
Development of a Multigram Synthetic Route to RM-581, an Orally Active Aminosteroid Derivative Against Several Types of Cancers


Abstract

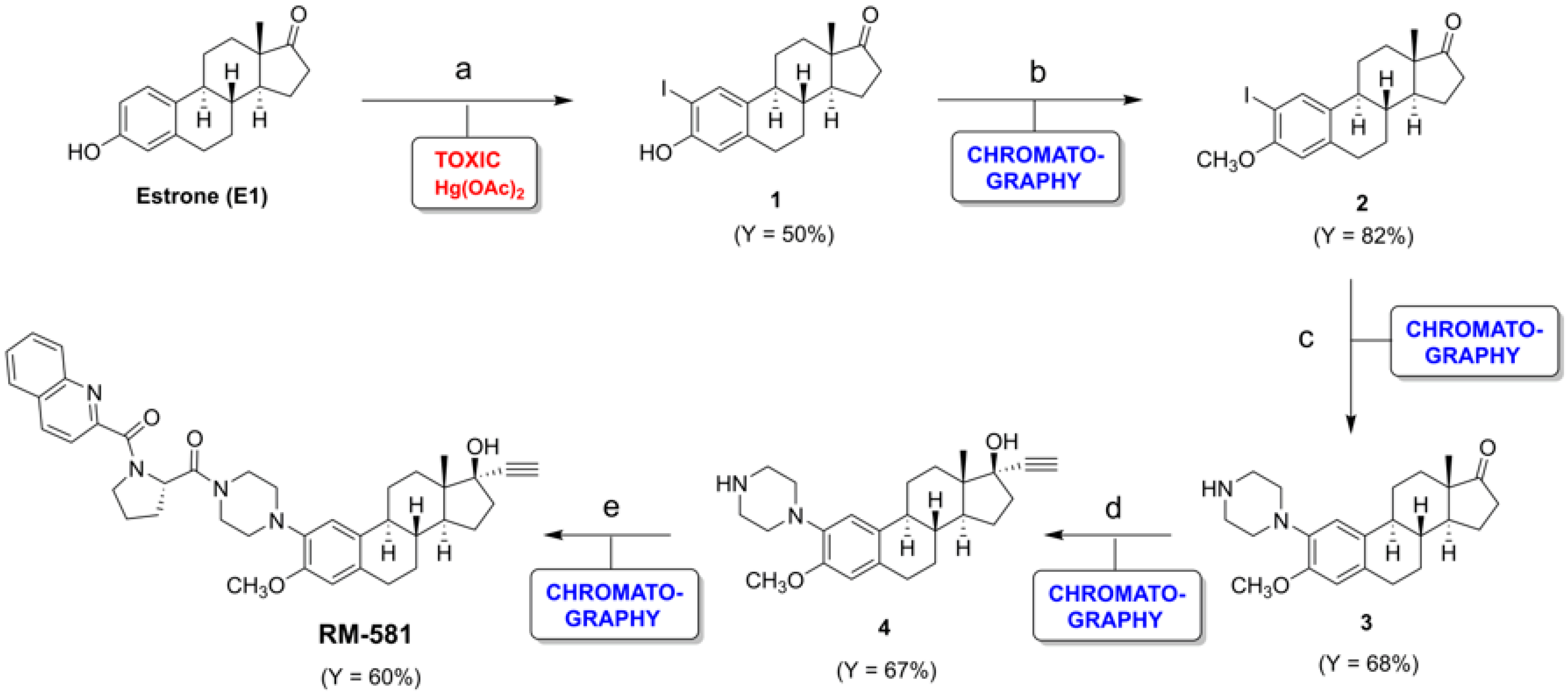
1. Introduction
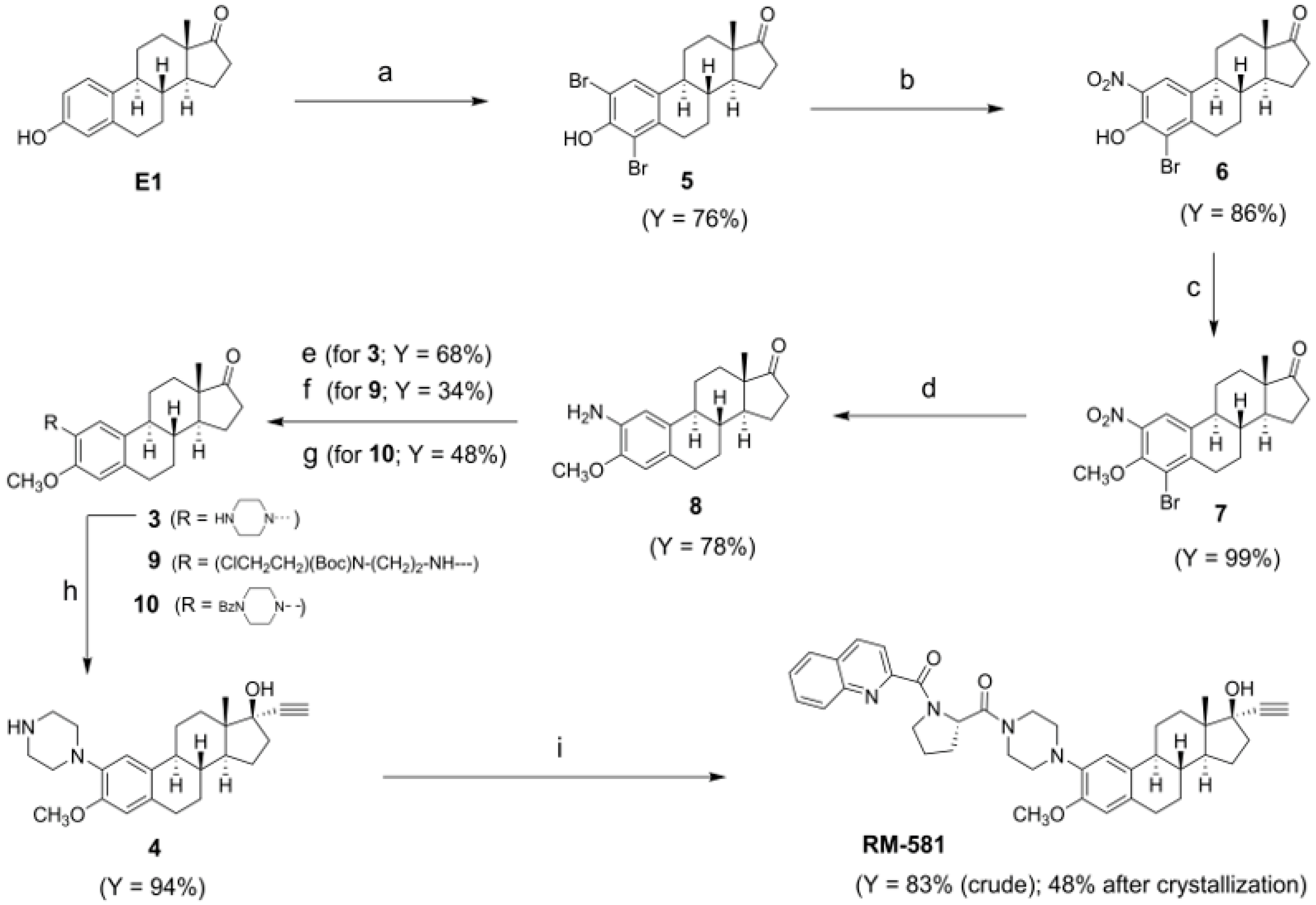
2. Results and Discussion
2.1. Efficient New Route to RM-581
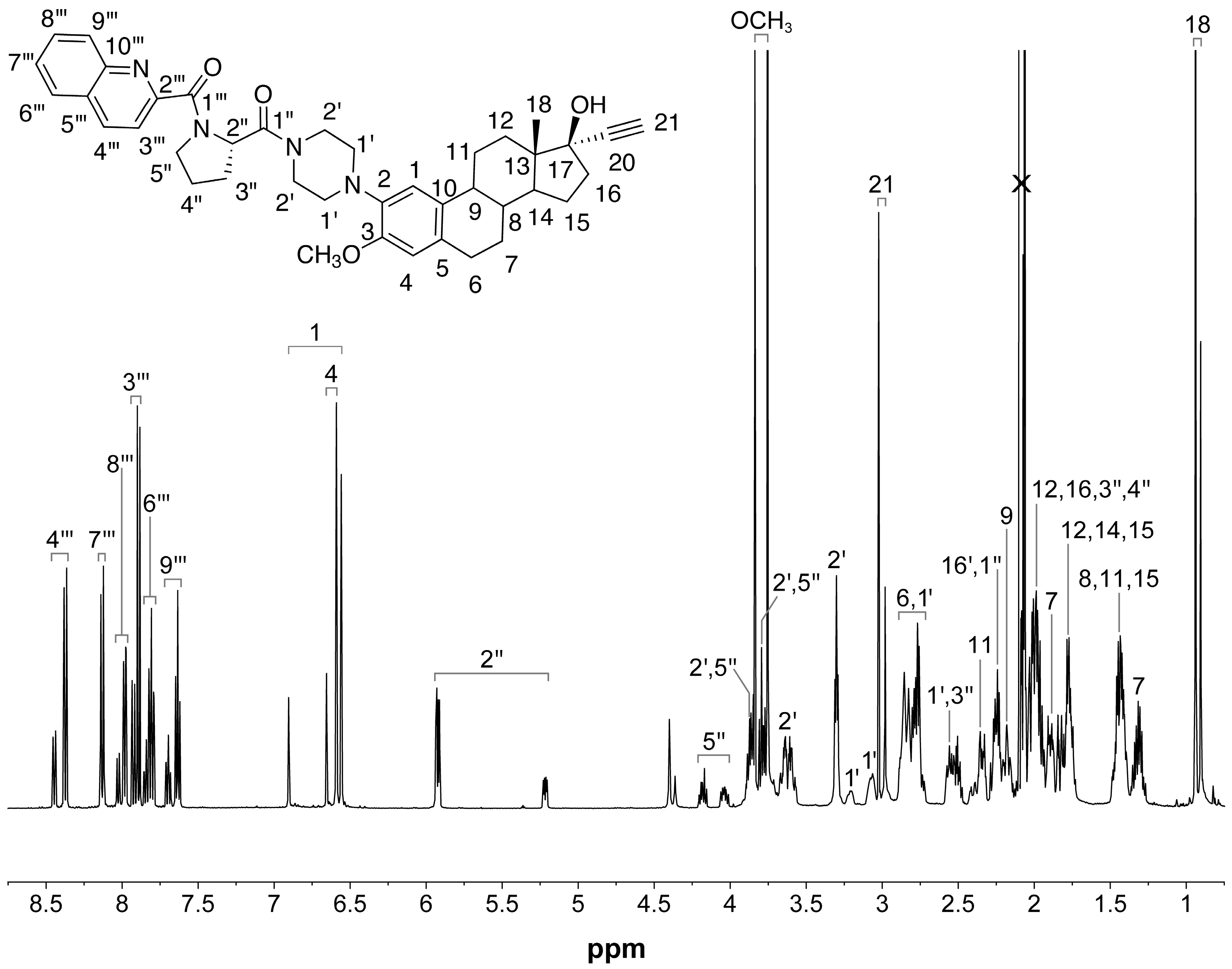
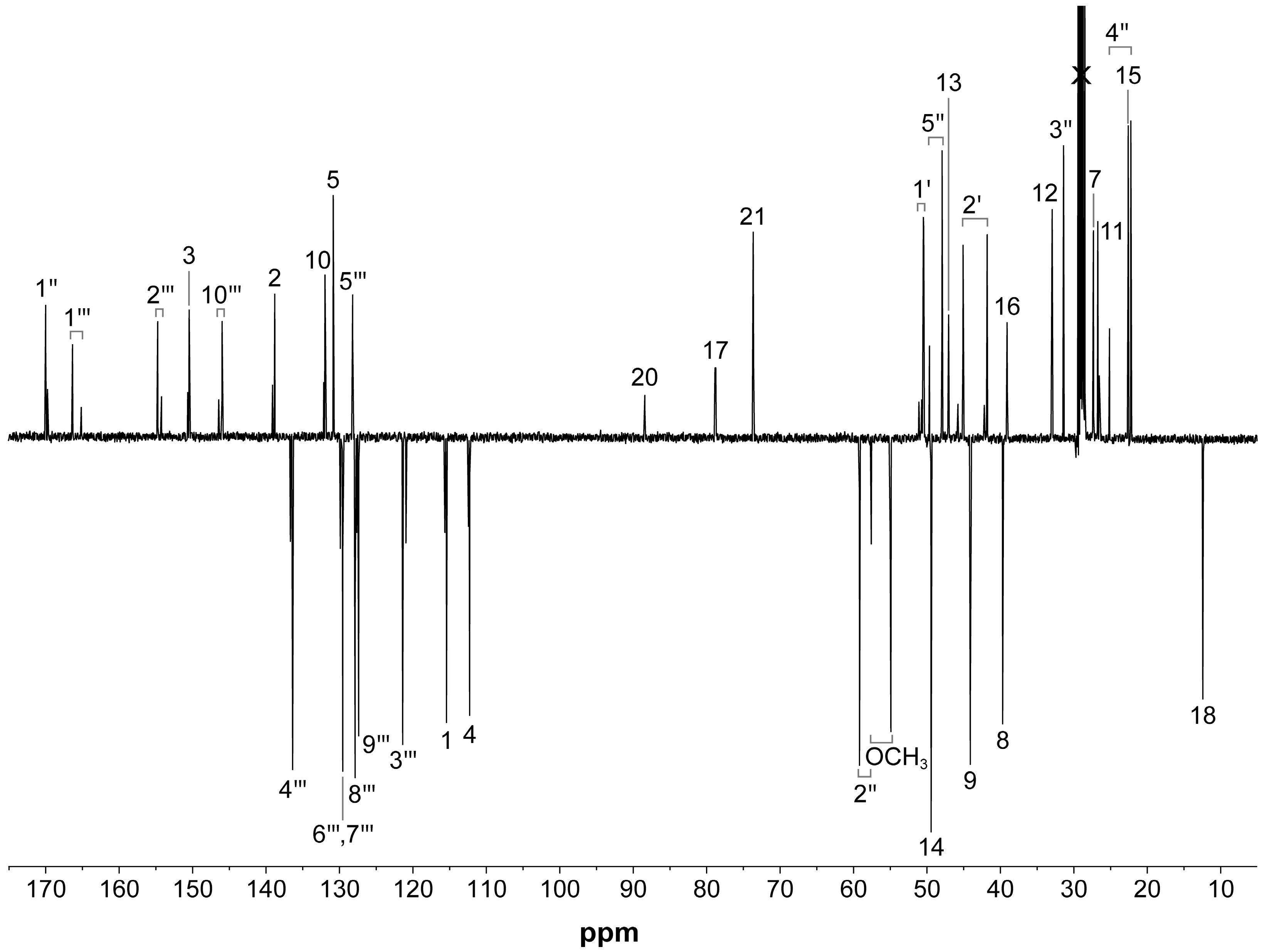
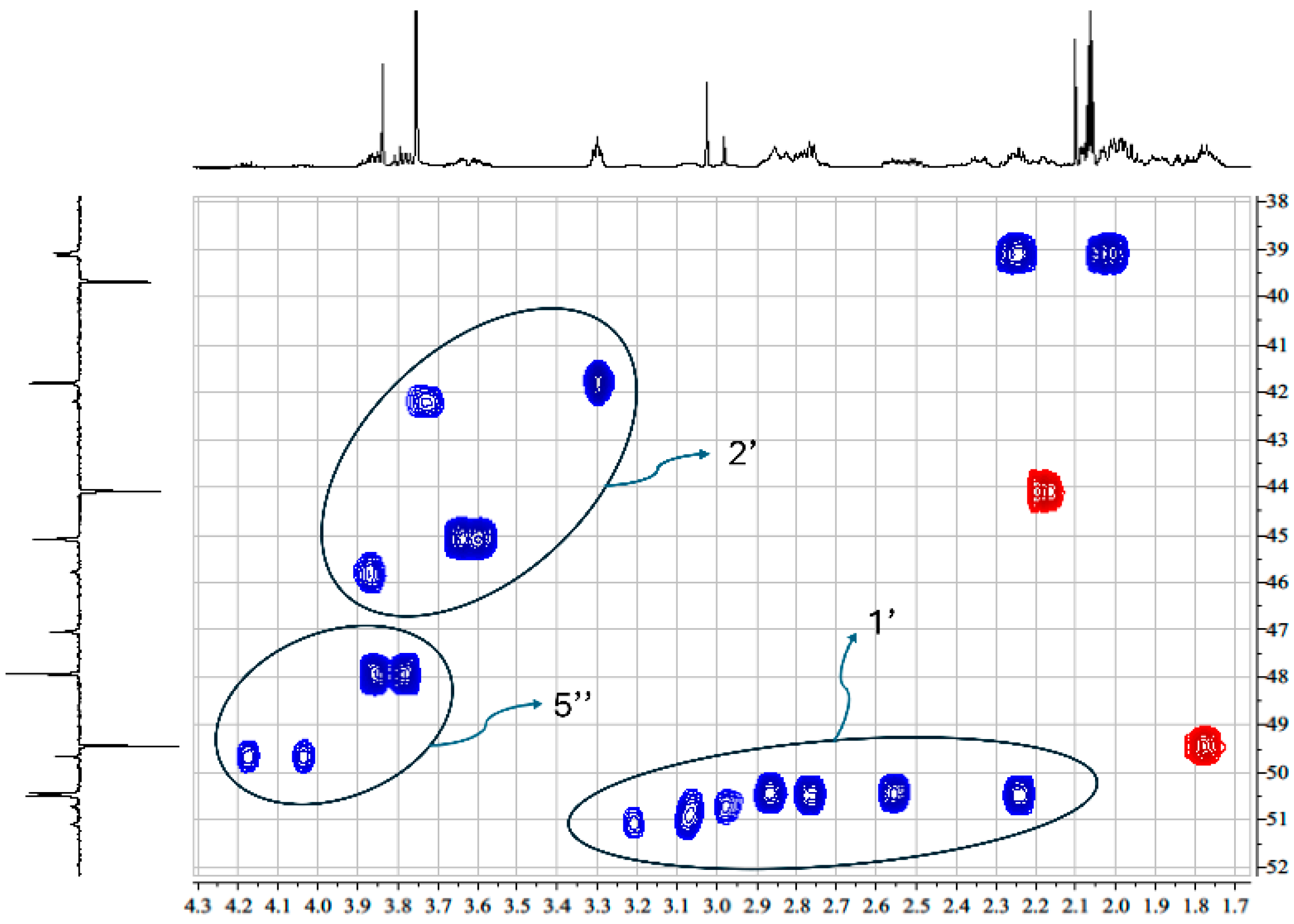
2.2. NMR Characterization of RM-581
3. Materials and Methods
3.1. General
3.2. Chemical Synthesis of RM-581
3.2.1. Synthesis of 2,4-Dibromo-3-hydroxyestra-1(10),2,4-trien-17-one (5)
3.2.2. Synthesis of 4-Bromo-3-hydroxy-2-nitroestra-1(10),2,4-trien-17-one (6)
3.2.3. Synthesis of 4-Bromo-3-methoxy-2-nitroestra-1(10),2,4-trien-17-one (7)
3.2.4. Synthesis of 2-Amino-3-methoxy-estra-1(10),2,4-trien-17-one (8)
3.2.5. Synthesis of 3-Methoxy-2-piperazin-1-yl-estra-1(10),2,4-trien-17-one (3)
3.2.6. Synthesis of 17α-Ethynyl-3-methoxy-2-piperazine-estra-1(10),2,4-trien-17β-ol (4)
