


Gating Mechanism for Biased Agonism at Angiotensin II Type 1 Receptors

 ,
,  ,
, 
Abstract
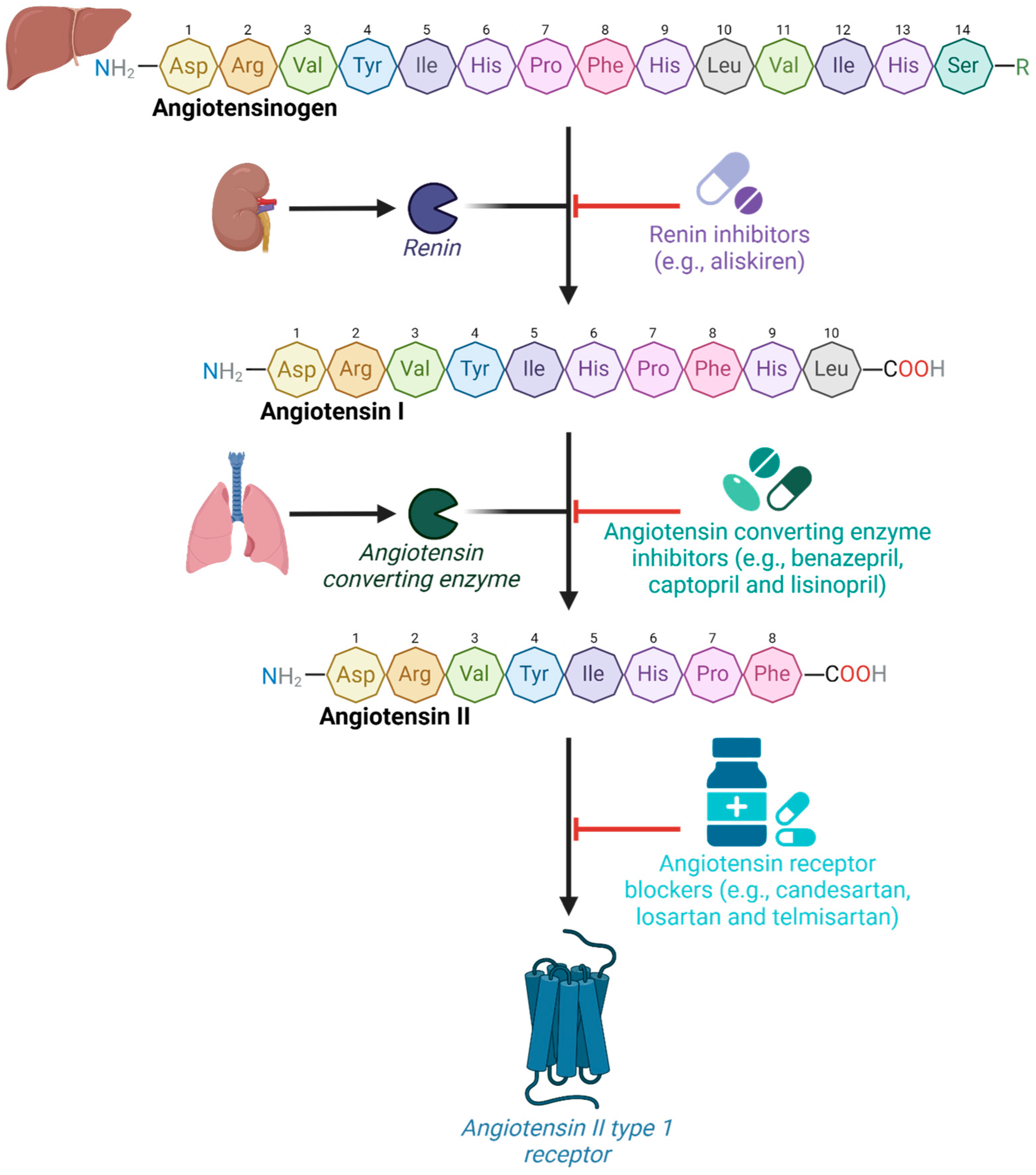
1. Introduction
2. Results and Discussion
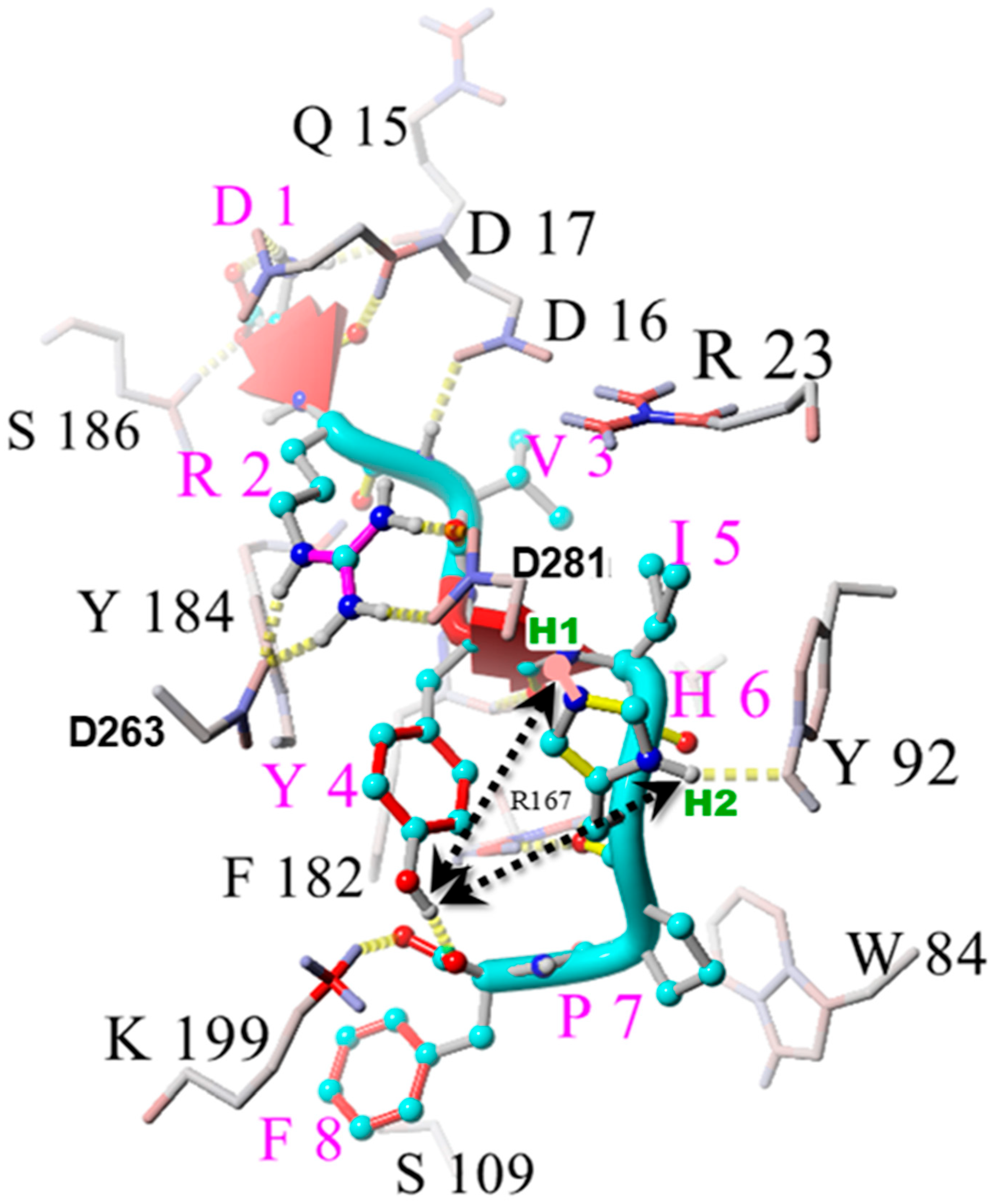
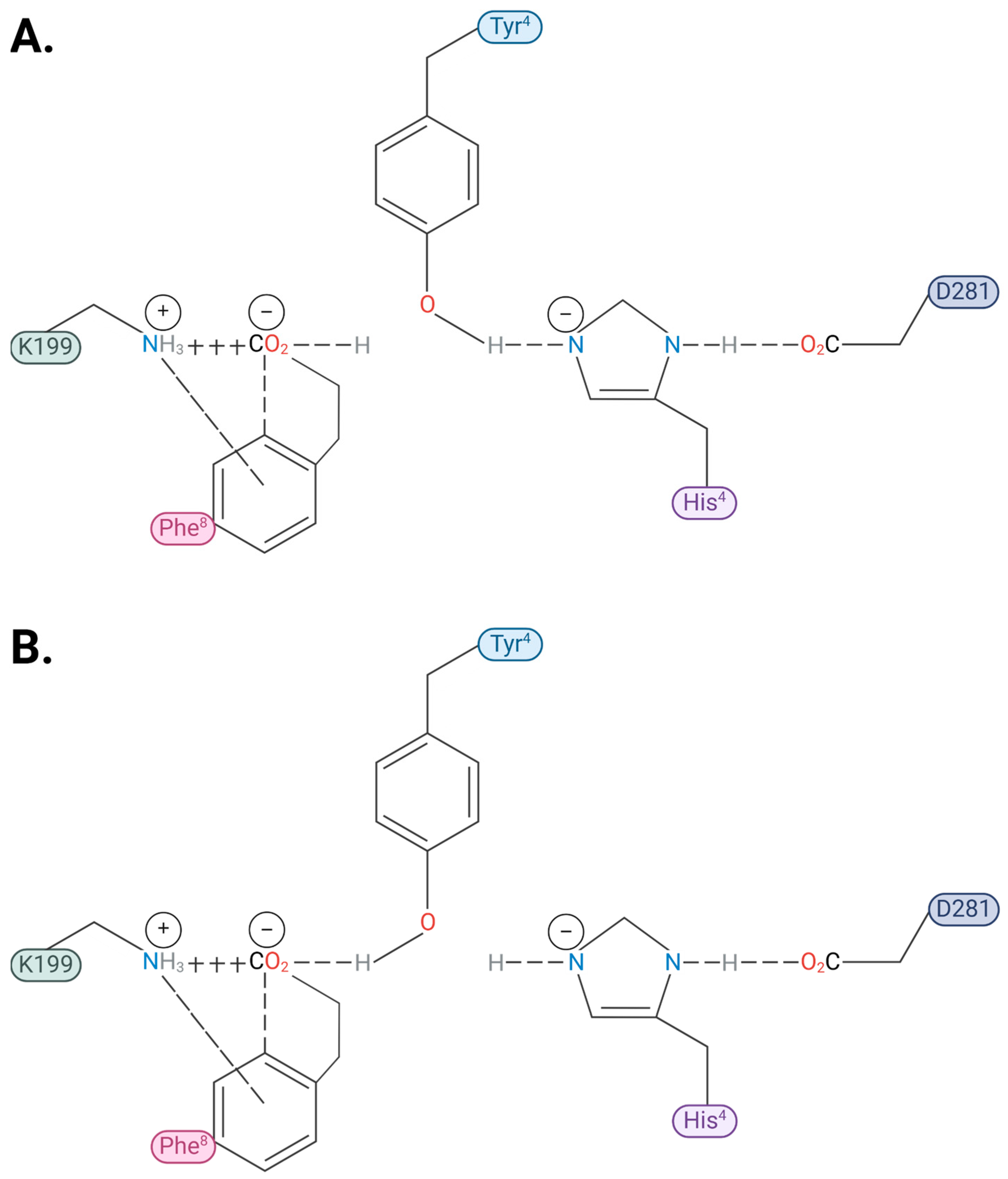
2.1. Charge Transfer Interactions in AngII Before and After Receptor Binding
2.2. Analysis by Amino Acid Position in AngII
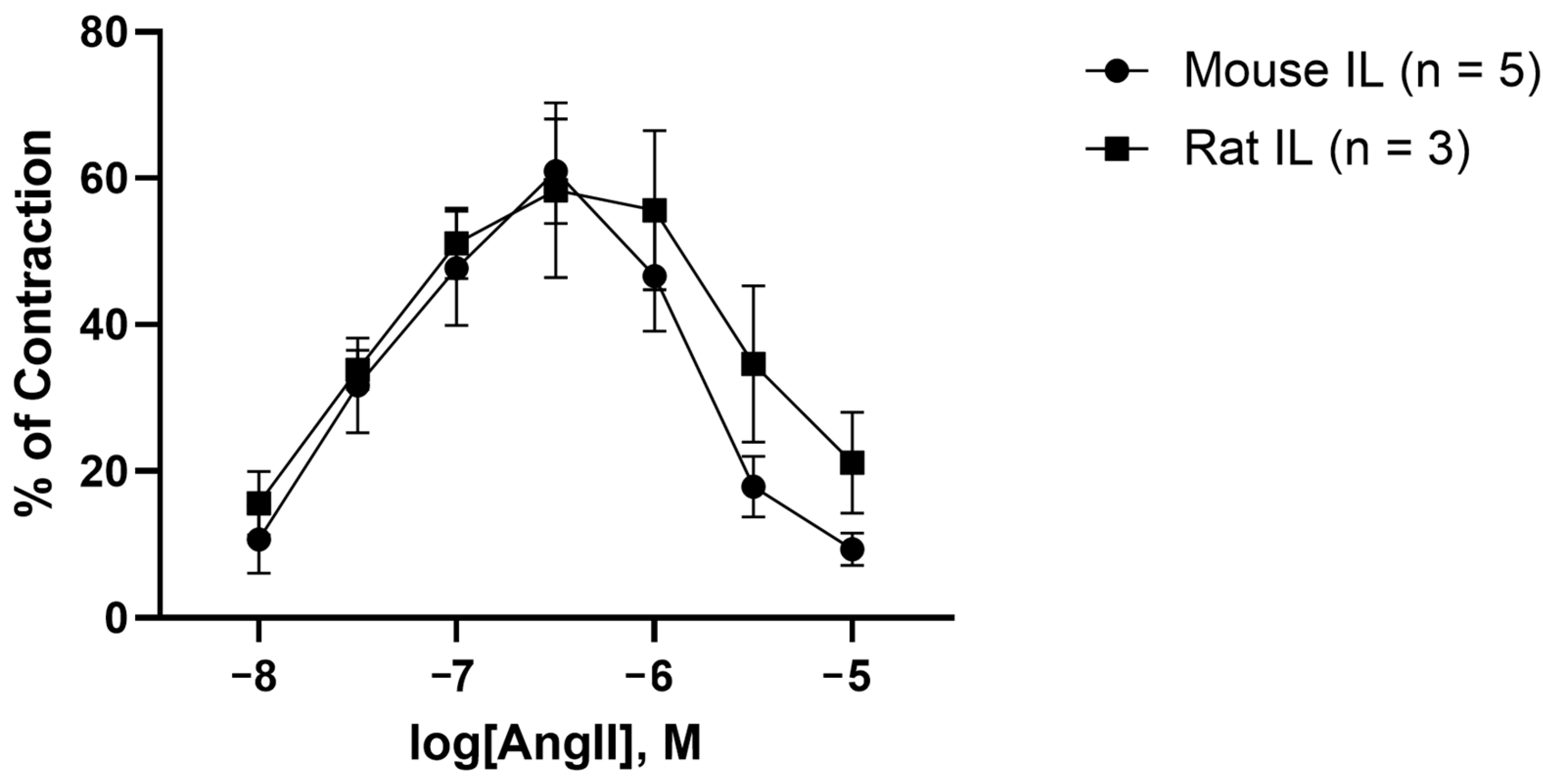
2.3. AngII Causes Biphasic Vasoactive Responses in Mouse and Rat Iliac Arteries
2.4. Tachyphylaxis, Receptor Desensitization, and Inverse Agonism by ARB Sartans
2.5. Sartans: Nonpeptide Mimetics of AngII
2.6. Bisartans: Second-Generation Nonpeptide Mimetics of AngII
3. Materials and Methods
3.1. Steered Molecular Dynamics
3.2. Calculation of Hydrogen Dissociation Energies
3.3. Ex Vivo Animal Experiments
3.3.1. Materials
3.3.2. Animals and Ethical Approval
3.3.3. Anesthesia, Humane Dispatchment, and Artery Collection
3.3.4. Isometric Tension Myography Studies
3.3.5. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AngII | Angiotensin II |
| ARB | Angiotensin receptor blocker |
| Arg | Arginine |
| Asp | Aspartic acid |
| AT1R | Angiotensin II type I receptor |
| Cat# | Catalog number |
| Cl− | Chloride |
| CRS | Charge relay system |
| H | Hydrogen |
| H-bond | Hydrogen bond |
| His | Histidine |
| HisNNH or HNNHis | His imidazole nitrogen (neutral, protonated) |
| Hr | Hour |
| Ile | Isoleucine |
| IL | Iliac artery |
| K+ | Potassium |
| Leu | Leucine |
| Lys | Lysine |
| N | Nitrogen |
| Na+ | Sodium |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| O | Oxygen |
| OH | Hydroxyl |
| Phe | Phenylalanine |
| PheCCOO−1e | Phe with deprotonated carboxylate group |
| Pro | Proline |
| R | Sidechain |
| RAS | Renin–angiotensin system |
| ROS | Reactive oxygen species |
| Sar | Sarcosine |
| SEM | Standard error of mean |
| Ser | Serine |
| SMD | Steered molecular dynamics |
| Tyr | Tyrosine |
| TyrO−1e | Tyr with deprotonated phenol OH group |
| UHF | Unrestricted Hartree–Fock |
| Val | Valine |
| VUAEC | Victoria University Animal Ethics Committee |
| Wk | Week |
| Å | Angstrom |
| β-arrestin | Beta-arrestin |
| NRG | Energy |
References
- Ruan, Y.; Yu, Y.; Wu, M.; Jiang, Y.; Qiu, Y.; Ruan, S. The renin-angiotensin-aldosterone system: An old tree sprouts new shoots. Cell. Signal. 2024, 124, 111426. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Barreto-Chaves, M.L. Beta-arrestins in the context of cardiovascular diseases: Focusing on angiotensin II type 1 receptor (AT1R). Cell. Signal. 2022, 92, 110253. [Google Scholar] [CrossRef]
- Tóth, A.D.; Turu, G.; Hunyady, L.; Balla, A. Novel mechanisms of G-protein-coupled receptors functions: AT1 angiotensin receptor acts as a signaling hub and focal point of receptor cross-talk. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Groban, L.; Wang, H.; Sun, X.; VonCannon, J.L.; Wright, K.N.; Ahmad, S. The renin–angiotensin system biomolecular cascade: A 2022 update of newer insights and concepts. Kidney Int. Suppl. 2022, 12, 36–47. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed]
- McFall, A.; Nicklin, S.A.; Work, L.M. The counter regulatory axis of the renin angiotensin system in the brain and ischaemic stroke: Insight from preclinical stroke studies and therapeutic potential. Cell. Signal. 2020, 76, 109809. [Google Scholar] [CrossRef]
- Wu, C.-H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-angiotensin system and cardiovascular functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef]
- Eguchi, S.; Kawai, T.; Scalia, R.; Rizzo, V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension 2018, 71, 804–810. [Google Scholar] [CrossRef]
- Dominici, F.P.; Gironacci, M.M.; Narvaez Pardo, J.A. Therapeutic opportunities in targeting the protective arm of the renin-angiotensin system to improve insulin sensitivity: A mechanistic review. Hypertens. Res. 2024, 12, 3397–3408. [Google Scholar] [CrossRef]
