
The Puzzle of the New Type of Intermediate in the Course of [2 + 2] Cycloaddition with the Participation of Conjugated Nitroalkenes: MEDT Computational Study



Abstract
1. Introduction
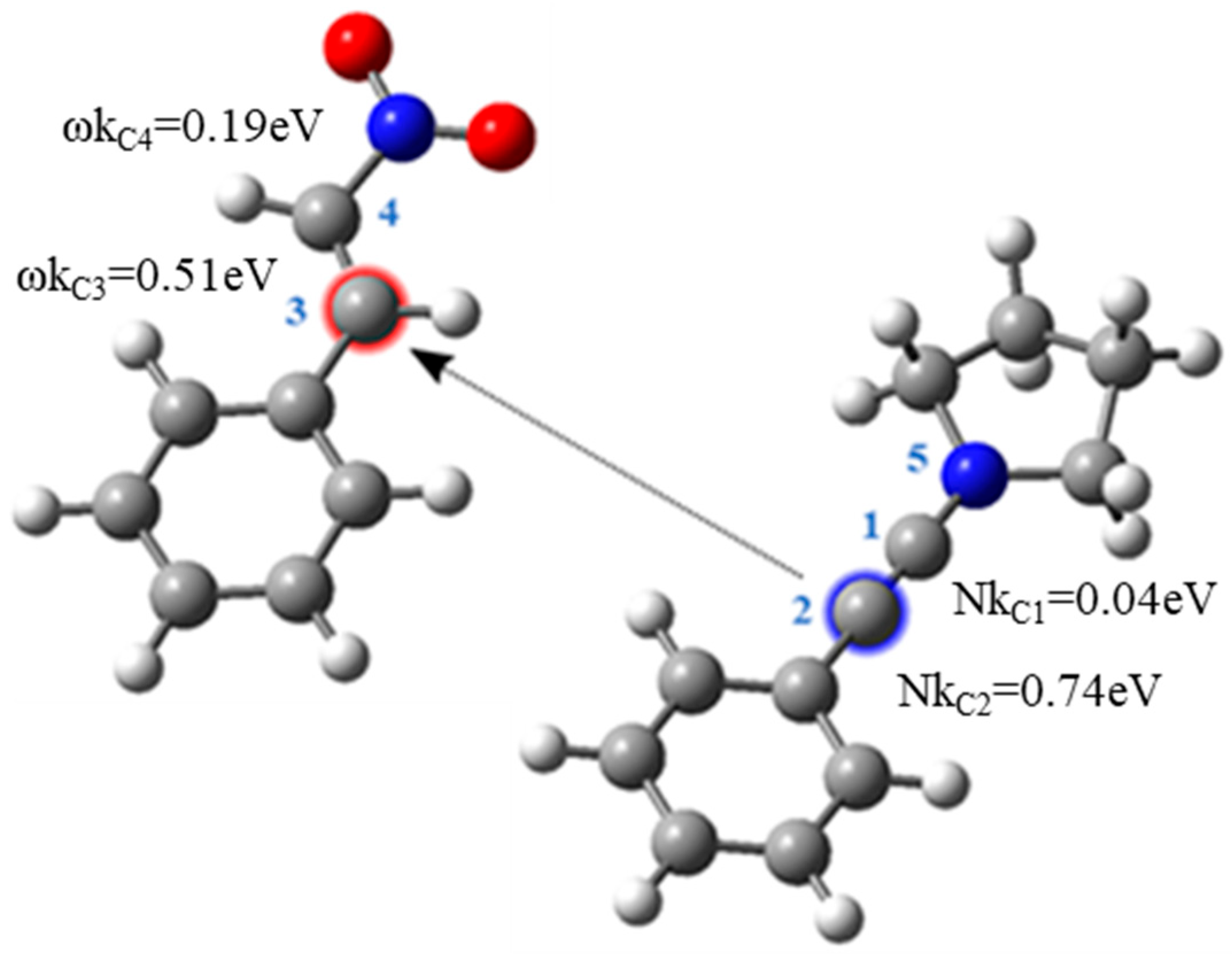
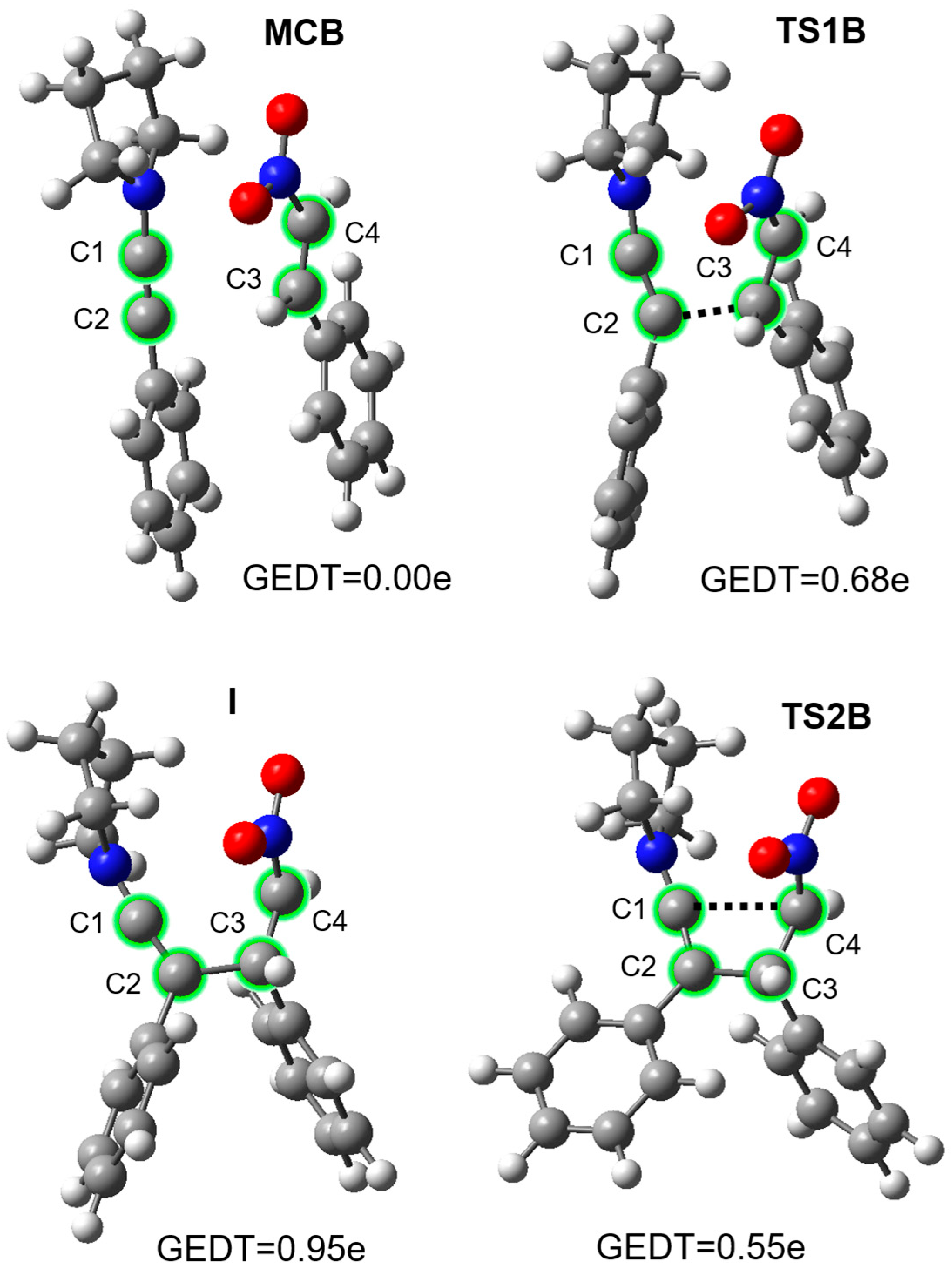
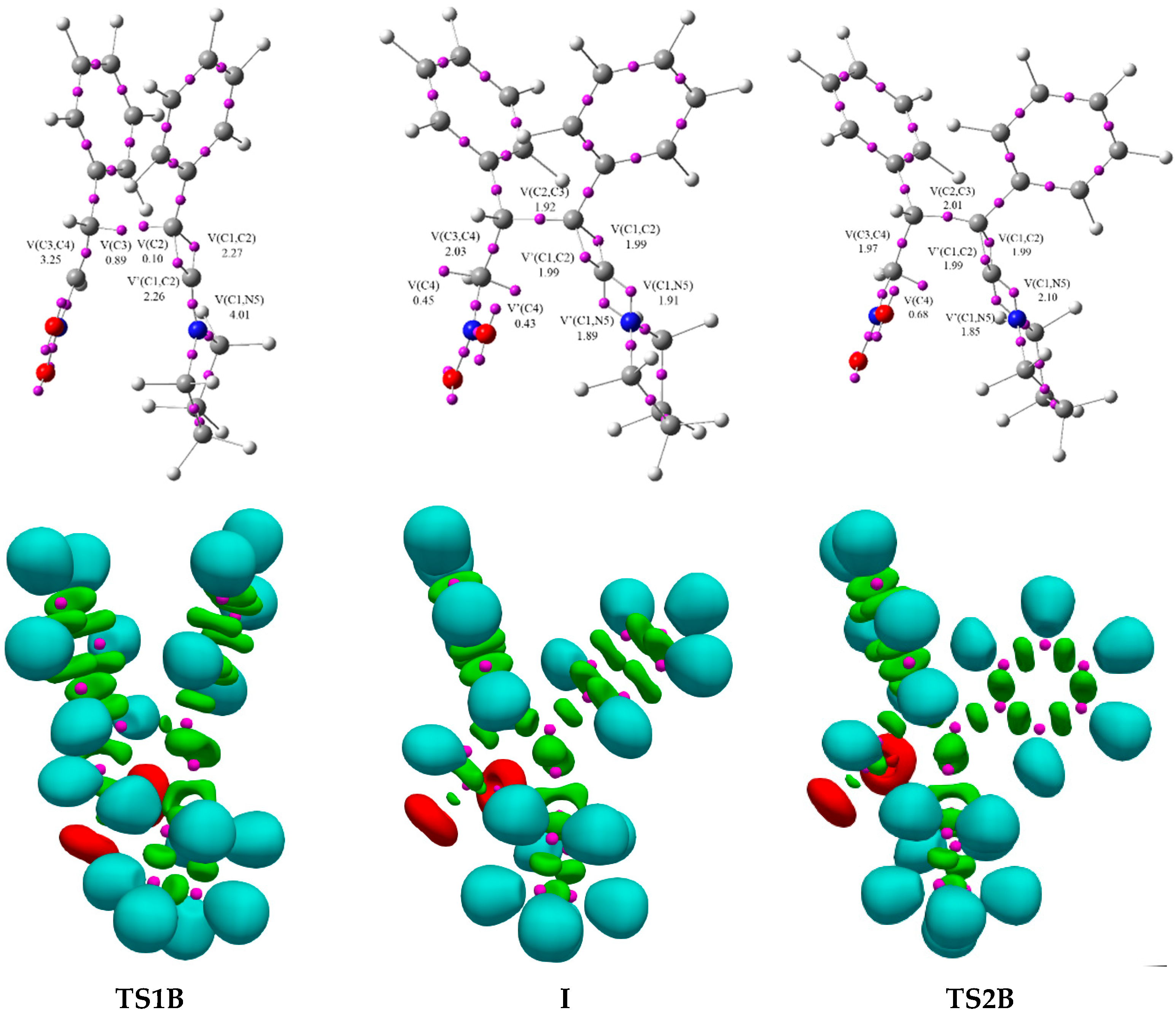
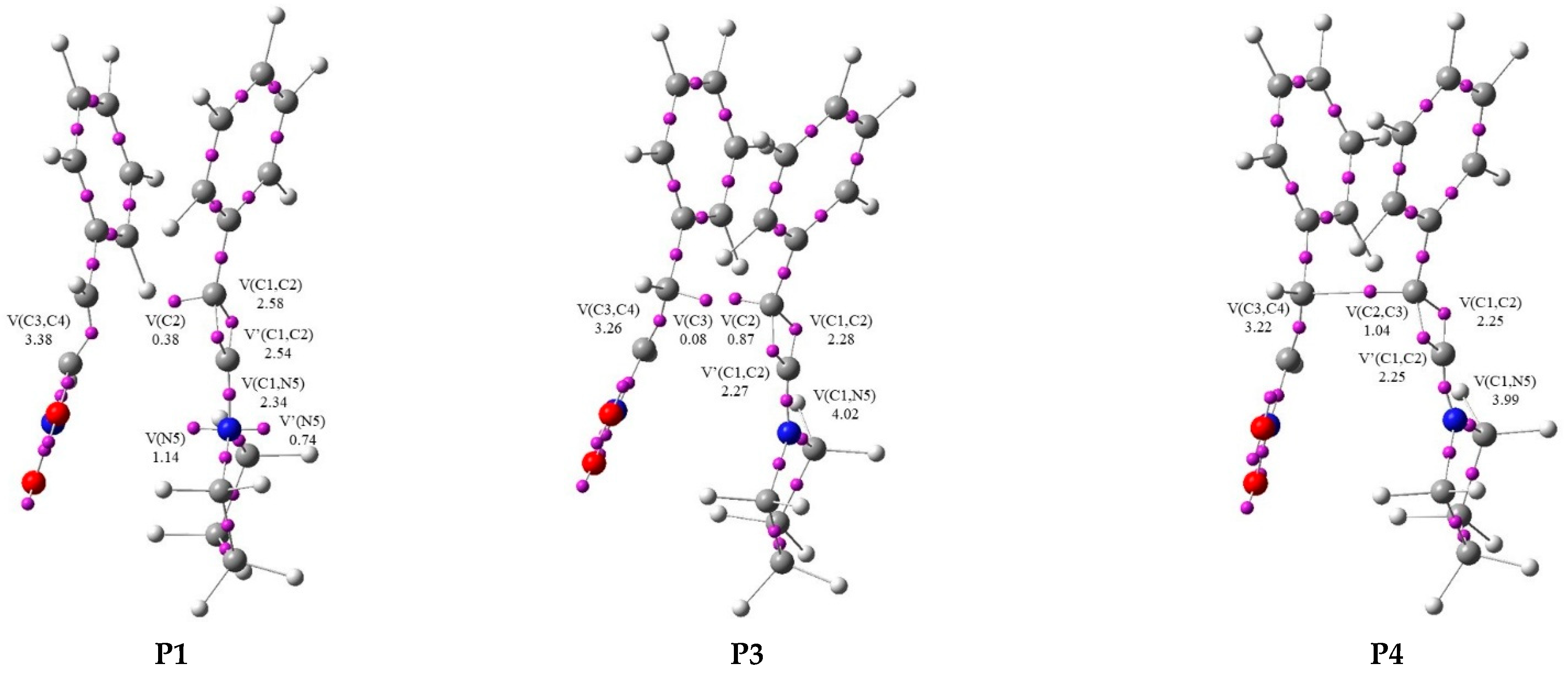
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, B.; Wang, J. Recent advances in metal-free catalytic enantioselective higher-order cycloadditions. Org. Chem. Front. 2024, 11, 1824–1842. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Ryachi, K.; Mohammad-Salim, H.; Al-Sadoon, M.K.; Zeroual, A.; de Julián-Ortiz, J.V.; El Idrissi, M.; Tounsi, A. Quantum study of the [3+2] cycloaddition of nitrile oxide and carvone oxime: Insights into toxicity, pharmacokinetics, and mechanism. Chem. Heterocycl. Compd. 2024, 60, 646–654. [Google Scholar] [CrossRef]
- Wróblewska, A.; Sadowski, M.; Jasiński, R. Selectivity and molecular mechanism of the Au(III)-catalyzed [3+2] cycloaddition reaction between (Z)-C,N-diphenylnitrone and nitroethene in the light of the molecular electron density theory computational study. Chem. Heterocycl. Compd. 2024, 60, 639–645. [Google Scholar] [CrossRef]
- Dresler, E. The Participation of Oleic Acid and its Esters in [3+2] Cycloaddition Reactions: A Mini-Review. Sci. Rad. 2024, 3, 53–61. [Google Scholar] [CrossRef]
- Sadowski, M.; Kula, K. Nitro-functionalized analogues of 1,3-Butadiene: An overview of characteristic, synthesis, chemical transformations and biological activity. Curr. Chem. Lett. 2024, 13, 15–30. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Jasiński, R. Molecular mechanism of Hetero Diels-Alder reactions between (E)-1,1,1-trifluoro-3-nitrobut-2-enes and enamine systems in the light of Molecular Electron Density Theory. J. Mol. Graph. Model. 2020, 101, 107714. [Google Scholar] [CrossRef]
- Domíngueza, G.; Pérez-Castells, J. Recent advances in [2+2+2] cycloaddition reactions. Chem. Soc. Rev. 2011, 40, 3430–3444. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A.; Abdallah, H.H.; Maiyelvaganan, K.R.; Prakash, M.; Hochlaf, M. Mechanistic study of the [2+2] cycloaddition reaction of cyclohexenone and its derivatives with vinyl acetate. Theor. Chem. Acc. 2020, 139, 19. [Google Scholar] [CrossRef]
- Sadowski, M.; Dresler, E.; Wróblewska, A.; Jasiński, R. A New Insight into the Molecular Mechanism of the Reaction between 2-Methoxyfuran and Ethyl (Z)-3-phenyl-2-nitroprop-2-enoate: An Molecular Electron Density Theory (MEDT) Computational Study. Molecules 2024, 29, 4876. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. On the energetic aspects and molecular mechanism of 3-nitro-substituted 2-isoxazolines formation via nitrile N-oxide [3+2] cycloaddition: An MEDT computational study. Molecules 2024, 29, 3042. [Google Scholar] [CrossRef] [PubMed]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the regioselectivity and the molecular mechanism of [3+2] cycloaddition reactions between nitrous oxide and conjugated nitroalkenes: DFT computational study. Molecules 2022, 27, 8441. [Google Scholar] [CrossRef] [PubMed]