3.2.7. Synthesis of {4-[17α-Ethynyl-17β-hydroxy-3-methoxyestra-1(10)2,4-trien-2-yl]piperazin-1-yl}[(2S)-1-(quinolin-2-ylcarbonyl)pyrrolidin-2-yl]-methanone (RM-581)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poirier, D.; Roy, J.; Maltais, R.; Weidmann, C.; Audet-Walsh, É. An aminosteroid derivative shows higher in vitro and in vivo potencies than gold standard drugs in androgen-dependent prostate cancer models. Cancers 2023, 15, 3033. [Google Scholar] [CrossRef]
- Burguin, A.; Roy, J.; Ouellette, G.; Maltais, R.; Bherer, J.; Diorio, C.; Poirier, D.; Durocher, F. Aminosteroid RM-581 decreases cell proliferation of all breast cancer molecular subtypes, alone and in combination with breast cancer treatments. J. Clin. Med. 2023, 12, 4241. [Google Scholar] [CrossRef]
- Maltais, R.; Roy, J.; Perreault, M.; Sato, S.; Lévesque, J.C.; Poirier, D. Induction of endoplasmic reticulum stress-mediated apoptosis by aminosteroid RM-581 efficiently blocks the growth of PC-3 cancer cells and tumors resistant or not to docetaxel. Int. J. Mol. Sci. 2021, 22, 11181. [Google Scholar] [CrossRef]