- Li, X.C.; Zhang, J.; Zhuo, J.L. The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol. Res. 2017, 125, 21–38. [Google Scholar] [CrossRef]
- Balakumar, P.; Jagadeesh, G. A century old renin–angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell. Signal. 2014, 26, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Ramya, K.; Suresh, R.; Kumar, H.Y.; Kumar, B.P.; Murthy, N.S. Decades-old renin inhibitors are still struggling to find a niche in antihypertensive therapy. A fleeting look at the old and the promising new molecules. Bioorg. Med. Chem. 2020, 28, 115466. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Suchard, M.A.; Krumholz, H.M.; Schuemie, M.J.; Shea, S.; Duke, J.; Pratt, N.; Reich, C.G.; Madigan, D.; You, S.C. Comparative first-line effectiveness and safety of ACE (angiotensin-converting enzyme) inhibitors and angiotensin receptor blockers: A multinational cohort study. Hypertension 2021, 78, 591–603. [Google Scholar] [CrossRef]
- Kahlon, T.; Carlisle, S.; Otero Mostacero, D.; Williams, N.; Trainor, P.; DeFilippis, A.P. Angiotensinogen: More than its downstream products: Evidence from population studies and novel therapeutics. Heart Fail. 2022, 10, 699–713. [Google Scholar]
- Sequeira-Lopez, M.L.S.; Gomez, R.A. Renin cells, the kidney, and hypertension. Circ. Res. 2021, 128, 887–907. [Google Scholar] [CrossRef]
- Bhandari, S.; Mehta, S.; Khwaja, A.; Cleland, J.G.; Ives, N.; Brettell, E.; Chadburn, M.; Cockwell, P. Renin–angiotensin system inhibition in advanced chronic kidney disease. N. Engl. J. Med. 2022, 387, 2021–2032. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-angiotensin system in pathogenesis of atherosclerosis and treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef]
- Zhu, D.; Shi, J.; Zhang, Y.; Wang, B.; Liu, W.; Chen, Z.; Tong, Q. Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS ONE 2011, 6, e16037. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Davisson, R.L. Angiotensin-II, the brain, and hypertension: An update. Hypertension 2015, 66, 920–926. [Google Scholar] [CrossRef]
- Goossens, G.; Blaak, E.; Arner, P.; Saris, W.; Van Baak, M. Angiotensin II: A hormone that affects lipid metabolism in adipose tissue. Int. J. Obes. 2007, 31, 382–384. [Google Scholar] [CrossRef]
- Tyurin-Kuzmin, P.A.; Kalinina, N.I.; Kulebyakin, K.Y.; Balatskiy, A.V.; Sysoeva, V.Y.; Tkachuk, V.A. Angiotensin receptor subtypes regulate adipose tissue renewal and remodelling. FEBS J. 2020, 287, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, L.; Sopontammarak, B.; Menikdiwela, K.R.; Moustaid-Moussa, N. Endoplasmic reticulum (ER) stress in part mediates effects of angiotensin II in pancreatic beta cells. Diabetes Metab. Syndr. Obes. 2020, 13, 2843–2853. [Google Scholar] [CrossRef]
- Shi, Z.; Stornetta, R.L.; Stornetta, D.S.; Abbott, S.B.; Brooks, V.L. The arcuate nucleus: A site of synergism between Angiotensin II and leptin to increase sympathetic nerve activity and blood pressure in rats. Neurosci. Lett. 2022, 785, 136773. [Google Scholar] [CrossRef]
- Garg, M.; Angus, P.W.; Burrell, L.M.; Herath, C.; Gibson, P.R.; Lubel, J.S. The pathophysiological roles of the renin–angiotensin system in the gastrointestinal tract. Aliment. Pharmacol. Ther. 2012, 35, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Ojeda, Z.A.; Trejo-Moreno, C.; Ferat-Osorio, E.; Méndez-Martínez, M.; Fragoso, G.; Rosas-Salgado, G. Role of Angiotensin II in Non-Alcoholic Steatosis Development. Arch. Med. Res. 2024, 55, 102986. [Google Scholar] [CrossRef]