- Poplata, S.; Tröster, A.; Zou, Y.Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Jia, Q.; Song, S.; Huang, X. [2 + 2]-Cycloaddition-derived cyclobutane natural products: Structural diversity, sources, bioactivities, and biomimetic syntheses. Nat. Prod. Rep. 2023, 40, 1094–1129. [Google Scholar] [CrossRef]
- Gan, M.M.; Yu, J.G.; Wang, Y.Y.; Han, Y.F. Template-Directed Photochemical [2 + 2] Cycloaddition in Crystalline Materials: A Useful Tool to Access Cyclobutane Derivatives. Cryst. Growth Des. 2018, 18, 553–565. [Google Scholar] [CrossRef]
- Pacansky, J.; Chang, J.S.; Brown, D.W.; Schwarz, W. The observation of zwitterions in the thermal reaction of ketenes with carbon-nitrogen double bonds. J. Org. Chem. 1982, 47, 2233–2244. [Google Scholar] [CrossRef]
- Liang, Y.; Jiao, L.; Zhang, S.; Xu, J. Microwave- and photoirradiation-induced staudinger reactions of cyclic imines and ketenes generated from alpha-diazoketones. A further investigation into the stereochemical process. J. Org. Chem. 2005, 70, 334–337. [Google Scholar] [CrossRef]
- Borodkin, G.I. Fluorination of heterocyclic compounds accompanied by molecular rearrangements. Chem. Heterocycl. Compd. 2024, 60, 323–335. [Google Scholar] [CrossRef]
- Smith, D.L.; Chidipudi, S.R.; Goundry, W.R.; Lam, H.W. Rhodium-Catalyzed [2 + 2] Cycloaddition of Ynamides with Nitroalkenes. Org. Lett. 2012, 14, 4934–4937. [Google Scholar] [CrossRef]
- Reinhoudt, D.N.; Kouwenhoven, C.G. [2 + 2] Cycloaddition reactions of heterocyclic compounds with ynamines. Recl. Trav. Chim. Pays-Bas 1976, 95, 67–73. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Wallbillich, G.E.H.; Montgomery, L.K. Cycloaddition. IV. Addition Products of 1, 1-Dichloro-2, 2-difluoroethylene to a Further Series of Dienes. J. Org. Chem. 1967, 32, 1290–1297. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Wallbillich, G.E.H.; Wingrove, A.S.; Swenton, J.S.; Mongomery, L.K.; Kramer, B.D. Cycloaddition. V. 2-Alkylbutadienes and 1, 1-dichloro-2, 2-difluoroethylene. Effect of diene conformation on mode of cycloaddition. J. Am. Chem. Soc. 1968, 90, 2049–2056. [Google Scholar] [CrossRef]
- Bartlett, P.D. 1, 2-and 1, 4-Cycloaddition to Conjugated Dienes: Dependence of products and rates on reactant structure tells something about competing reaction mechanisms. Science 1968, 159, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Siadati, S.A. Beyond the alternatives that switch the mechanism of the 1, 3-dipolar cycloadditions from concerted to stepwise or vice versa: A literature review. Prog. React. Kinet. Mech. 2016, 41, 331–344. [Google Scholar] [CrossRef]