- Perreault, M.; Maltais, R.; Roy, J.; Picard, S.; Popa, I.; Bertrand, N.; Poirier, D. Induction of endoplasmic reticulum stress by aminosteroid derivative RM-581 leads to tumor regression in PANC-1 xenograft model. Investig. New Drugs 2019, 37, 431–440. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Ellison, L.F.; Saint-Jacques, N. Five-year cancer survival by stage at diagnosis in Canada. Health Rep. 2023, 34, 3–15. [Google Scholar] [CrossRef]
- Kenmogne, L.C.; Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. The aminosteroid derivative RM-133 shows in vitro and in vivo antitumor activity in human ovarian and pancreatic cancers. PLoS ONE 2015, 10, e0144890. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Biological activity and structural diversity of steroids containing aromatic rings, phosphate groups, or halogen atoms. Molecules 2023, 28, 5549. [Google Scholar] [CrossRef]
- Hanson, J.R. Steroids: Partial synthesis in medicinal chemistry. Nat. Prod. Rep. 2010, 27, 887–899. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef]
- Brito, V.; Alves, G.; Almeida, P.; Silvestre, S. Highlights on steroidal arylidene derivatives as a source of pharmacologically active compounds: A review. Molecules 2021, 26, 2032. [Google Scholar] [CrossRef]