- Ahmadian, E.; Pennefather, P.S.; Eftekhari, A.; Heidari, R.; Eghbal, M.A. Role of renin-angiotensin system in liver diseases: An outline on the potential therapeutic points of intervention. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1279–1288. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Penninger, J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006, 6, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, H.; Lan, T.; Jia, R.; Cao, M.; Zhou, L.; Zhao, Z.; Pan, W. Physiological and pathological roles of Ang II and Ang-(1-7) in the female reproductive system. Front. Endocrinol. 2022, 13, 1080285. [Google Scholar] [CrossRef]
- Irani, R.A.; Xia, Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. In Seminars in Nephrology; WB Saunders: Philadelphia, PA, USA, 2011; pp. 47–58. [Google Scholar]
- Gianzo, M.; Subirán, N. Regulation of male fertility by the renin-angiotensin system. Int. J. Mol. Sci. 2020, 21, 7943. [Google Scholar] [CrossRef]
- Guimond, M.-O.; Hallberg, M.; Gallo-Payet, N.; Wallinder, C. Saralasin and sarile are AT2 receptor agonists. ACS Med. Chem. Lett. 2014, 5, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Karnik, S.S. Structural perspectives on the mechanism of signal activation, ligand selectivity and allosteric modulation in angiotensin receptors: IUPHAR Review 34. Br. J. Pharmacol. 2022, 179, 4461–4472. [Google Scholar] [CrossRef]
- Asada, H.; Inoue, A.; Kadji, F.M.N.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T. The crystal structure of angiotensin II type 2 receptor with endogenous peptide hormone. Structure 2020, 28, 418–425.e414. [Google Scholar] [CrossRef]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J. Molecular mechanism of biased signaling in a prototypical G-protein-coupled receptor. Biophys. J. 2020, 118, 162a. [Google Scholar] [CrossRef]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.; Suomivuori, C.-M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef]
- Ma, Z.; Viswanathan, G.; Sellig, M.; Jassal, C.; Choi, I.; Garikipati, A.; Xiong, X.; Nazo, N.; Rajagopal, S. β-Arrestin–Mediated Angiotensin II Type 1 Receptor Activation Promotes Pulmonary Vascular Remodeling in Pulmonary Hypertension. Basic Transl. Sci. 2021, 6, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, N.M.; Nakagawa, P.; Grobe, J.L.; Sigmund, C.D. Insights into the role of angiotensin-II AT1 receptor-dependent β-arrestin signaling in cardiovascular disease. Hypertension 2024, 81, 6–16. [Google Scholar] [CrossRef]
- Zamel, I.A.; Palakkott, A.; Ashraf, A.; Iratni, R.; Ayoub, M.A. Interplay between angiotensin II type 1 receptor and thrombin receptor revealed by bioluminescence resonance energy transfer assay. Front. Pharmacol. 2020, 11, 1283. [Google Scholar] [CrossRef]
- Duarte, D.A.; Parreiras-e-Silva, L.T.; Oliveira, E.B.; Bouvier, M.; Costa-Neto, C.M. Angiotensin II type 1 receptor tachyphylaxis is defined by agonist residence time. Hypertension 2022, 79, 115–125. [Google Scholar] [CrossRef]
- Kee, T.R.; Khan, S.A.; Neidhart, M.B.; Masters, B.M.; Zhao, V.K.; Kim, Y.K.; McGill Percy, K.C.; Woo, J.-A.A. The multifaceted functions of β-arrestins and their therapeutic potential in neurodegenerative diseases. Exp. Mol. Med. 2024, 56, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Erol, I.; Cosut, B.; Durdagi, S. Toward understanding the impact of dimerization interfaces in angiotensin II type 1 receptor. J. Chem. Inf. Model. 2019, 59, 4314–4327. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 2019, 176, 479–490.e412. [Google Scholar] [CrossRef]
- Moore, G.J.; Scanlon, M.N. Methods for analyzing and interpreting cooperativity in dose-response curves--I. Antagonist effects on angiotensin receptors in smooth muscle. Gen. Pharmacol. 1989, 20, 193–198. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Bigam, G.; Zhou, N.; Moore, G.J.I. 1H-NMR studies of [Sar1] angiotensin II conformation by nuclear Overhauser effect spectroscopy in the rotating frame (ROESY): Clustering of the aromatic rings in dimethylsulfoxide. Peptides 1990, 11, 359–366. [Google Scholar] [CrossRef]