- Jasiński, R.; Dresler, E. On the question of zwitterionic intermediates in the [3+2] cycloaddition reactions: A critical review. Organics 2020, 1, 49. [Google Scholar] [CrossRef]
- Jasiński, R. On the question of stepwise [4+2] cycloaddition reactions and their stereochemical aspects. Symmetry 2012, 13, 1911. [Google Scholar] [CrossRef]
- Siadati, S.A.; Rezazadeh, S. The extraordinary gravity of three atom 4π-components and 1,3-dienes to C20-nXn fullerenes; a new gate to the future of Nano technology. Sci. Radices 2022, 1, 46–68. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and mechanism of 1,3-ipolar cycloadditions. Angew. Chem. Int. Ed. 1963, 2, 633–645. [Google Scholar] [CrossRef]
- Huisgen, R. Mechanism of 1,3-dipolar cycloadditions. A Reply. J. Org. Chem. 1968, 33, 2291–2297. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. 76. Concerted nature of 1, 3-dipolar cycloadditions and the question of diradical intermediates. J. Org. Chem. 1976, 41, 403–419. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Domingo, L.R.; Ríos-Gutiérrez, M.; Jasiński, R. Understanding the mechanism of the decomposition reaction of nitroethyl benzoate through the Molecular Electron Density Theory. Theor. Chem. Acc. 2017, 136, 129. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Jasiński, R. Unexpected molecular mechanism of trimethylsilyl bromide elimination from 2-(trimethylsilyloxy)-3-bromo-3-methyl-isoxazolidines. Theor. Chem. Acc. 2019, 138, 81. [Google Scholar] [CrossRef]
- Sadowski, M.; Dresler, E.; Zawadzińska, K.; Wróblewska, A.; Jasiński, R. Syn-Propanethial S-Oxide as an Available Natural Building Block for the Preparation of Nitro-Functionalized, Sulfur-Containing Five-Membered Heterocycles: An MEDT Study. Molecules 2024, 29, 4892. [Google Scholar] [CrossRef] [PubMed]
- Dresler, E.; Woliński, P.; Wróblewska, A.; Jasiński, R. On the question of zwitterionic intermediates in the [3+2] cycloaddition reactions between aryl azides and ethyl propiolate. Molecules 2023, 28, 8152. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. A molecular electron density theory study of the higher-order cycloaddition reactions of tropone with electron-rich ethylenes. The role of the Lewis acid catalyst in the mechanism and pseudocyclic selectivity. New J. Chem. 2022, 46, 294–308. [Google Scholar] [CrossRef]
- Kula, K.; Jasiński, R. Synthesis of bis(het)aryl systems via domino reaction involving (2E,4E)-2,5-dinitrohexa-2,4-diene: DFT mechanistic considerations. Chem. Heterocycl. Compd. 2024, 60, 600–610. [Google Scholar] [CrossRef]
