Application of VUV/Sulfite Defluorination System for the Simple Detection of Perfluoroalkyl Substances


Abstract

1. Introduction
2. Results and Discussion
2.1. Defluorination of PFAS by VUV/Sulfite
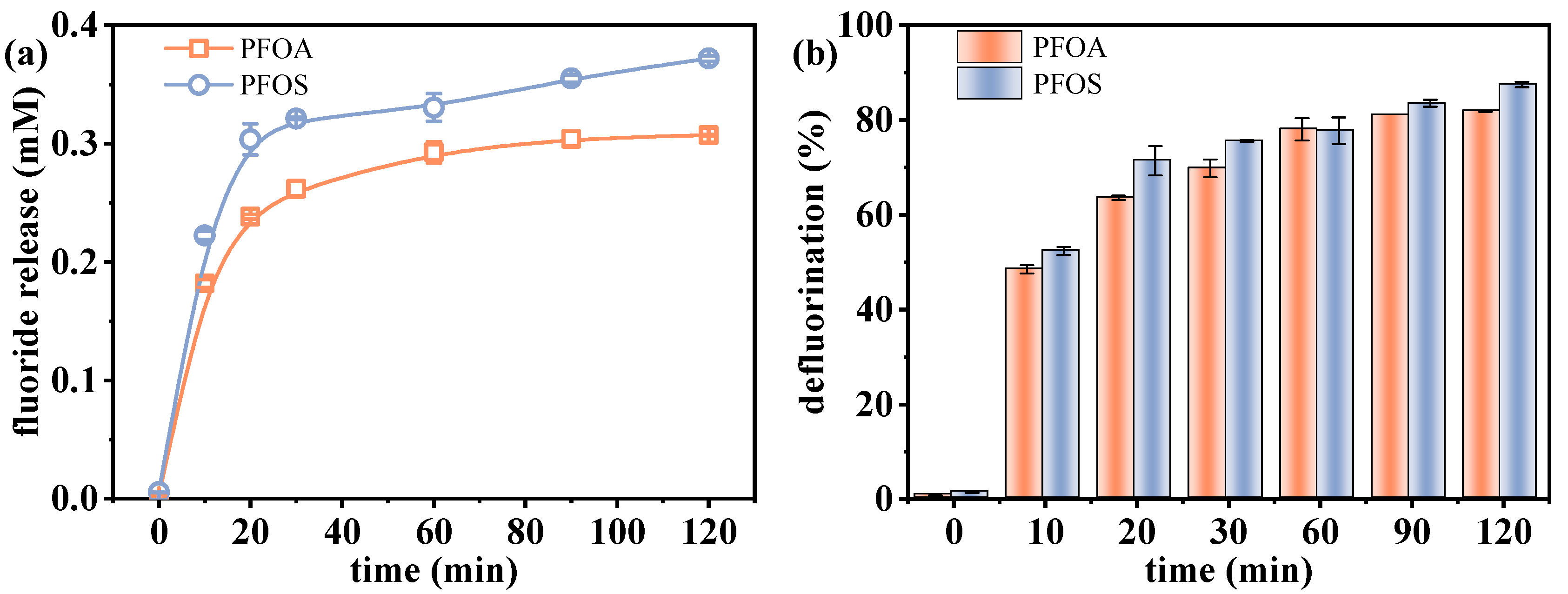
2.1.1. Effect of Irradiation Time on the Defluorination of PFAS
2.1.2. Effect of Initial pH on the Defluorination
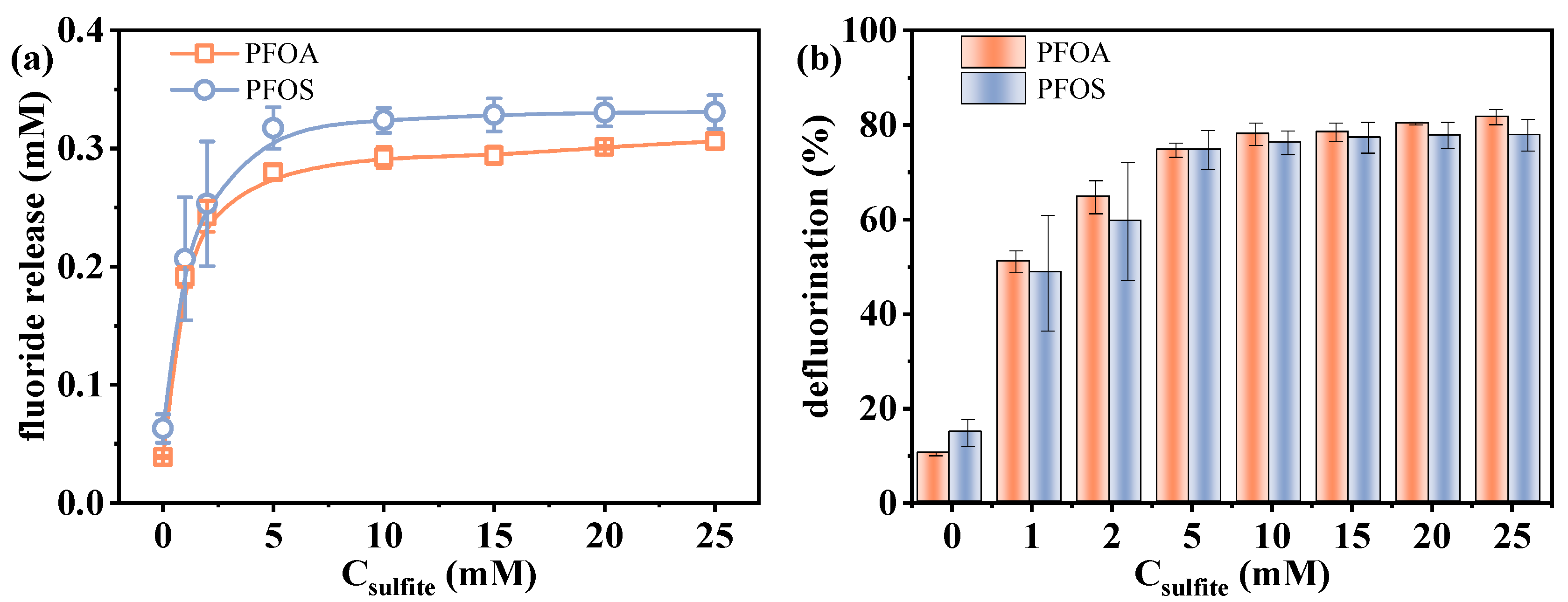
2.1.3. Effect of Sulfite Concentration on the Defluorination
2.2. Effect of Ions on the Defluorination of PFAS
2.2.1. Effect of Anions
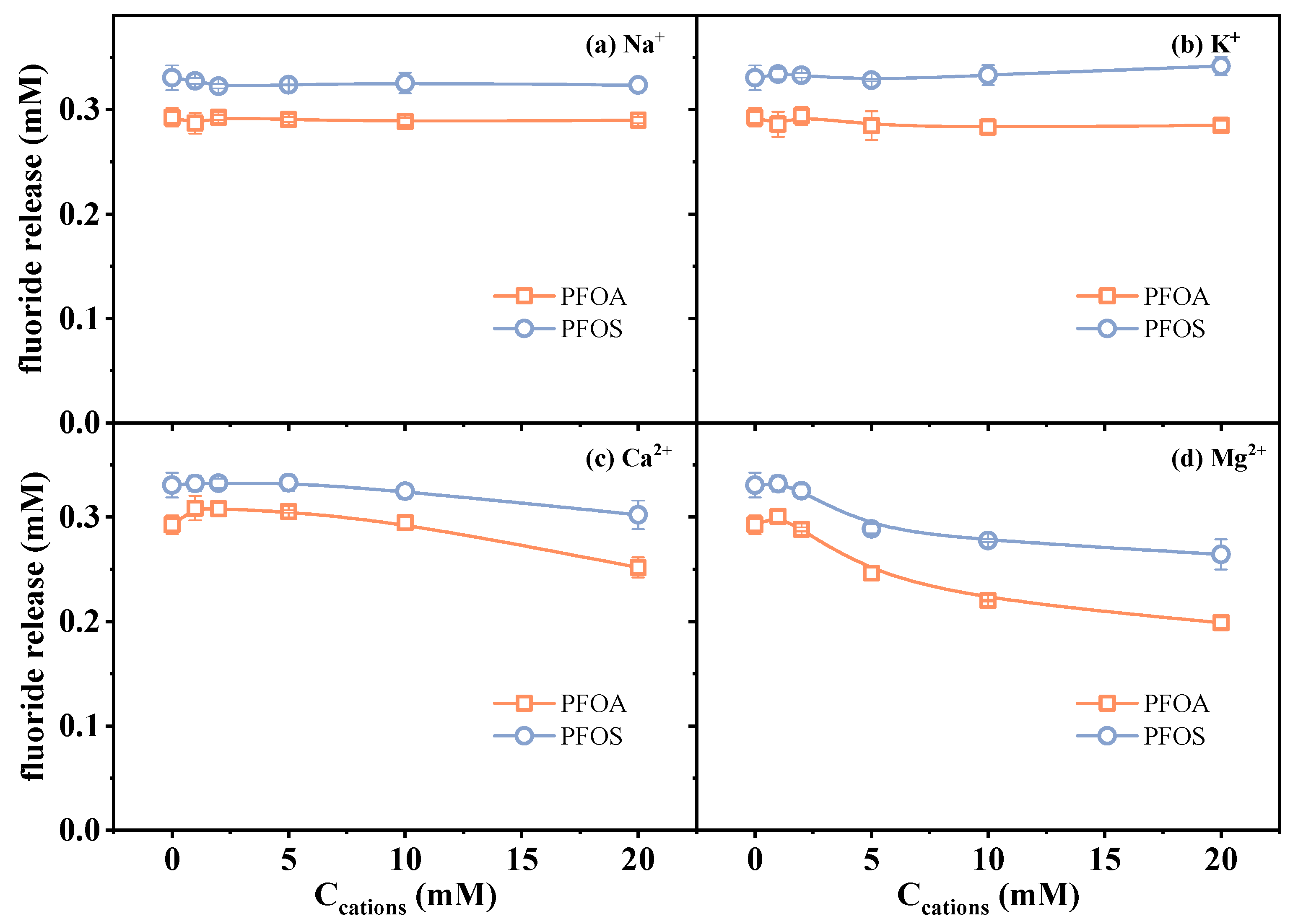
2.2.2. Effect of Cations
2.3. Application of VUV/Sulfite System in PFAS Quantification Analysis
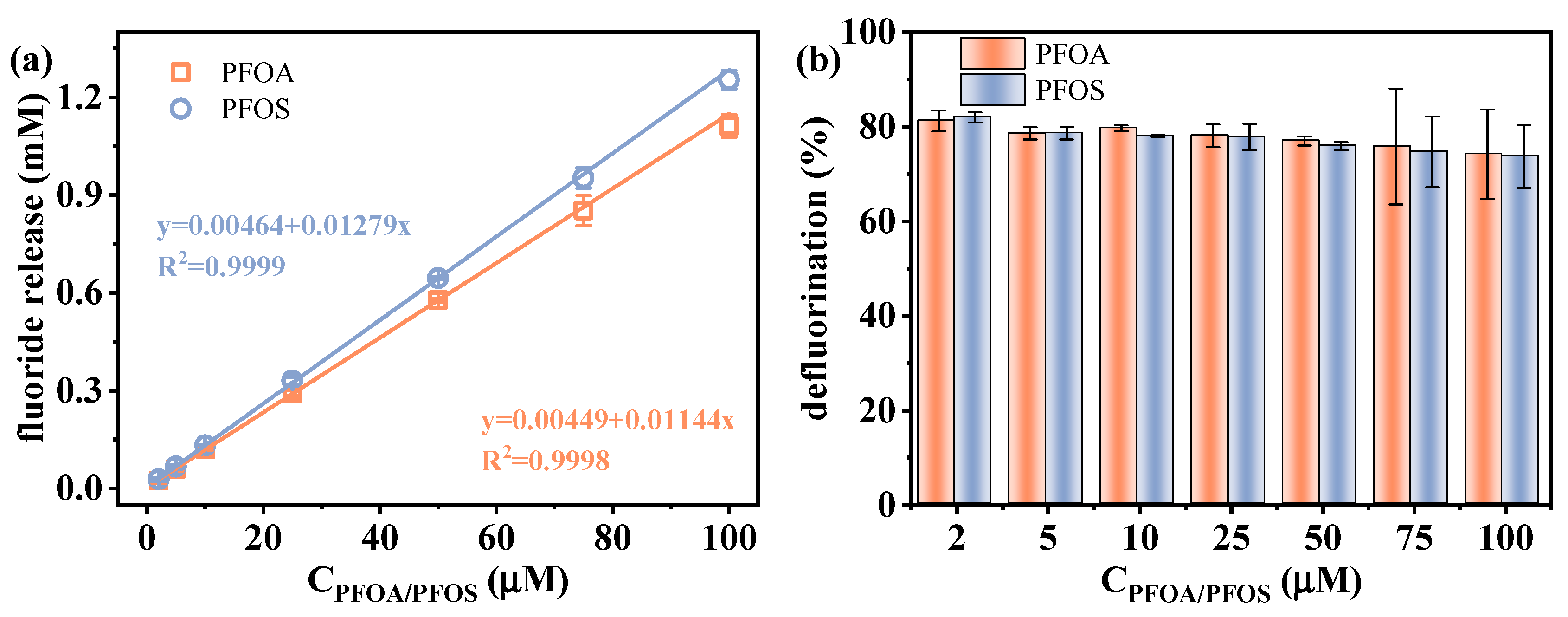
2.3.1. Establishment of Calibration Curves
2.3.2. Application in Adsorption Experiment
3. Materials and Methods
3.1. Chemicals and Sediments
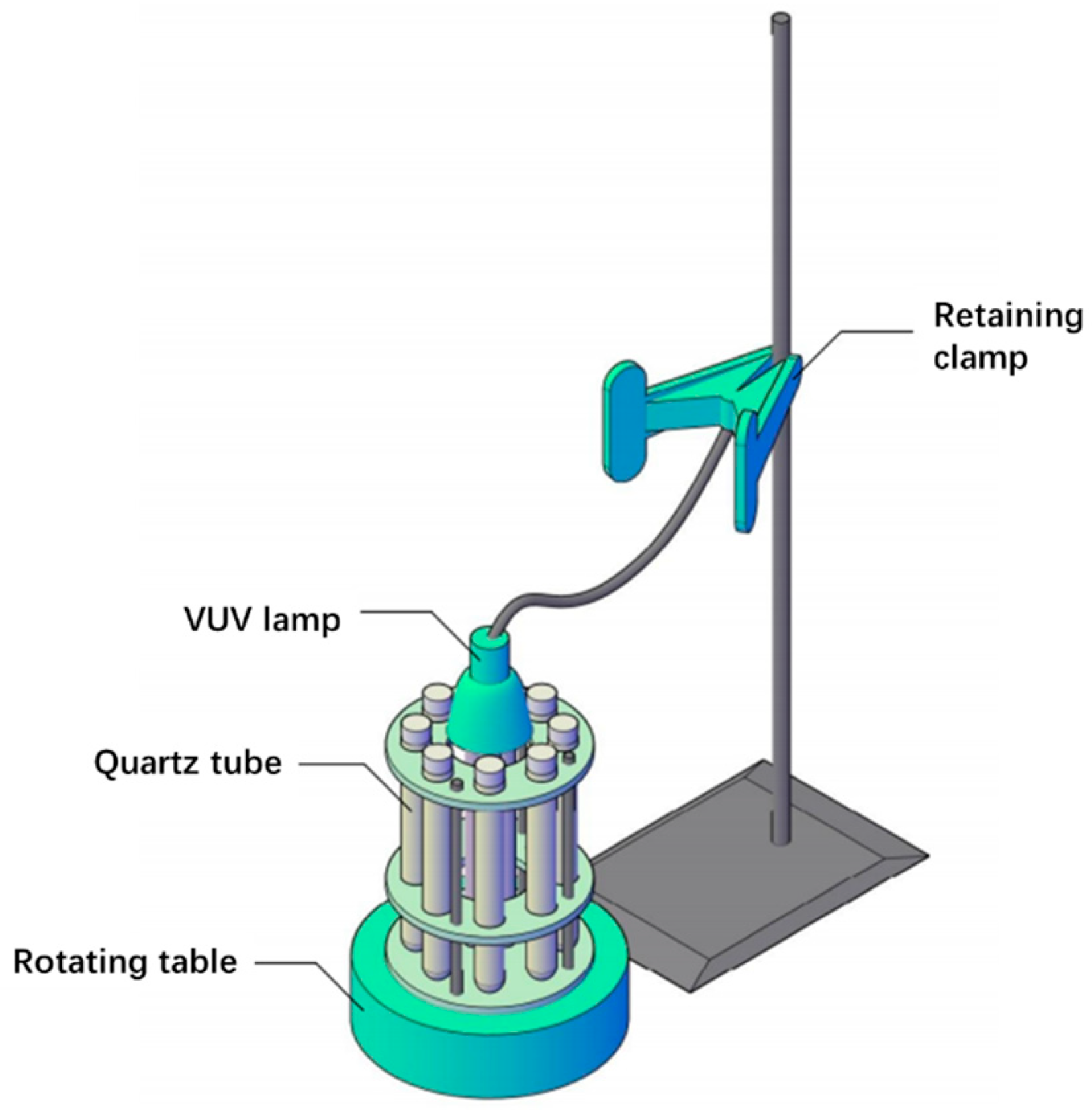
3.2. Photochemical Reaction
3.3. Simulated Adsorption Experiments
3.4. Analytical Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cousins, I.T.; Johansson, J.H.; Salter, M.E.; Sha, B.; Scheringer, M. Outside the safe operating space of a new planetary boundary for per- and polyfluoroalkyl substances (PFAS). Environ. Sci. Technol. 2022, 56, 11172–11179. [Google Scholar] [CrossRef]
- Milinovic, J.; Lacorte, S.; Vidal, M.; Rigol, A. Sorption behaviour of perfluoroalkyl substances in soils. Sci. Total Environ. 2015, 511, 63–71. [Google Scholar] [CrossRef]
- Li, F.; Duan, J.; Tian, S.; Ji, H.; Zhu, Y.; Wei, Z.; Zhao, D. Short-chain per- and polyfluoroalkyl substances in aquatic systems: Occurrence, impacts and treatment. Chem. Eng. J. 2020, 380, 122506. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, J.; Tang, S.; Qiu, J.; Luo, Y.; Yang, C.; Lai, X.; Wang, Q.; Cao, H. Per- and polyfluoroalkyl substances (PFAS) in consumer products: An overview of the occurrence, migration, and exposure assessment. Molecules 2025, 30, 994. [Google Scholar] [CrossRef]