- Turner, R.J.; Matsoukas, J.M.; Moore, G.J. Fluorescence properties of angiotensin II analogues in receptor-simulating environments: Relationship between tyrosinate fluorescence lifetime and biological activity. Biochim. Biophys. Acta (BBA)-Biomembr. 1991, 1065, 21–28. [Google Scholar] [CrossRef]
- MOORE, G.J. Kinetics of acetylation-deacetylation of angiotensin II: Intramolecular interactions of the tyrosine and histidine side-chains. Int. J. Pept. Protein Res. 1985, 26, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.; Chontzopoulou, E.; Routsi, E.A.; Stavrakaki, I.G.; Petsas, E.; Zoupanou, N.; Kakava, M.G.; Tzeli, D.; Mavromoustakos, T.; Kiriakidi, S. Exploring Hypertension: The Role of AT1 Receptors, Sartans, and Lipid Bilayers. ACS Omega 2024, 45, 44876–44890. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zaidi, S.A.; Xu, L.; Zhan, Y.; Chen, A.; Guo, J.; Huang, X.P.; Roth, B.L.; Katritch, V. Structural insights into angiotensin receptor signaling modulation by balanced and biased agonists. EMBO J. 2023, 42, e112940. [Google Scholar] [CrossRef]
- Moore, G.J.; Smitht, J.R.; Baylis, B.W.; Matsoukas, J.M. Design and pharmacology of peptide mimetics. Adv. Pharmacol. 1995, 33, 91–141. [Google Scholar]
- Fowler, P.W.; Moore, G.J. Calculation of the magnitude and orientation of electrostatic interactions between small aromatic rings in peptides and proteins: Implications for angiotensin II. Biochem. Biophys. Res. Commun. 1988, 153, 1296–1300. [Google Scholar] [CrossRef]
- Bovy, P.R.; Getman, D.P.; Matsoukas, J.M.; Moore, G.J. Influence of polyfluorination of the phenylalanine ring of angiotensin II on conformation and biological activity. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1991, 1079, 23–28. [Google Scholar] [CrossRef]
- Hondrelis, J.; Matsoukas, J.; Cordopatis, P.; Ganter, R.C.; Franklin, K.J.; Moore, G.J. Synthesis and biological activities of angiotensin II and sarmesin analogues containing cyclohexylalanine. Int. J. Pept. Protein Res. 1991, 37, 21–26. [Google Scholar] [CrossRef]
- van Geel, P.P.; Pinto, Y.M.; Voors, A.A.; Buikema, H.; Oosterga, M.; Crijns, H.J.; van Gilst, W.H. Angiotensin II type 1 receptor A1166C gene polymorphism is associated with an increased response to angiotensin II in human arteries. Hypertension 2000, 35, 717–721. [Google Scholar] [CrossRef]
- Voors, A.A.; van Geel, P.P.; Buikema, H.; Oosterga, M.; van Veldhuisen, D.J.; van Gilst, W.H. High angiotensin II responsiveness is associated with decreased endothelium-dependent relaxation in human arteries. J. Renin-Angiotensin-Aldosterone Syst. 2005, 6, 145–150. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, P.; Asselbergs, F.W.; Buikema, H.; Voors, A.A.; van Veldhuisen, D.J.; van Gilst, W.H. Effects of C-reactive protein and cholesterol on responsiveness in vitro of the internal thoracic artery to angiotensin II in patients having coronary artery bypass grafting. Am. J. Cardiol. 2006, 98, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dirksen, W.P.; Babu, G.J.; Periasamy, M. Differential vasoconstrictions induced by angiotensin II: Role of AT1 and AT2 receptors in isolated C57BL/6J mouse blood vessels. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, H2797–H2803. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Dirksen, W.P.; Morris, M.; Periasamy, M. AT1b receptor predominantly mediates contractions in major mouse blood vessels. Circ. Res. 2003, 93, 1089–1094. [Google Scholar] [CrossRef]
- Xie-Zukauskas, H.; Das, J.; Short, B.L.; Gutkind, J.S.; Ray, P.E. Heparin inhibits angiotensin II-induced vasoconstriction on isolated mouse mesenteric resistance arteries through Rho-A-and PKA-dependent pathways. Vasc. Pharmacol. 2013, 58, 313–318. [Google Scholar] [CrossRef]
