
Enhancing Oral Absorption of Quercetin Through Multifactorial Synergies in Crystal Dispersion Systems

Abstract

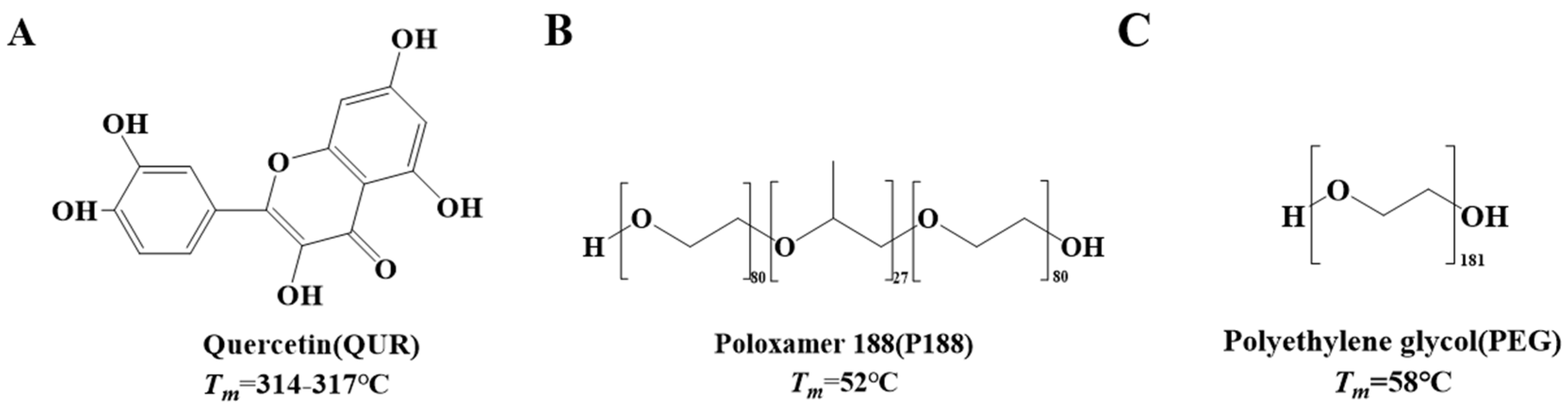
1. Introduction
2. Results and Discussion
2.1. Determination of Solubility Parameter and Interaction Parameter χ Value
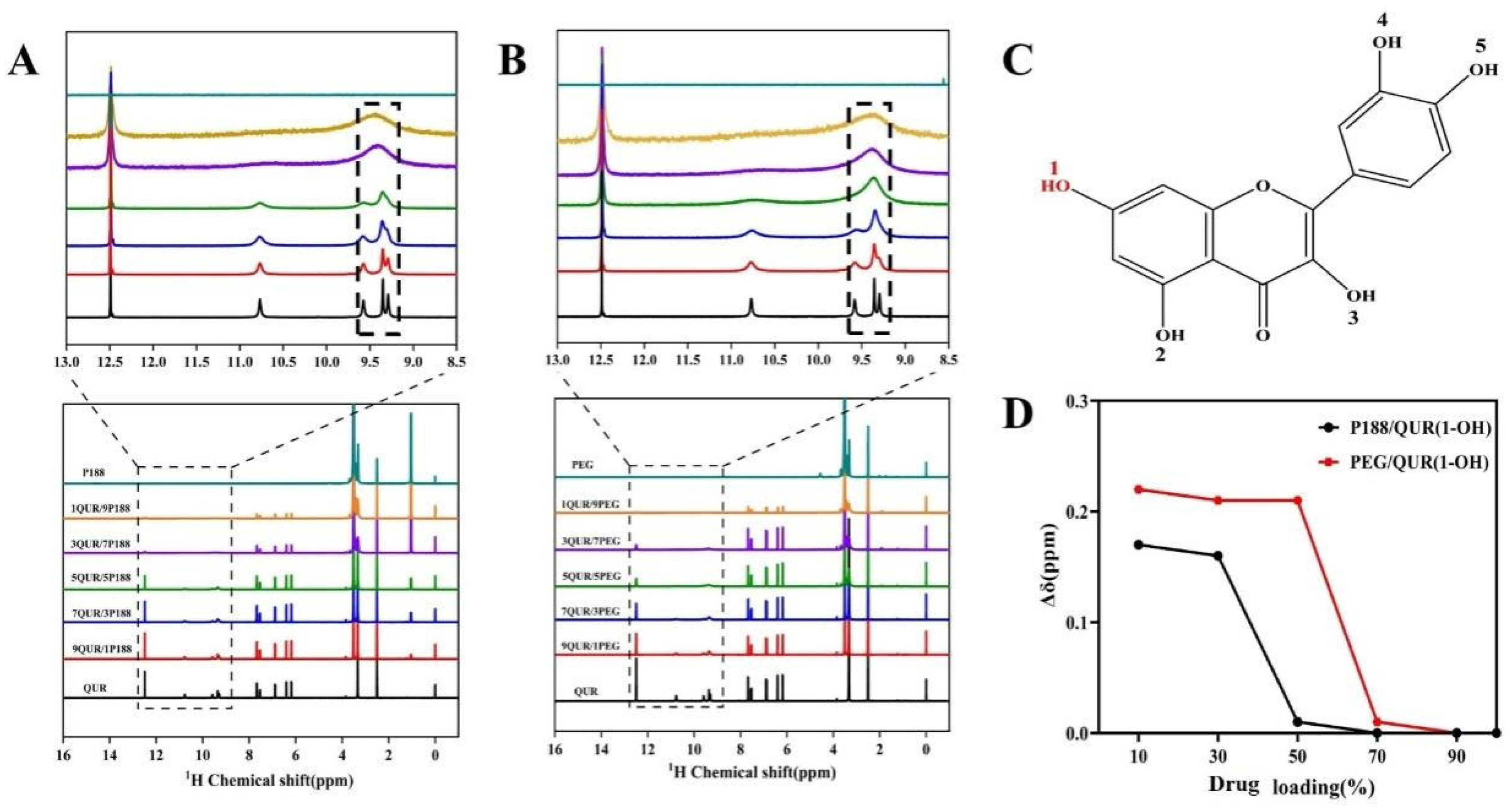
2.2. Intermolecular Interactions
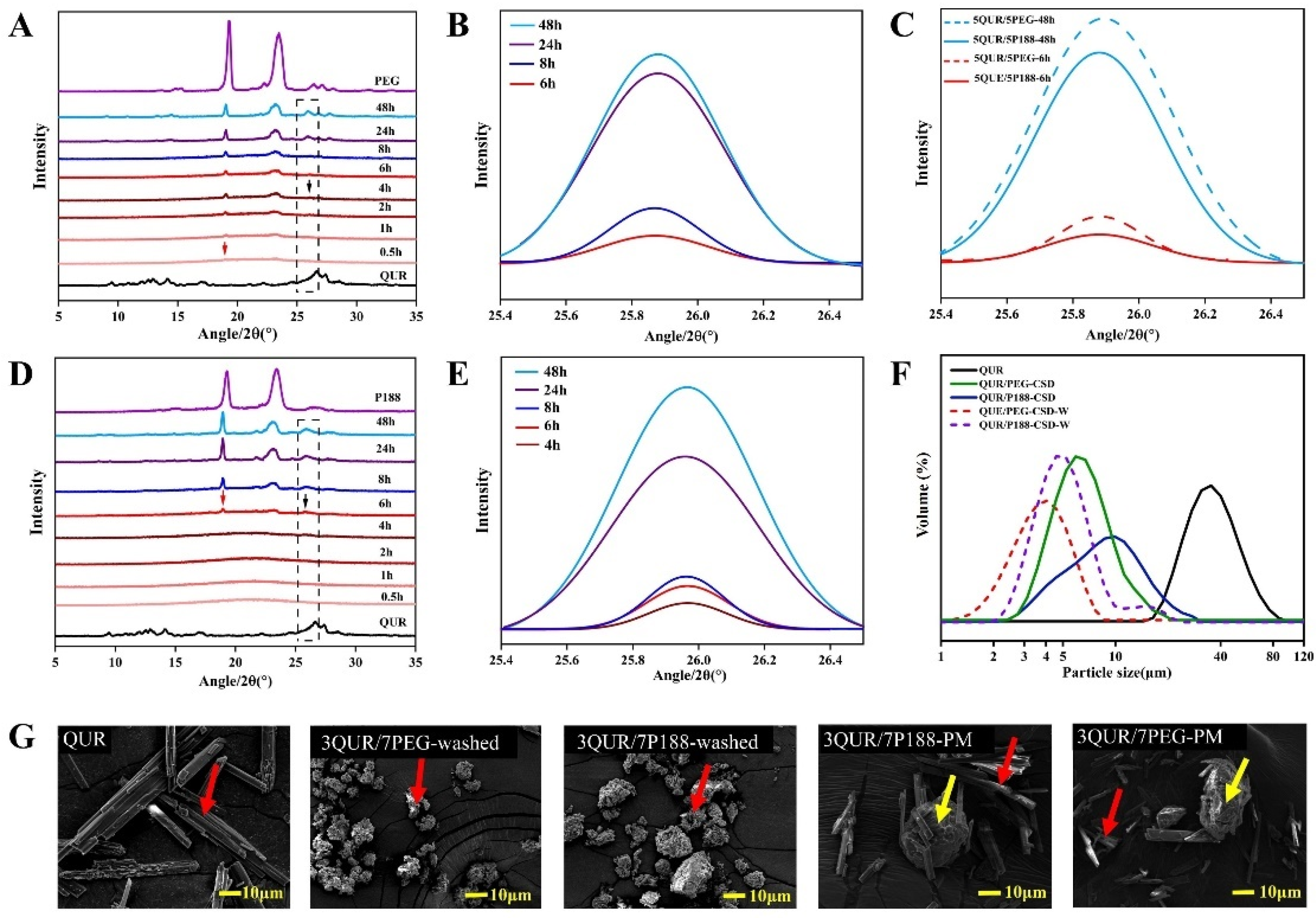
2.3. Crystallization Kinetics and Crystalline Size
2.4. In Vitro Dissolution Rate
2.4.1. Equilibrium Solubility of QUR
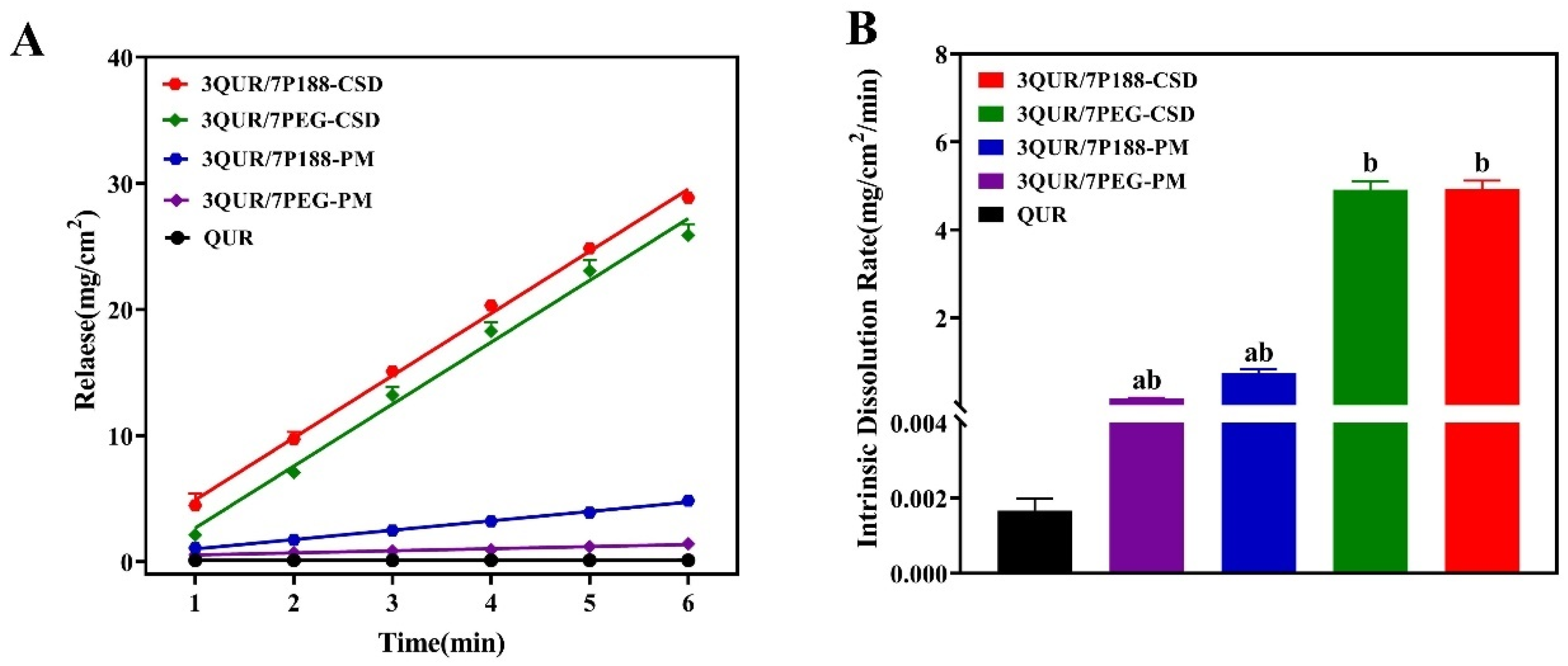
2.4.2. Intrinsic Dissolution Rate (IDR)
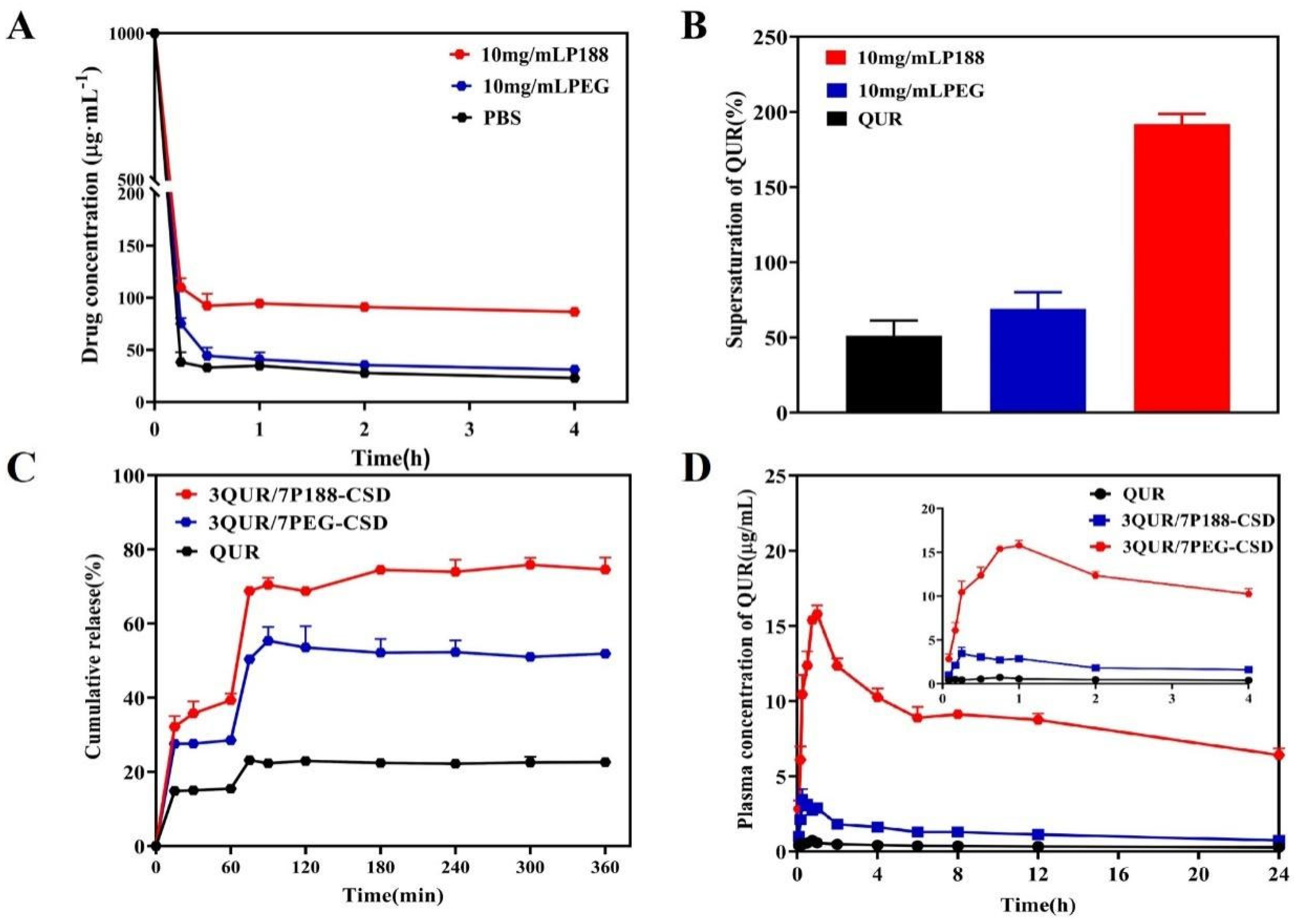
2.4.3. Effect of Polymers on QUR’s Ability to Maintain Supersaturation
2.4.4. pH Conversion Two-Step Dissolution
2.5. Pharmacokinetics
2.6. Cell Permeability
2.7. In Vivo Single Pass Intestinal Perfusion
3. Materials and Methods
3.1. Reagent and Materials
3.2. Preparation of CSD and Physical Mixing (PM)
3.3. Scanning Electron Microscopy (SEM)
3.4. Powders X-Ray Diffraction (PXRD)
3.5. Laser Particle Size Analyzer (PSA)
3.6. Hansen Solubility Parameters of Drug and Polymer
3.7. 1H-Nuclear Magnetic Resonance (1H-NMR)
3.8. Determination of Interaction Parameter χ
3.9. Dissolution Kinetics In Vitro
3.10. Effects of Different Polymers on Supersaturated QUR
3.11. Pharmacokinetic Analysis
3.12. Caco-2 Cell Experiments