- Zhao, L.; Bian, J.; Zhang, Y.; Zhu, L.; Liu, Z. Comparison of the sorption behaviors and mechanisms of perfluorosulfonates and perfluorocarboxylic acids on three kinds of clay minerals. Chemosphere 2014, 114, 51–58. [Google Scholar] [CrossRef]
- Sharma, B.M.; Bharat, G.K.; Tayal, S.; Larssen, T.; Bečanová, J.; Karásková, P.; Whitehead, P.G.; Futter, M.N.; Butterfield, D.; Nizzetto, L. Perfluoroalkyl substances (PFAS) in river and ground/drinking water of the Ganges River basin: Emissions and implications for human exposure. Environ. Pollut. 2016, 208, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Kirchgeorg, T.; Dreyer, A.; Gabrielli, P.; Gabrieli, J.; Thompson, L.G.; Barbante, C.; Ebinghaus, R. Seasonal accumulation of persistent organic pollutants on a high altitude glacier in the Eastern Alps. Environ. Pollut. 2016, 218, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kannan, K. Perfluorinated acids in air, rain, snow, surface runoff, and lakes: Relative importance of pathways to contamination of urban lakes. Environ. Sci. Technol. 2007, 41, 8328–8334. [Google Scholar] [CrossRef]
- Tang, A.; Zhang, X.; Li, R.; Tu, W.; Guo, H.; Zhang, Y.; Li, Z.; Liu, Y.; Mai, B. Spatiotemporal distribution, partitioning behavior and flux of per- and polyfluoroalkyl substances in surface water and sediment from Poyang Lake, China. Chemosphere 2022, 295, 133855. [Google Scholar] [CrossRef]
- Liu, T.; Qian, X.; Wang, S.; Wang, H.; Wei, S.; Chen, H. Occurrence and transport of perfluoroalkyl acids (PFAAs) in a Yangtze River water diversion project during water diversion and flooding. Water Res. 2021, 205, 117662. [Google Scholar] [CrossRef]
- Rehman, A.U.; Crimi, M.; Andreescu, S. Current and emerging analytical techniques for the determination of PFAS in environmental samples. Trends Environ. Anal. Chem. 2023, 37, e00198. [Google Scholar] [CrossRef]