- Ameur, S.; Barhoumi, A.; El Abdallaoui, H.A.; Syed, A.; Belghiti, M.E.; Elgorban, A.M.; Wong, L.S.; Wang, S.; El Idrissi, M.; Zeroual, A.; et al. Molecular docking, exploring diverse selectivities and mechanistic insights in the cycloaddition reaction between 3-benzoylpyrrolo [1,2-a]quinoxaline-1,2,4(5H)-triones and butyl vinyl ether. Chem. Heterocycl. Compd. 2024, 60, 584–591. [Google Scholar] [CrossRef]
- Aitouna, A.O.; Syed, A.; Alfagham, A.T.; Mazoir, N.; de Julián-Ortiz, J.V.; Elgorban, A.M.; El Idrissi, M.; Wong, L.S.; Zeroual, A. Investigating the chemical reactivity and molecular docking of 2-diazo-3,3,3-trifluoro-1-nitropropane with phenyl methacrylate using computational methods. Chem. Heterocycl. Compd. 2024, 60, 592–599. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Zeroual, A.; Syed, A.; Bahkali, A.H. Docking Survey, ADME, Toxicological Insights, and Mechanistic Exploration of the Diels-Alder Reaction between Hexachlorocyclopentadiene and Dichloroethylene. J. Comput. Chem. 2025, 46, e70092. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Understanding of the stability of acyclic nitronic acids in the light of Molecular Electron Density Theory. J. Mol. Graph. Model. 2024, 129, 108754. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Dresler, E.; Sadowski, M.; Demchuk, O.M. Reactivity of the ethyl oleate in the [3+2] cycloaddition to arylonitrile N-oxide: A reexamination. Sci. Radices 2024, 3, 108–121. [Google Scholar] [CrossRef]
- Kula, K.; Zawadzińska, K. Local nucleophile-electrophile interactions in [3+2] cycloaddition reactions between benzonitrile N-oxide and selected conjugated nitroalkenes in the light of MEDT computational study. Curr. Chem. Lett. 2021, 10, 9–16. [Google Scholar] [CrossRef]
- Domingo, L.R. 1999–2024, a Quarter Century of the Parr’s Electrophilicity ω Index. Sci. Radices 2024, 3, 157–186. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Electrophilicity w and Nucleophilicity N Scales for Cationic and Anionic Species. Sci. Radices 2025, 4, 1–17. [Google Scholar] [CrossRef]
- Zawadzińska-Wrochniak, K.; Kula, K.; Ríos-Gutiérrez, M.; Gostyński, B.; Krawczyk, T.; Jasiński, R. A Comprehensive Study of the Synthesis, Spectral Characteristics, Quantum–Chemical Molecular Electron Density Theory, and In Silico Future Perspective of Novel CBr3-Functionalyzed Nitro-2-Isoxazolines Obtained via (3 + 2) Cycloaddition of (E)-3,3,3-Tribromo-1-Nitroprop-1-ene. Molecules 2025, 30, 2149. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the molecular mechanism of thermal and LA-catalysed Diels-Alder reaction between cyclopentadiene and isopropyl 3-nitroprop-2-enate. Molecules 2023, 28, 5289. [Google Scholar] [CrossRef] [PubMed]