- Liu, X.; Viswanadhapalli, S.; Kumar, S.; Lee, T.-K.; Moore, A.; Ma, S.; Chen, L.; Hsieh, M.; Li, M.; Sareddy, G.R.; et al. Targeting lipa independent of its lipase activity is a therapeutic strategy in solid tumors via induction of endoplasmic reticulum stress. Nat. Cancer 2022, 3, 866–884. [Google Scholar] [CrossRef]
- Chung, V.; Mizrahi, J.D.; Pant, S. Novel therapies for pancreatic cancer. JCO Oncol. Pract. 2024, 21, 613–619. [Google Scholar] [CrossRef]
- Ozay, Z.I.; Swami, U.; Agarwal, N. Challenges in the management of advanced prostate cancer. JCO Oncol. Pract. 2025, 21, 447–449. [Google Scholar] [CrossRef]
- Xiong, N.; Wu, H.; Yu, Z. Advancements and challenges in triple-negative breast cancer: A comprehensive review of therapeutic and diagnostic strategies. Front. Oncol. 2024, 14, 1405491. [Google Scholar] [CrossRef]
- Perreault, M.; Maltais, R.; Roy, J.; Dutour, R.; Poirier, D. Design of a mestranol 2-N-piperazino-substituted derivative showing potent and selective in vitro and in vivo activities in MCF-7 breast cancer models. ChemMedChem 2017, 12, 177–182. [Google Scholar] [CrossRef]