- Aborode, A.T.; Adesola, R.O.; Idris, I.; Sakariyau Adio, W.; Olapade, S.; Oluwafisayo, G.; Onifade, I.A.; Fakorede, S.; Bakare-Abidola, T.; Olaoye, J.; et al. Challenges associated with PFAS detection method in Africa. Environ. Health Insights 2025, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, Y.; Yang, R.; Jiang, Y.; Meng, M.; Liu, Z.; Ni, L.; Wu, W.; Liu, H. Adsorption for perfluorooctanoic acid with graphitic-phase carbon nitride and its HPLC fluorescence determination. Can. J. Chem. Eng. 2020, 98, 394–403. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Pan, H.; Wei, Y.; Yang, Y.; Xu, N.; Chen, B.; Mohseni, M.; Esfahani, E.B. Total organic fluorine (TOF) analysis by completely converting TOF into fluoride with vacuum ultraviolet. J. Hazard. Mater. 2022, 429, 128389. [Google Scholar] [CrossRef]
- Whitehead, H.D.; Venier, M.; Wu, Y.; Eastman, E.; Urbanik, S.; Diamond, M.L.; Shalin, A.; Schwartz-Narbonne, H.; Bruton, T.A.; Blum, A.; et al. Fluorinated compounds in North American cosmetics. Environ. Sci. Technol. Lett. 2021, 8, 538–544. [Google Scholar] [CrossRef]
- Gauthier, J.R.; Mabury, S.A. Experimentally determined aqueous diffusion coefficients of PFAS using 19F NMR diffusion-ordered spectroscopy. ACS ES&T Water 2024, 4, 4615–4624. [Google Scholar] [CrossRef]
- D’Agostino, L.A.; Mabury, S.A. Identification of novel fluorinated surfactants in aqueous film forming foams and commercial surfactant concentrates. Environ. Sci. Technol. 2014, 48, 121–129. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, T.; Yu, S.; Yi, P.; Li, L. Influence of UV lamp, sulfur(IV) concentration, and pH on bromate degradation in UV/sulfite systems: Mechanisms and applications. Water Res. 2017, 111, 288–296. [Google Scholar] [CrossRef]
- Wang, X.; Liu, H.; Shan, C.; Zhang, W.; Pan, B. A novel combined process for efficient removal of Se(VI) from sulfate-rich water: Sulfite/UV/Fe(III) coagulation. Chemosphere 2018, 211, 867–874. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, C.; Li, F.; Chen, J.; Zhou, Q. Photo-reductive defluorination of perfluorooctanoic acid in water. Water Res. 2010, 44, 2939–2947. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Banayan Esfahani, E.; Mohseni, M. Fluence-based photo-reductive decomposition of PFAS using vacuum UV (VUV) irradiation: Effects of key parameters and decomposition mechanism. J. Environ. Chem. Eng. 2022, 10, 107050. [Google Scholar] [CrossRef]