- Gadanec, L.K.; McSweeney, K.R.; Kubatka, P.; Caprnda, M.; Gaspar, L.; Prosecky, R.; Dragasek, J.; Kruzliak, P.; Apostolopoulos, V.; Zulli, A. Angiotensin II constricts mouse iliac arteries: Possible mechanism for aortic aneurysms. Mol. Cell. Biochem. 2024, 479, 233–242. [Google Scholar] [CrossRef]
- McSweeney, K.R.; Gadanec, L.K.; Kubatka, P.; Caprnda, M.; Gaspar, L.; Prosecky, R.; Delev, D.; Kruzliak, P.; Apostolopoulos, V.; Zulli, A. Cisplatin treatment reduces contraction to angiotensin II by altering expression of angiotensin II receptors: A pilot study. Mol. Cell. Biochem. 2023, 478, 2907–2916. [Google Scholar] [CrossRef]
- Habiyakare, B.; Alsaadon, H.; Mathai, M.L.; Hayes, A.; Zulli, A. Reduction of angiotensin A and alamandine vasoactivity in the rabbit model of atherogenesis: Differential effects of alamandine and A ng (1–7). Int. J. Exp. Pathol. 2014, 95, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Gadanec, L.K.; Swiderski, J.; Apostolopoulos, V.; Kelaidonis, K.; Vidali, V.P.; Canko, A.; Moore, G.J.; Matsoukas, J.M.; Zulli, A. Existence of quantum pharmacology in Sartans: Evidence in isolated rabbit iliac arteries. Int. J. Mol. Sci. 2023, 24, 17559. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.M.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Kelaidonis, K.; Ligielli, I.; Moschovou, K.; Georgiou, N.; Plotas, P.; Chasapis, C.T. Diminazene aceturate reduces angiotensin II constriction and interacts with the spike protein of severe acute respiratory syndrome coronavirus 2. Biomedicines 2022, 10, 1731. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.J.; Pires, J.M.; Kelaidonis, K.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Matsoukas, J.M. Receptor interactions of angiotensin II and angiotensin receptor blockers—Relevance to COVID-19. Biomolecules 2021, 11, 979. [Google Scholar] [CrossRef]
- Loot, A.E.; Roks, A.J.; Henning, R.H.; Tio, R.A.; Suurmeijer, A.J.; Boomsma, F.; van Gilst, W.H. Angiotensin-(1–7) attenuates the development of heart failure after myocardial infarction in rats. Circulation 2002, 105, 1548–1550. [Google Scholar] [CrossRef]
- Moreira, J.D.; Pernomian, L.; Gomes, M.S.; Pernomian, L.; Moreira, R.P.; do Prado, A.F.; da Silva, C.H.; de Oliveira, A.M. Acute restraint stress increases carotid reactivity in type-I diabetic rats by enhancing Nox4/NADPH oxidase functionality. Eur. J. Pharmacol. 2015, 765, 503–516. [Google Scholar] [CrossRef]
- Pernomian, L.; Gomes, M.S.; Restini, C.B.A.; de Oliveira, A.M. Mas-Mediated Antioxidant Effects Restore the Functionality of Angiotensin Converting Enzyme 2-Angiotensin-(1–7)-Mas Axis in Diabetic Rat Carotid. BioMed Res. Int. 2014, 2014, 640329. [Google Scholar] [CrossRef]
- Bian, J.; Zhang, S.; Yi, M.; Yue, M.; Liu, H. The mechanisms behind decreased internalization of angiotensin II type 1 receptor. Vasc. Pharmacol. 2018, 103, 1–7. [Google Scholar] [CrossRef]
- Turu, G.; Balla, A.; Hunyady, L. The role of β-arrestin proteins in organization of signaling and regulation of the AT1 angiotensin receptor. Front. Endocrinol. 2019, 10, 519. [Google Scholar] [CrossRef]
- Kim, H.J.; Jang, J.H.; Zhang, Y.H.; Yoo, H.Y.; Kim, S.J. Fast relaxation and desensitization of angiotensin II contraction in the pulmonary artery via AT1R and Akt-mediated phosphorylation of muscular eNOS. Pflügers Arch.-Eur. J. Physiol. 2019, 471, 1317–1330. [Google Scholar] [CrossRef]
- Hussain, M.B.; Püntmann, V.O.; Mayr, M.; Khong, T.; Singer, D.R. The role of oxidant stress in angiotensin II-mediated contraction of human resistance arteries in the state of health and the presence of cardiovascular disease. Vasc. Pharmacol. 2006, 45, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Zulli, A.; Hare, D.L.; Buxton, B.F.; Widdop, R.E. Vasoactive role for angiotensin II type 2 receptors in human radial artery. Int. J. Immunopathol. Pharmacol. 2014, 27, 79–85. [Google Scholar] [CrossRef]