- Numazawa, M.; Kimura, K. Novel and regiospecific synthesis of 2-amino estrogens via zincke nitration. Steroids 1983, 41, 675. [Google Scholar] [CrossRef]
- Maltais, R.; Hospital, A.; Delhomme, A.; Roy, J.; Poirier, D. Chemical synthesis, NMR analysis and evaluation on a cancer xenograft model (HL-60) of the aminosteroid derivative RM-133. Steroids 2014, 82, 68–76. [Google Scholar] [CrossRef]
- Augustine, J.K.; Vairaperumal, V.; Narasimhan, S.; Alagarsamy, P.; Radhakrishnan, A. Propylphosphonic anhydride (t3p®): An efficient reagent for the one-pot synthesis of 1,2,4-oxadiazoles, 1,3,4-oxadiazoles, and 1,3,4-thiadiazoles. Tetrahedron 2009, 65, 9989–9996. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Comu: A third generation of uronium-type coupling reagents. J. Pept. Sci. 2010, 16, 6–9. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Subirós Funosas, R.; Prohens, R.; Albericio, F. Comu: A safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chem. Eur. J. 2009, 15, 9404–9416. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Provencher, L.; Gauthier, S. Topical, Antiandrogenic Steroids. WO 2004/089971 A1, 21 October 2004. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent (A, B, or C) (Equivalent) 1 | Solvent (Base) | Time (h) | Temp (°C) | Chromato. Purification | Isolated Yield (%) | HPLC Purity (%) |
|---|---|---|---|---|---|---|---|
| 1 | A (4.0) | 1-Butanol (--) | 27 | 150 | Yes | 47 | 23.0 |
| 2 | A (1.05) | 1-Butanol (--) | 96 | 135 | No | 68 | 97.9 |
| 3 | A (1.05) | Ethylene glycol (--) | 27 | 150 | No | 25 | 88.4 |
| 4 | A (1.05) | DMSO (--) | 24 | 135 | No | 0 2 | ND 3 |
| 5 | A (4.0) | DMSO (--) | 24 | 135 | No | 0 2 | ND 3 |
| 6 | B (1.4) | 1-Butanol (K2CO3) | 12 | 150 | Yes | 0 (34 of 9) | ND 3 |
| 7 | C (4.0) | 1-Butanol (--) | 8 | 150 | Yes | 0 (48 of 10) | ND 3 |
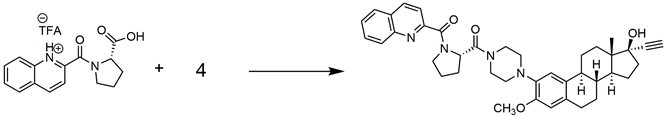 | |||
|---|---|---|---|
| Entry | Coupling Reagent | Equivalent | RM-581 Yield (%) |
| 1 | COMU | 1.2 | 83 |
| 2 | HBTU | 1.2 | 80 |