- Wu, S.; Shen, L.; Lin, Y.; Yin, K.; Yang, C. Sulfite-based advanced oxidation and reduction processes for water treatment. Chem. Eng. J. 2021, 414, 128872. [Google Scholar] [CrossRef]
- Liu, S.; Chen, G.; Shi, Q.; Gan, J.; Jin, B.; Men, Y.; Liu, H. Promotive effects of chloride and sulfate on the near-complete destruction of perfluorocarboxylates (PFCAs) in brine via hydrogen-tuned 185-nm UV photolysis: Mechanisms and kinetics. Environ. Sci. Technol. 2024, 58, 10347–10356. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Liu, X.; Cui, F.; Zhao, Z. Efficient reductive and oxidative decomposition of haloacetic acids by the vacuum-ultraviolet/sulfite system. Water Res. 2022, 210, 117974. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, T.; Kim, J.; Kim, M.K.; Eom, S.; Choi, Y.; Zoh, K.D. Reductive degradation mechanism of perfluorooctanoic acid (PFOA) during vacuum ultraviolet (VUV) reactions combining with sulfite and iodide. Chemosphere 2024, 348, 140759. [Google Scholar] [CrossRef]
- Li, S.; Xu, J.; Chen, W.; Yu, Y.; Liu, Z.; Li, J.; Wu, F. Multiple transformation pathways of p-arsanilic acid to inorganic arsenic species in water during UV disinfection. J. Environ. Sci. 2016, 47, 39–48. [Google Scholar] [CrossRef]
- Luo, T.; Le Crom, S.; Luong, N.T.; Hanna, K.; Boily, J.F. Goethite-bound copper controls the fate of antibiotics in aquatic environments. ACS ES&T Water 2024, 4, 638–647. [Google Scholar] [CrossRef]