3.13. In Vivo Single Pass Intestinal Perfusion Experiment
3.14. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Biasutto, L.; Marotta, E.; Garbisa, S.; Zoratti, M.; Paradisi, C. Determination of Quercetin and Resveratrol in Whole Blood—Implications for Absorption Studies. Molecules 2010, 15, 6570–6579. [Google Scholar] [CrossRef]
- Bischoff, S.C. Quercetin: Potentials in the prevention and therapy of disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 733–740. [Google Scholar] [CrossRef]
- Gilley, A.D.; Arca, H.C.; Nichols BL, B.; Mosquera-Giraldo, L.I.; Taylor, L.S.; Edgar, K.J.; Neilson, A.P. Novel cellulose-based amorphous solid dispersions enhance quercetin solution concentrations in vitro. Carbohydr. Polym. 2017, 157, 86–93. [Google Scholar] [CrossRef]
- Attar, E.S.; Chaudhari, V.H.; Deokar, C.G.; Dyawanapelly, S.; Devarajan, P.V. Nano Drug Delivery Strategies for an Oral Bioenhanced Quercetin Formulation. Eur. J. Drug Metab. Pharmacokinet. 2023, 48, 495–514. [Google Scholar] [CrossRef]
- Li, B.; Konecke, S.; Harich, K.; Wegiel, L.; Taylor, L.S.; Edgar, K.J. Solid dispersion of quercetin in cellulose derivative matrices influences both solubility and stability. Carbohydr. Polym. 2013, 92, 2033–2040. [Google Scholar] [CrossRef]