- Marchetti, J.; Helou, C.; Chollet, C.; Rajerison, R.; Alhenc-Gelas, F. Regulation of Cardiovascular Signaling by Kinins and Products of Similar Converting Enzyme Systems ACE and non-ACE mediated effect of angiotensin I on intracellular calcium mobilization in rat glomerular arterioles. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1933–H1941. [Google Scholar] [CrossRef] [PubMed]
- Moltzer, E.; Verkuil, A.V.; van Veghel, R.; Danser, A.J.; van Esch, J.H. Effects of Angiotensin Metabolites in the Coronary Vascular Bed of the Spontaneously Hypertensive Rat: Loss of Angiotensin II Type 2 Receptor–Mediated Vasodilation. Hypertension 2010, 55, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, N.; Chen, X.; Gao, Q.; Zhou, X.; Zhang, Y.; Liu, B.; Sun, M.; Xu, Z. Prenatal hypoxia induced dysfunction in cerebral arteries of offspring rats. J. Am. Heart Assoc. 2017, 6, e006630. [Google Scholar] [CrossRef]
- Dinh, Q.N.; Drummond, G.R.; Kemp-Harper, B.K.; Diep, H.; De Silva, T.M.; Kim, H.A.; Vinh, A.; Robertson, A.A.; Cooper, M.A.; Mansell, A. Pressor response to angiotensin II is enhanced in aged mice and associated with inflammation, vasoconstriction and oxidative stress. Aging 2017, 9, 1595. [Google Scholar] [CrossRef]
- Moltzer, E.; te Riet, L.; Swagemakers, S.M.; van Heijningen, P.M.; Vermeij, M.; van Veghel, R.; Bouhuizen, A.M.; van Esch, J.H.; Lankhorst, S.; Ramnath, N.W. Impaired vascular contractility and aortic wall degeneration in fibulin-4 deficient mice: Effect of angiotensin II type 1 (AT1) receptor blockade. PLoS ONE 2011, 6, e23411. [Google Scholar] [CrossRef]
- Suchard, M.A.; Schuemie, M.J.; Krumholz, H.M.; You, S.C.; Chen, R.; Pratt, N.; Reich, C.G.; Duke, J.; Madigan, D.; Hripcsak, G. Comprehensive comparative effectiveness and safety of first-line antihypertensive drug classes: A systematic, multinational, large-scale analysis. Lancet 2019, 394, 1816–1826. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Ntallis, C.; Chasapis, C.T.; Kelaidonis, K.; Matsoukas, M.-T.; Plotas, P.; Apostolopoulos, V.; Moore, G.; Tsiodras, S.; Paraskevis, D. Molecular epidemiology of SARS-CoV-2: The dominant role of arginine in mutations and infectivity. Viruses 2023, 15, 309. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Durdagi, S.; Spyridaki, K.; Tůmová, T.; Slaninová, J.; Giannopoulos, P.; Vlahakos, D.; Liapakis, G.; Mavromoustakos, T. The discovery of new potent non-peptide Angiotensin II AT1 receptor blockers: A concise synthesis, molecular docking studies and biological evaluation of N-substituted 5-butylimidazole derivatives. Eur. J. Med. Chem. 2012, 55, 358–374. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Koukoulitsa, C.; Tůmová, T.; Slaninová, J.; Kalavrizioti, D.; Spyridaki, K.; Afantitis, A.; Melagraki, G.; Siafaka, A. Rational design, efficient syntheses and biological evaluation of N, N′-symmetrically bis-substituted butylimidazole analogs as a new class of potent Angiotensin II receptor blockers. Eur. J. Med. Chem. 2013, 62, 352–370. [Google Scholar] [CrossRef]
- Kelaidonis, K.; Ligielli, I.; Letsios, S.; Vidali, V.P.; Mavromoustakos, T.; Vassilaki, N.; Moore, G.J.; Hoffmann, W.; Węgrzyn, K.; Ridgway, H. Computational and enzymatic studies of sartans in SARS-CoV-2 Spike RBD-ACE2 binding: The role of tetrazole and perspectives as antihypertensive and COVID-19 therapeutics. Int. J. Mol. Sci. 2023, 24, 8454. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Georgiou, N.; Tzeli, D.; Mavromoustakos, T.; Moore, G.J.; Kelaidonis, K.; Matsoukas, M.-T.; Tsiodras, S.; Swiderski, J.; Gadanec, L.K. Density functional theory and enzyme studies support interactions between angiotensin receptor blockers and angiotensin converting enzyme-2: Relevance to coronavirus 2019. Bioorg. Chem. 2024, 150, 107602. [Google Scholar] [CrossRef]