- Oliveira, C.; Lima, D.L.D.; Silva, C.P.; Calisto, V.; Otero, M.; Esteves, V.I. Photodegradation of sulfamethoxazole in environmental samples: The role of pH, organic matter and salinity. Sci. Total Environ. 2019, 648, 1403–1410. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Z.; Gao, J.; Yu, Y.; Men, Y.; Gu, C.; Liu, J. Accelerated degradation of perfluorosulfonates and perfluorocarboxylates by UV/sulfite + iodide: Reaction mechanisms and system efficiencies. Environ. Sci. Technol. 2022, 56, 3699–3709. [Google Scholar] [CrossRef]
- Lorpaiboon, W.; Ho, J. High-level quantum chemical prediction of C-F bond dissociation energies of perfluoroalkyl substances. J. Phys. Chem. A 2023, 127, 7943–7953. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Bergmann, U.; Leiviskä, T. Reductive degradation of perfluorooctanoic acid in complex water matrices by using the UV/sulfite process. Water Res. 2021, 205, 117676. [Google Scholar] [CrossRef] [PubMed]
- Bentel, M.J.; Yu, Y.; Xu, L.; Li, Z.; Wong, B.M.; Men, Y.; Liu, J. Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: Structural dependence and implications to PFAS remediation and management. Environ. Sci. Technol. 2019, 53, 3718–3728. [Google Scholar] [CrossRef]
- Song, Z.; Tang, H.; Wang, N.; Zhu, L. Reductive defluorination of perfluorooctanoic acid by hydrated electrons in a sulfite-mediated UV photochemical system. J. Hazard. Mater. 2013, 262, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Luo, T.; Xu, J.; Li, J.; Wu, F.; Brigante, M.; Mailhot, G. Enhanced oxidation of aniline using Fe(III)-S(IV) system: Role of different oxysulfur radicals. Chem. Eng. J. 2019, 362, 183–189. [Google Scholar] [CrossRef]
- Luo, T.; Wang, Z.; Wang, Y.; Liu, Z.; Pozdnyakov, I.P. Different role of bisulfite/sulfite in UVC-S(IV)-O2 system for arsenite oxidation in water. Molecules 2019, 24, 2307. [Google Scholar] [CrossRef]
- Gu, Y.; Liu, T.; Wang, H.; Han, H.; Dong, W. Hydrated electron based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system. Sci. Total Environ. 2017, 607–608, 541–548. [Google Scholar] [CrossRef]
- Xin, X.; Kim, J.; Ashley, D.C.; Huang, C.H. Degradation and defluorination of per- and polyfluoroalkyl substances by direct photolysis at 222 nm. ACS ES&T Water 2023, 3, 2776–2785. [Google Scholar] [CrossRef]
