mediated endocytosis. These mechanisms were validated by in vitro and in vivo experiments with CUR as a model drug, demonstrating the nanocarrier’s potential for small-molecule delivery. Additionally, the intrinsic antioxidant properties of the fullerene core, which synergistically contribute to therapeutic efficacy, highlight its expanding applications in biomedicine. The therapeutic strategy capitalizes on the inherent antioxidant properties of C60 coupled with enhanced membrane penetration afforded by the delivery system, aiming to develop an oral formulation for intestinal disorders. Our study has yielded encouraging results, demonstrating efficient gastrointestinal delivery of curcumin. However, as illustrated by the data, the carrier exhibits limited regional absorption specificity along the intestinal tract. Future investigations will focus on achieving site-specific release (e.g., colon or inflammatory loci) through targeted modifications of the nanocarrier or fabrication of enteric-coated microcapsules.
While nanocarriers have achieved notable progress in drug delivery, significant gaps remain in terms of successful clinical translation. Comprehensive investigations are required to elucidate the structure/physiological activity relationships of nanocarrier systems, address discrepancies between animal models and the human gastrointestinal tract environment, and evaluate potential side effects arising from their interactions with biological systems. Additionally, the stringent precision demanded in nanopharmaceutical manufacturing poses challenges in maintaining consistency across large-scale production batches. Overcoming these barriers will be critical for realizing the clinical potential of nanocarriers in therapeutic applications. Future research can combine the QbD concept with interdisciplinary technologies (such as AI-driven process optimization) and refer to the successful experience of liposomes/micelles to accelerate their clinical transformation.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}