- Kakran, M.; Sahoo, N.G.; Li, L.; Judeh, Z. Fabrication of quercetin nanoparticles by anti-solvent precipitation method for enhanced dissolution. Powder Technol. 2012, 223, 59–64. [Google Scholar] [CrossRef]
- Patra, A.; Satpathy, S.; Shenoy, A.; Bush, J.; Kazi, M.; Hussain, M.D. Formulation and evaluation of mixed polymeric micelles of quercetin for treatment of breast, ovarian, and multidrug resistant cancers. Int. J. Nanomed. 2018, 13, 2869–2881. [Google Scholar] [CrossRef]
- Arbain, N.H.; Salim, N.; Masoumi HR, F.; Wong, T.W.; Basri, M.; Abdul Rahman, M.B. In vitro evaluation of the inhalable quercetin loaded nanoemulsion for pulmonary delivery. Drug Deliv. Transl. Res. 2019, 9, 497–507. [Google Scholar] [CrossRef]
- Tran, T.H.; Guo, Y.; Song, D.; Bruno, R.S.; Lu, X. Quercetin-Containing Self-Nanoemulsifying Drug Delivery System for Improving Oral Absorption. J. Pharm. Sci. 2014, 103, 840–852. [Google Scholar] [CrossRef]
- Han, J.; Tong, M.; Li, S.; Yu, X.; Hu, Z.; Zhang, Q.; Xu, R.; Wang, J. Surfactant-free amorphous solid dispersion with high dissolution for absorption enhancement of hydrophobic drugs: A case of quercetin. Drug Dev. Ind. Pharm. 2021, 47, 153–162. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Hu, C. Study on the crystallinity of PEG on the crystalline size of flavonoids in a crystalline dispersion system. Eur. J. Pharm. Biopharm. 2024, 205, 114536. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Liu, Y.; Guo, Y.; Hu, C. Influence of Intermolecular Interactions on Crystallite Size in Crystalline Solid Dispersions. Pharmaceutics 2023, 15, 2493. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Q.; Liu, Y.; Hu, C. Study on the regulation mechanism of effective glass transition temperature on the crystallization of crystalline solid dispersion. Drug Deliv. Transl. Res. 2023, 13, 2677–2689. [Google Scholar] [CrossRef]
- Hu, C.; Liu, Z.; Liu, C.; Zhang, Y.; Fan, H.; Qian, F. Improvement of Antialveolar Echinococcosis Efficacy of Albendazole by a Novel Nanocrystalline Formulation with Enhanced Oral Absorption. ACS Infect. Dis. 2020, 6, 802–810. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Z.; Qian, F. Crystallization of bifonazole and acetaminophen within the matrix of semicrystalline, PEO-PPO-PEO triblock copolymers. Mol. Pharm. 2015, 12, 590–599. [Google Scholar] [CrossRef]
- Zhang, F.; Stühn, B. Composition fluctuation and domain spacing of low molar weight PEO–PPO–PEO triblock copolymers in the melt, during crystallization and in the solid state. Colloid Polym. Sci. 2006, 284, 823–833. [Google Scholar] [CrossRef]
- Zhang, F.; Stühn, B. Crystallization and melting behavior of low molar weight PEO–PPO–PEO triblock copolymers. Colloid Polym. Sci. 2006, 285, 371–379. [Google Scholar] [CrossRef]