- Maza, W.A.; Breslin, V.M.; Plymale, N.T.; Desario, P.A.; Epshteyn, A.; Owrutsky, J.C.; Pate, B.B. Nanosecond transient absorption studies of the pH-dependent hydrated electron quenching by HSO3−. Photochem. Photobiol. Sci. 2019, 18, 1526–1532. [Google Scholar] [CrossRef]
- Olawade, D.B.; Ijiwade, J.O.; Fapohunda, O.; Ige, A.O.; Olajoyetan, D.O.; Wada, O.Z. Predictive modeling of PFAS behavior and degradation in novel treatment scenarios: A review. Process Saf. Environ. Prot. 2025, 196, 106869. [Google Scholar] [CrossRef]
- Lado Ribeiro, A.R.; Moreira, N.F.F.; Li Puma, G.; Silva, A.M.T. Impact of water matrix on the removal of micropollutants by advanced oxidation technologies. Chem. Eng. J. 2019, 363, 155–173. [Google Scholar] [CrossRef]
- Huang, Y.; Sheng, B.; Wang, Z.; Liu, Q.; Yuan, R.; Xiao, D.; Liu, J. Deciphering the degradation/chlorination mechanisms of maleic acid in the Fe(II)/peroxymonosulfate process: An often overlooked effect of chloride. Water Res. 2018, 145, 453–463. [Google Scholar] [CrossRef]
- Li, X.; Shen, J.; Sun, Z.; Zhang, W.; Ma, F.; Gu, Q. Insights into the impacts of chloride ions on the oxidation of 2,4-dinitrotoluene using ferrous activated persulfate: Removal efficiency, reaction mechanism, transformation pathway, and toxicity assessment. Chemosphere 2023, 317, 137887. [Google Scholar] [CrossRef] [PubMed]
- Milh, H.; Yu, X.; Cabooter, D.; Dewil, R. Degradation of ciprofloxacin using UV-based advanced removal processes: Comparison of persulfate-based advanced oxidation and sulfite-based advanced reduction processes. Sci. Total Environ. 2021, 764, 144510. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, H.; Luo, T.; Liu, Z.; Xia, J.; Zhang, X. Phototransformation of p-arsanilic acid in aqueous media containing nitrogen species. Chemosphere 2018, 212, 777–783. [Google Scholar] [CrossRef]