| 3 | T3P | 2 | 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maltais, R.; de Sainte Maresville, D.; Desrosiers, V.; Poirier, D. Development of a Multigram Synthetic Route to RM-581, an Orally Active Aminosteroid Derivative Against Several Types of Cancers. Molecules 2025, 30, 2441. https://doi.org/10.3390/molecules30112441
Maltais R, de Sainte Maresville D, Desrosiers V, Poirier D. Development of a Multigram Synthetic Route to RM-581, an Orally Active Aminosteroid Derivative Against Several Types of Cancers. Molecules. 2025; 30(11):2441. https://doi.org/10.3390/molecules30112441
Chicago/Turabian StyleMaltais, René, Doriane de Sainte Maresville, Vincent Desrosiers, and Donald Poirier. 2025. "Development of a Multigram Synthetic Route to RM-581, an Orally Active Aminosteroid Derivative Against Several Types of Cancers" Molecules 30, no. 11: 2441. https://doi.org/10.3390/molecules30112441
APA StyleMaltais, R., de Sainte Maresville, D., Desrosiers, V., & Poirier, D. (2025). Development of a Multigram Synthetic Route to RM-581, an Orally Active Aminosteroid Derivative Against Several Types of Cancers. Molecules, 30(11), 2441. https://doi.org/10.3390/molecules30112441