- Wang, T.; Guo, Y.; He, Y.; Ren, T.; Yin, L.; Fawcett, J.P.; Gu, J.; Sun, H. Impact of molecular weight on the mechanism of cellular uptake of polyethylene glycols (PEGs) with particular reference to P-glycoprotein. Acta Pharm. Sin. B 2020, 10, 2002–2009. [Google Scholar] [CrossRef]
- Mukesh, S.; Joshi, P.; Bansal, A.K.; Kashyap, M.C.; Mandal, S.K.; Sathe, V.; Sangamwar, A.T. Amorphous Salts Solid Dispersions of Celecoxib: Enhanced Biopharmaceutical Performance and Physical Stability. Mol. Pharm. 2021, 18, 2334–2348. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Hussain, M.A. Drug–Polymer Solubility and Miscibility: Stability Consideration and Practical Challenges in Amorphous Solid Dispersion Development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; OReilly, N.J. Understanding the generation and maintenance of supersaturation during the dissolution of amorphous solid dispersions using modulated DSC and 1H NMR. Int. J. Pharm. 2018, 536, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180, 112528. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Fan, N.; Zhang, G.; Sun, J.; He, Z.; Li, J. Quercetin amorphous solid dispersions prepared by hot melt extrusion with enhanced solubility and intestinal absorption. Pharm. Dev. Technol. 2020, 25, 472–481. [Google Scholar] [CrossRef]