- Xu, J.; Marsac, R.; Wei, C.; Wu, F.; Boily, J.F.; Hanna, K. Cobinding of pharmaceutical compounds at mineral surfaces: Mechanistic modeling of binding and cobinding of nalidixic acid and niflumic acid at goethite surfaces. Environ. Sci. Technol. 2017, 51, 11617–11624. [Google Scholar] [CrossRef]
- Cheng, W.; Zhou, L.; Marsac, R.; Boily, J.F.; Hanna, K. Effects of organic matter–goethite interactions on reactive transport of nalidixic acid: Column study and modeling. Environ. Res. 2020, 191, 110187. [Google Scholar] [CrossRef] [PubMed]
- Fovet, Y.; Gal, J.Y. Formation constants β2 of calcium and magnesium fluorides at 25 °C. Talanta 2000, 53, 617–626. [Google Scholar] [CrossRef]
- Fennell, B.D.; Odorisio, A.; Mckay, G. Quantifying hydrated electron transformation kinetics in UV-advanced reduction processes using the Re-,UV method. Environ. Sci. Technol. 2022, 56, 10329–10338. [Google Scholar] [CrossRef]
- Tipplook, M.; Hisama, K.; Koyama, M.; Fujisawa, K.; Hayashi, F.; Sudare, T.; Teshima, K. Cation-doped nanocarbons for enhanced perfluoroalkyl substance removal: Exotic bottom-up solution plasma synthesis and characterization. ACS Appl. Mater. Interfaces 2024, 16, 61832–61845. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, L.; Yang, L.; Liu, Z.; Zhang, Y. Distribution and desorption of perfluorinated compounds in fractionated sediments. Chemosphere 2012, 88, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, Y.; Zhao, X.; Du, P.; Liu, S.; Lv, J.; Xu, F.; Meng, W.; Xu, J. Distribution, source characterization and inventory of perfluoroalkyl substances in Taihu Lake, China. Chemosphere 2015, 127, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Fagbayigbo, B.O.; Opeolu, B.O.; Fatoki, O.S.; Olatunji, O.S.; Akharame, M.O.; Human, I.S. Sorption and partitioning of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) onto sediments of Diep and Plankenburg river systems Western Cape, South Africa. Environ. Technol. Innov. 2022, 25, 102110. [Google Scholar] [CrossRef]