- Ridgway, H.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Hoffmann, W.; Węgrzyn, K.; Vassilaki, N.; Mpekoulis, G.; Zouridakis, M. Novel benzimidazole angiotensin receptor blockers with anti-SARS-CoV-2 activity equipotent to that of nirmatrelvir: Computational and enzymatic studies. Expert Opin. Ther. Targets 2024, 28, 437–459. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, K.; Fatseas, P.; Melissari, E.; Vlahakos, D.; Roumelioti, P.; Mavromoustakos, T.; Mihailescu, S.; Paredes-Carbajal, M.C.; Mascher, D.; Matsoukas, J. Design and synthesis of novel biologically active thrombin receptor non-peptide mimetics based on the pharmacophoric cluster Phe/Arg/NH2 of the Ser42-Phe-Leu-Leu-Arg46 motif sequence: Platelet aggregation and relaxant activities. J. Med. Chem. 2004, 47, 3338–3352. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Panagiotopoulos, D.; Keramida, M.; Mavromoustakos, T.; Yamdagni, R.; Wu, Q.; Moore, G.J.; Saifeddine, M.; Hollenberg, M.D. Synthesis and contractile activities of cyclic thrombin receptor-derived peptide analogues with a Phe-Leu-Leu-Arg motif: Importance of the Phe/Arg relative conformation and the primary amino group for activity. J. Med. Chem. 1996, 39, 3585–3591. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, M.; Apostolopoulos, V.; Matsoukas, J.; Wolf, W.M.; Blaskovich, M.A.; Bojarska, J.; Ziora, Z.M. Tetrazoles: A multi-potent motif in drug design. Eur. J. Med. Chem. 2024, 279, 116870. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Garcia-Revilla, J.; Soto, M.S.; Perryman, R.; Symington, J.; Quarles, C.C.; Healey, D.R.; Guo, Y.; Orta-Vázquez, M.L.; Mateos-Cordero, S. Arginine deprivation alters microglial polarity and synergizes with radiation to eradicate non-arginine-auxotrophic glioblastoma tumors. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Perryman, R. Inhibition of the angiotensin II type 2 receptor AT2R is a novel therapeutic strategy for GBM. Proc. Natl. Acad. Sci. USA 2022, 199, e2116289119. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Mohan, B.; Cui, X.; Chua, K.; Islam, M. Molecular dynamics simulation of gas-phase ozone reactions with sabinene and benzene. J. Mol. Graph. Model. 2017, 74, 241–250. [Google Scholar] [CrossRef]
- Ridgway, H.; Orbell, J.; Gray, S. Chlorination of oxybenzone and prediction of transformation products using non-equilibrium “forced” molecular dynamics. Desalination Water Treat. 2018, 114, 31–50. [Google Scholar] [CrossRef]
- Leusch, F.D.; Neale, P.A.; Busetti, F.; Card, M.; Humpage, A.; Orbell, J.D.; Ridgway, H.F.; Stewart, M.B.; Van de Merwe, J.P.; Escher, B.I. Transformation of endocrine disrupting chemicals, pharmaceutical and personal care products during drinking water disinfection. Sci. Total Environ. 2019, 657, 1480–1490. [Google Scholar] [CrossRef]
- Samain, E.; Bouillier, H.; Rucker-Martin, C.; Mazoit, J.-X.; Marty, J.; Renaud, J.-F.; Dagher, G. Isoflurane Alters Angiotensin II–Induced Ca2+ Mobilization in Aortic Smooth Muscle Cells from Hypertensive Rats: Implication of Cytoskeleton. J. Am. Soc. Anesthesiol. 2002, 97, 642–651. [Google Scholar] [CrossRef]
- Yu, J.; Ogawa, K.; Tokinaga, Y.; Iwahashi, S.; Hatano, Y. The vascular relaxing effects of sevoflurane and isoflurane are more important in hypertensive than in normotensive rats. Can. J. Anesth. 2004, 51, 979. [Google Scholar] [CrossRef]
- Ishikawa, A.; Ogawa, K.; Tokinaga, Y.; Uematsu, N.; Mizumoto, K.; Hatano, Y. The mechanism behind the inhibitory effect of isoflurane on angiotensin II-induced vascular contraction is different from that of sevoflurane. Anesth. Analg. 2007, 105, 97–102. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Artery Type | AngII DRC | AngIImax | Shape of Contraction | Reference |
|---|---|---|---|---|---|
| Patients undergoing coronary artery bypass graft surgery | Internal mammary artery | 10−10.0–10−6.0 M | 10−6.5 M | Parabola | [55] |
| Patients undergoing coronary artery bypass graft surgery | Internal mammary artery | 10−10.0–10−6.0 M | 10−7.0 M | Parabola | [56] |