- Woliński, P.; Kącka-Zych, A.; Wróblewska, A.; Wielgus, E.; Dolot, R.; Jasiński, R. Fully selective synthesis of spirocyclic-1,2-oxazine N-oxides via non-catalysed Hetero Diels-Alder reactions with the participation of cyanofunctionalysed conjugated nitroalkenes. Molecules 2023, 28, 4586. [Google Scholar] [CrossRef] [PubMed]
- Zawadzińska, K.; Gadocha, Z.; Pabian, K.; Wróblewska, A.; Wielgus, E.; Jasiński, R. The First Examples of [3+2] Cycloadditions with the Participation of (E)-3,3,3-Tribromo-1-Nitroprop-1-Ene. Materials 2022, 15, 7584. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzinska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the question of the formation nitro-functionalized 2,4-pyrazole analogs on the basis of nitrylimine molecular systems and 3,3,3-trichloro-1-nitroprop-1-ene. Molecules 2022, 27, 8409. [Google Scholar] [CrossRef]
- Elbouhi, M.; Tabti, K.; Ouabane, M.; Alaqarbeh, M.; Elkamel, K.; Lakhlifi, T.; Sbai, A.; Bouachrine, M. A computational exploration of the antioxidant potential of conjugated quinazolinone Schiff bases. Chem. Heterocycl. Compd. 2024, 60, 627–638. [Google Scholar] [CrossRef]
- Wróbel, D.; Graja, A. Photoinduced electron transfer processes in fullerene–organic chromophore systems. Coord. Chem. Rev. 2011, 255, 2555–2577. [Google Scholar] [CrossRef]
- Baharfar, M.; Hillier, A.C.; Mao, G. Charge-Transfer Complexes: Fundamentals and Advances in Catalysis, Sensing, and Optoelectronic Applications. Adv. Mater. 2024, 36, 2406083. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Aitouna, A.O.; Rossafi, B.; El Abdallaoui, H.E.A.; Zeroual, A. Molecular Electron Denstity Study on the [3+2] cycloaddition reaction between diphenylnitrylimine and cinnamaldehyde. Sci. Radices 2025, 4, 18–28. [Google Scholar] [CrossRef]
- Woliński, P.; Dreler, E.; Jasiński, R. A new mechanistic insight into the molecular mechanisms of the addition reactions of 2-aryl-3-nitro-2H-chromenes to pyrazoles and cyclopentadienes. New. J. Chem. 2025, 49, 8442–8453. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.J.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 16 Rev A.1; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jasiński, R. A reexamination of molecular mechanism of Diels-Alder reaction between tetrafluoroethene and cyclopentadiene. React. Kinet. Mech. Catal. 2016, 119, 49–57. [Google Scholar] [CrossRef]
- Jasiński, R.; Kącka, A. A polar nature of benzoic acids extrusion from nitroalkyl benzoates: DFT mechanistic study. J. Mol. Model. 2015, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Radices 2023, 2, 1–24. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Jasiński, R. A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalyzed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett. 2015, 56, 532–535. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016.