- Chinese Standard of GB 7484-87. Available online: https://www.codeofchina.com/standard/GBT7484-1987.html (accessed on 28 April 2025).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Blank (μM) | Measurement ± SD (μM) | LOD of PFAS (μM) | PFAS (μM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|---|---|
| PFOA | 0 | 0.0185 ± 0.0183 0.0048 ± 0.0164 | 0.0549 | 25 | 100.8 | 2.95% |
| PFOS | 0.0492 | 101.9 | 3.63% |
| Sediments | Compounds | Equation | R2 |
|---|---|---|---|
| S1 | PFOA | y = 0.01017x + 0.0012 | 0.9988 |
| PFOS | y = 0.01290x − 0.0064 | 0.9649 | |
| S2 | PFOA | y = 0.00997x + 0.0018 | 0.9941 |
| PFOS | y = 0.00816x + 0.0015 | 0.9986 | |
| S3 | PFOA | y = 0.01047x + 0.0062 | 0.9974 |
| PFOS | y = 0.00520x + 0.0030 | 0.9980 |
| Sediments | foc (%) | PFOA | PFOS | ||||
|---|---|---|---|---|---|---|---|
| logKd | logKoc | R2 | logKd | logKoc | R2 | ||
| S1 | 5.18 | 1.00 | 2.29 | 0.9966 | 0.70 | 1.98 | 0.9241 |
| S2 | 6.35 | 1.05 | 2.25 | 0.9898 | 1.88 | 3.07 | 0.9898 |
| S3 | 5.08 | 0.99 | 2.28 | 0.9919 | 1.26 | 2.55 | 0.9878 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, S.; Chen, Y.; Mei, X.; Jin, L.; Wu, F.; Xu, J. Application of VUV/Sulfite Defluorination System for the Simple Detection of Perfluoroalkyl Substances. Molecules 2025, 30, 2475. https://doi.org/10.3390/molecules30112475
Tao S, Chen Y, Mei X, Jin L, Wu F, Xu J. Application of VUV/Sulfite Defluorination System for the Simple Detection of Perfluoroalkyl Substances. Molecules. 2025; 30(11):2475. https://doi.org/10.3390/molecules30112475
Chicago/Turabian StyleTao, Shiyong, Yilin Chen, Xiao Mei, Luyao Jin, Feng Wu, and Jing Xu. 2025. "Application of VUV/Sulfite Defluorination System for the Simple Detection of Perfluoroalkyl Substances" Molecules 30, no. 11: 2475. https://doi.org/10.3390/molecules30112475
APA StyleTao, S., Chen, Y., Mei, X., Jin, L., Wu, F., & Xu, J. (2025). Application of VUV/Sulfite Defluorination System for the Simple Detection of Perfluoroalkyl Substances. Molecules, 30(11), 2475. https://doi.org/10.3390/molecules30112475