| Healthy volunteers and patients undergoing elective cardiac revascularization surgery | Resistance arteries isolated from subcutaneous and gluteal adipose tissue | 10−10.0–10−7.5 M | 10−8.0 M and 10−7.5 M | Mix; however, reduced contraction responses to AngII when repeated on the same vessel 1 h after initial DRC, possibly through tachyphylaxis | [73] |
| Patients undergoing coronary artery bypass graft surgery with high and low cholesterol and C-reactive protein levels | Internal thoracic artery | 10−10.0–10−6.0 M | 10−7.0 M | Parabola | [57] |
| Patients undergoing coronary artery bypass graft surgery | Radial artery | 10−10.0–10−6.0 M | 10−6.0 M | Sigmoidal | [74] |
| Healthy and atherogenic male New Zealand White rabbits | Abdominal aorta | 10−9.0–10−6.0 M | 10−7.5 M | Parabola | [63] |
| Healthy male New Zealand White rabbits | Iliac artery | 10−11.0–10−5.0 M | 10−8.0 M | Parabola | [64,65,66] |
| Male Sprague–Dawley rats 1, 3, and 9 wks post-coronary artery ligation-induced myocardial infarction | Aorta | 10−10.0–10−6.0 M | Sham: 10−7.5 M Myocardial infarction: 10−7.5 M and 10−7.0 M | Parabola | [67] |
| Rat | Juxta-medullary glomerular afferent and efferent arterioles | 10−12.5–10−5.5 M | 10−7.0 M | Sigmoidal | [75] |
| Normoglycemic and streptozotocin-induced Type I diabetic male Wistar rats | Carotid artery | 10−9.0–10−3.0 M | 10−8.0 M | Parabola | [69] |
| Male normotensive Wistar and spontaneous hypertensive rats | Abdominal aorta and iliac artery | 10−10.0–10−6.0 M | 10−6.0 M | Sigmoidal | [76] |
| Normoglycemic and streptozotocin-induced Type I diabetic male Wistar rats | Carotid artery | 10−11.0–10−6.0 M | 10−7.0 M | Parabola | [68] |
| Control and prenatal hypoxia exposure Sprague–Dawley rats | Middle cerebral artery | 10−11.0–10−5.0 M | 10−5.0 M | Sigmoidal | [77] |
| Male C57BL/6J mice | Abdominal aorta | 10−10.0–10−6.0 M | 10−6.5 M | Parabola | [58] |
| Female AT1Ra+/+ and AT1Ra−/− mice | Abdominal aorta and femoral artery | 10−10.0–10−6.0 M | 10−6.5 M | Parabola | [59] |
| Male FVB/N mice | Mesenteric artery | 10−10.0–10−7.5 M | 10−8.0 M | Parabola | [60] |
| Old and young male C57BL/6J mice | Mesenteric artery | 10−11.0–10−8.0 M | 10−8.0 M | Sigmoidal | [78] |
| Fibulin-4+/+, Fibulin-4+/-, and Fibullin-4-/- mice | Descending thoracic aorta, abdominal aorta, and iliac artery | 10−10.0–10−6.0 M | 10−6.0 M | Sigmoidal; however, it was reported that descending and ascending thoracic aortae did not respond to AngII at any concentration | [79] |
| Control and cisplatin-induced acute kidney injury male C57/BL6 mice | Brachiocephalic artery, iliac artery, and abdominal and thoracic aorta | 10−8.0–10−5.0 M | 10−6.5 M | Parabola; however, it was reported that the brachiocephalic artery and abdominal and thoracic aorta did not respond to AngII at any concentration | [61,62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, G.J.; Ridgway, H.; Gadanec, L.K.; Apostolopoulos, V.; Zulli, A.; Matsoukas, J.M. Gating Mechanism for Biased Agonism at Angiotensin II Type 1 Receptors. Molecules 2025, 30, 2399. https://doi.org/10.3390/molecules30112399
Moore GJ, Ridgway H, Gadanec LK, Apostolopoulos V, Zulli A, Matsoukas JM. Gating Mechanism for Biased Agonism at Angiotensin II Type 1 Receptors. Molecules. 2025; 30(11):2399. https://doi.org/10.3390/molecules30112399
Chicago/Turabian StyleMoore, Graham J., Harry Ridgway, Laura Kate Gadanec, Vasso Apostolopoulos, Anthony Zulli, and John M. Matsoukas. 2025. "Gating Mechanism for Biased Agonism at Angiotensin II Type 1 Receptors" Molecules 30, no. 11: 2399. https://doi.org/10.3390/molecules30112399
APA StyleMoore, G. J., Ridgway, H., Gadanec, L. K., Apostolopoulos, V., Zulli, A., & Matsoukas, J. M. (2025). Gating Mechanism for Biased Agonism at Angiotensin II Type 1 Receptors. Molecules, 30(11), 2399. https://doi.org/10.3390/molecules30112399