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization. In Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: Clifton Park, NY, USA, 2015. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | structure | R | σR [45] |
| 1a | H | 0.00 | |
| 1b | NMe2 | −0.72 | |
| 1c | NO2 | 0.78 |
| Reaction | Solvent | Transition | ΔH | ΔS | ΔG |
|---|---|---|---|---|---|
| 1a + 2 | n-Pentane | 1a + 2→MCB | −11.5 | −44.9 | 1.8 |
| 1a + 2→TS1B | 3.8 | −55.2 | 20.3 | ||
| 1a + 2→I | −1.3 | −51.1 | 13.9 | ||
| 1a + 2→TS2B | −1.3 | −51.7 | 14.1 | ||
| 1a + 2→3a | −44.3 | −52.6 | −28.6 | ||
| 1a + 2→TSrot | 0.8 | −53.8 | 16.9 | ||
| 1a + 2→I′ | −0.6 | −53.4 | 15.4 | ||
| 1a + 2→TSC | 0.9 | −53.5 | 16.8 | ||
| 1a + 2→4a | −43.7 | −55.3 | −27.2 | ||
| Acetone | 1a + 2→MCB | −9.6 | −44.5 | 3.6 | |
| 1a + 2→TS1B | 2.0 | −53.9 | 18.1 | ||
| 1a + 2→I | −9.5 | −50.5 | 5.6 | ||
| 1a + 2→TS2B | −7.6 | −51.1 | 7.6 | ||
| 1a + 2→3a | −42.2 | −52.5 | −26.6 | ||
| 1a + 2→TSrot | −7.3 | −53.1 | 8.5 | ||
| 1a + 2→I′ | −8.2 | −53.9 | 7.8 | ||
| 1a + 2→TSC | −4.4 | −55.1 | 12.0 | ||
| 1a + 2→4a | −41.9 | −55.2 | −25.5 | ||
| Nitromethane | 1a + 2→MCB | −9.5 | −44.6 | 3.8 | |
| 1a + 2→TS1B | 1.9 | −54.4 | 18.1 | ||
| 1a + 2→I | −10.1 | −49.2 | 4.6 | ||
| 1a + 2→TS2B | −8.1 | −51.4 | 7.3 | ||
| 1a + 2→3a | −42.2 | −51.5 | −26.8 | ||
| 1a + 2→TSrot | −8.0 | −52.2 | 7.5 | ||
| 1a + 2→I′ | −8.8 | −53.1 | 7.0 | ||
| 1a + 2→TSC | −4.9 | −55.0 | 11.5 | ||
| 1a + 2→4a | −41.9 | −55.2 | −25.4 | ||
| 1b + 2 | n-Pentane | 1b + 2→MCB | −12.7 | −45.9 | 0.9 |
| 1b + 2→TS1B | 6.0 | −55.3 | 22.4 | ||
| 1b + 2→I | 1.5 | −53.3 | 17.4 | ||
| 1b + 2→TS2B | 1.3 | −54.7 | 17.6 | ||
| 1b + 2→3b | −41.2 | −54.0 | −25.1 | ||
| 1c + 2 | n-Pentane | 1c + 2→MCB | −14.5 | −47.7 | −0.3 |
| 1c + 2→TS1B | 1.1 | −54.4 | 17.3 | ||
| 1c + 2→I | −4.5 | −53.6 | 11.4 | ||
| 1c + 2→TS2B | −4.1 | −54.0 | 12.0 | ||
| 1c + 2→3c | −46.8 | −53.3 | −31.0 |
| Reaction | Solvent | Structure | Interatomic Distances r [Å] | Bond Development l | GEDT | ||||
|---|---|---|---|---|---|---|---|---|---|
| C1-C2 | C2-C3 | C3-C4 | C4-C1 | C2-C3 | C4-C1 | [e] | |||
| 1 + 2a | n-Pentane | 2 | 1.210 | ||||||
| 1a | 1.332 | ||||||||
| MCB | 1.211 | 3.209 | 1.333 | 3.336 | 0.00 | ||||
| TS1B | 1.251 | 1.975 | 1.404 | 2.905 | 0.737 | 0.68 | |||
| I | 1.288 | 1.564 | 1.489 | 2.756 | 0.95 | ||||
| TS2B | 1.308 | 1.525 | 1.506 | 2.426 | 0.384 | 0.55 | |||
| 3a | 1.356 | 1.520 | 1.571 | 1.501 | |||||
| TSrot | 1.288 | 1.554 | 1.490 | 2.757 | 0.75 | ||||
| I′ | 1.298 | 1.541 | 1.541 | 2.688 | 0.52 | ||||
| TSC | 1.308 | 1.521 | 1.512 | 2.418 | 0.388 | 0.51 | |||
| 4a | 1.358 | 1.520 | 1.580 | 1.499 | |||||
| Acetone | 2 | 1.213 | |||||||
| 1a | 1.334 | ||||||||
| MCB | 1.211 | 3.185 | 1.335 | 3.295 | 0.00 | ||||
| TS1B | 1.245 | 2.077 | 1.397 | 2.923 | 0.654 | 0.64 | |||
| I | 1.294 | 1.543 | 1.496 | 2.759 | 0.80 | ||||
| TS2B | 1.312 | 1.515 | 1.511 | 2.312 | 0.460 | 0.68 | |||
| 3a | 1.358 | 1.521 | 1.571 | 1.502 | |||||
| TSrot | 1.295 | 1.543 | 1.497 | 2.720 | 0.60 | ||||
| I′ | 1.296 | 1.532 | 1.502 | 2.797 | 0.62 | ||||
| TSC | 1.308 | 1.521 | 1.512 | 2.418 | 0.388 | 0.61 | |||
| 4a | 1.361 | 1.520 | 1.580 | 1.500 | |||||
| Nitromethane | 2 | 1.213 | |||||||
| 1a | 1.334 | ||||||||
| MCB | 1.211 | 3.182 | 1.335 | 3.293 | 0.00 | ||||
| TS1B | 1.245 | 2.084 | 1.396 | 2.924 | 0.649 | 0.64 | |||
| I | 1.294 | 1.542 | 1.496 | 2.758 | 0.80 | ||||
| TS2B | 1.312 | 1.514 | 1.512 | 2.301 | 0.468 | 0.71 | |||
| 3a | 1.358 | 1.521 | 1.571 | 1.502 | |||||
| TSrot | 1.295 | 1.542 | 1.497 | 2.722 | 0.60 | ||||
| I′ | 1.296 | 1.532 | 1.502 | 2.800 | 0.63 | ||||
| TSC | 1.313 | 1.513 | 1.517 | 2.277 | 0.481 | 0.61 | |||
| 4a | 1.361 | 1.520 | 1.580 | 1.500 | |||||
| 1 + 2b | n-Pentane | 1b | 1.338 | ||||||
| MCB | 1.211 | 3.442 | 1.340 | 3.537 | 0.00 | ||||
| TS1B | 1.252 | 1.960 | 1.410 | 2.900 | 0.749 | 0.69 | |||
| I | 1.288 | 1.567 | 1.490 | 2.733 | 1.07 | ||||
| TS2B | 1.307 | 1.528 | 1.506 | 2.450 | 0.367 | 0.50 | |||
| 3b | 1.356 | 1.521 | 1.574 | 1.501 | |||||
| 1 + 2c | n-Pentane | 1c | 1.329 | ||||||
| MCB | 1.211 | 3.425 | 1.330 | 3.410 | 0.00 | ||||
| TS1B | 1.252 | 1.976 | 1.402 | 2.917 | 0.730 | 0.73 | |||
| I | 1.294 | 1.556 | 1.492 | 2.715 | 0.64 | ||||
| TS2B | 1.309 | 1.524 | 1.506 | 2.424 | 0.386 | 0.39 | |||
| 3c | 1.357 | 1.519 | 1.569 | 1.501 | |||||
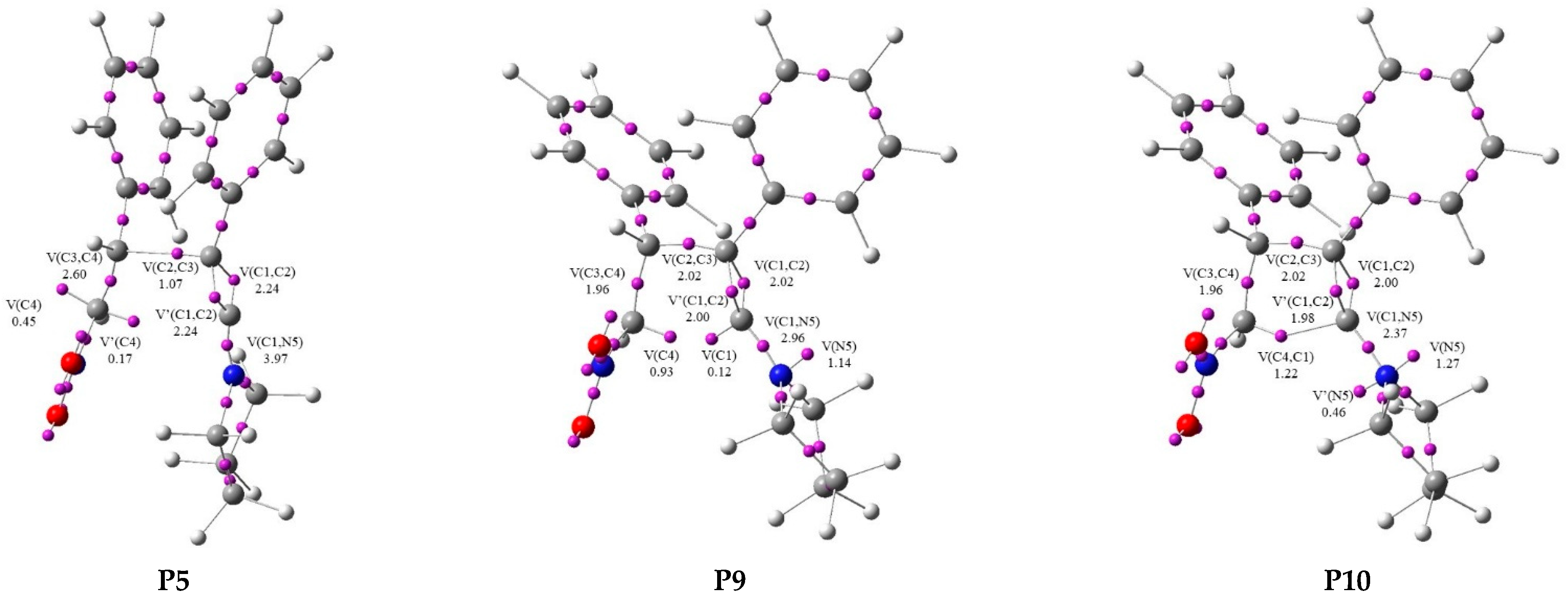
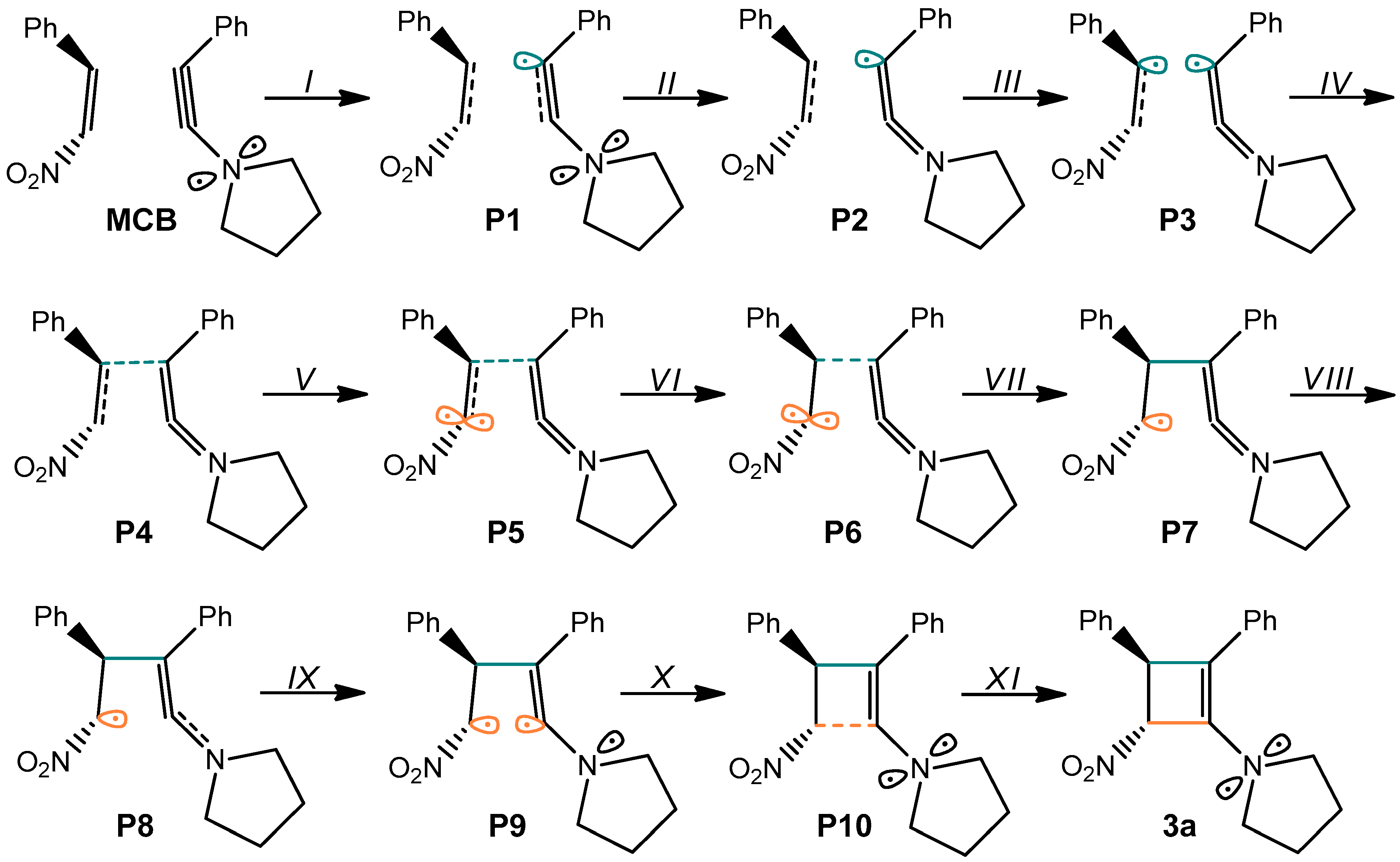
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Points | 1a | 2 | MCB | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | 3a |
| Phases | I | II | III | IV | V | VI | VII | VIII | IX | X | XI | |||
| d(C2–C3) | 2.681 | 2.417 | 2.125 | 2.098 | 2.057 | 2.044 | 1.863 | 1.519 | 1.513 | 1.511 | 1.511 | 1.514 | ||
| d(C4–C1) | 3.076 | 2.994 | 2.931 | 2.926 | 2.919 | 2.917 | 2.879 | 2.389 | 2.247 | 2.103 | 2.055 | 1.622 | ||
| ΔE a | −9.0 | −5.3 | 0.4 | 2.5 | −1.3 | −6.7 | −9.5 | −7.5 | −14.2 | −25.3 | −36.8 | −41.6 | ||
| V(C1,C2) | 2.80 | 2.87 | 2.58 | 2.30 | 2.28 | 2.25 | 2.24 | 2.15 | 1.98 | 1.99 | 2.02 | 2.00 | 1.85 | |
| V′(C1,C2) | 2.79 | 2.65 | 2.54 | 2.29 | 2.27 | 2.25 | 2.24 | 2.14 | 1.98 | 1.99 | 2.00 | 1.98 | 1.82 | |
| V(C1) | 0.12 | |||||||||||||
| V(C2) | 0.38 | 0.84 | 0.87 | |||||||||||
| V(C3) | 0.08 | |||||||||||||
| V(C2,C3) | 1.04 | 1.07 | 1.40 | 2.00 | 2.02 | 2.02 | 2.02 | 1.99 | ||||||
| V(C3,C4) | 1.74 | 3.41 | 3.38 | 3.34 | 3.26 | 3.22 | 2.60 | 2.31 | 1.98 | 1.97 | 1.96 | 1.96 | 1.89 | |
| V′(C3,C4) | 1.74 | |||||||||||||
| V(C4) | 0.45 | 0.55 | 0.66 | 0.76 | 0.93 | |||||||||
| V′(C4) | 0.17 | 0.25 | ||||||||||||
| V(C4,C1) | 1.22 | 1.95 | ||||||||||||
| V(C1,N5) | 2.10 | 2.22 | 2.34 | 4.03 | 4.02 | 3.99 | 3.97 | 2.00 | 2.00 | 3.98 | 2.96 | 2.37 | 1.84 | |
| V′(C1,N5) | 1.87 | 1.88 | ||||||||||||
| V(N5) | 1.33 | 1.34 | 1.14 | 1.14 | 1.27 | 1.87 | ||||||||
| V′(N5) | 0.98 | 0.74 | 0.74 | 0.46 | 0.65 | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasiński, R.; Kącka-Zych, A. The Puzzle of the New Type of Intermediate in the Course of [2 + 2] Cycloaddition with the Participation of Conjugated Nitroalkenes: MEDT Computational Study. Molecules 2025, 30, 2410. https://doi.org/10.3390/molecules30112410
Jasiński R, Kącka-Zych A. The Puzzle of the New Type of Intermediate in the Course of [2 + 2] Cycloaddition with the Participation of Conjugated Nitroalkenes: MEDT Computational Study. Molecules. 2025; 30(11):2410. https://doi.org/10.3390/molecules30112410
Chicago/Turabian StyleJasiński, Radomir, and Agnieszka Kącka-Zych. 2025. "The Puzzle of the New Type of Intermediate in the Course of [2 + 2] Cycloaddition with the Participation of Conjugated Nitroalkenes: MEDT Computational Study" Molecules 30, no. 11: 2410. https://doi.org/10.3390/molecules30112410
APA StyleJasiński, R., & Kącka-Zych, A. (2025). The Puzzle of the New Type of Intermediate in the Course of [2 + 2] Cycloaddition with the Participation of Conjugated Nitroalkenes: MEDT Computational Study. Molecules, 30(11), 2410. https://doi.org/10.3390/molecules30112410