- Tao, J.; Sun, Y.; Zhang, G.G.; Yu, L. Solubility of small-molecule crystals in polymers: D-mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm. Res. 2009, 26, 855–864. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Wang, S.; Liu, C.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Initial Drug Dissolution from Amorphous Solid Dispersions Controlled by Polymer Dissolution and Drug-Polymer Interaction. Pharm. Res. 2016, 33, 2445–2458. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chen, Z.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Drug-polymer-water interaction and its implication for the dissolution performance of amorphous solid dispersions. Mol. Pharm. 2015, 12, 576–589. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Zhang, L.; Qian, F. Dissolution Characteristics of Fast-Crystallizing β-Lapachone within Different Semicrystalline Microstructures of Polyethylene Glycol or Poly(ethylene oxide)–Poly(propylene oxide)–Poly(ethylene oxide) Triblock Copolymer. Cryst. Growth Des. 2016, 16, 5367–5376. [Google Scholar] [CrossRef]
- Vetter, T.; Mazzotti, M.; Brozio, J. Slowing the Growth Rate of Ibuprofen Crystals Using the Polymeric Additive Pluronic F127. Cryst. Growth Des. 2011, 11, 3813–3821. [Google Scholar] [CrossRef]
- Fischer, S.M.; Brandl, M.; Fricker, G. Effect of the non-ionic surfactant Poloxamer 188 on passive permeability of poorly soluble drugs across Caco-2 cell monolayers. Eur. J. Pharm. Biopharm. 2011, 79, 416–422. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Na, Y.; Bang, K.; Lee, H.; Wang, M.; Huh, H.; Cho, C. A novel composition of ticagrelor by solid dispersion technique for increasing solubility and intestinal permeability. Int. J. Pharm. 2019, 555, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xing, M.; Wang, K.; Wei, Q.; Zhao, J.; Wang, Y.; Ji, K.; Song, S. Application of Fucoidan in Caco-2 Model Establishment. Pharmaceuticals 2022, 15, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, W.; Bu, T.; Zhao, R.; Niu, R.; Liu, L.; Li, J.; Wu, J.; Liu, D. Digestion, absorption, and transport properties of soy-fermented douchi hypoglycemic peptides VY and SFLLR under simulated gastrointestinal digestion and Caco-2 cell monolayers. Food Res. Int. 2023, 164, 23–40. [Google Scholar] [CrossRef]
- Catalan-Figueroa, J.; Garcia, M.A.; Contreras, P.; Boisset, C.B.; Gonzalez, P.M.; Fiedler, J.L.; Perez, M.F.; Morales, J.O. Poloxamer 188-Coated Ammonium Methacrylate Copolymer Nanocarriers Enhance Loperamide Permeability across Pgp-Expressing Epithelia. Mol. Pharm. 2021, 18, 743–750. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Alhalaweh, A.; Velaga, S.P. Hansen solubility parameter as a tool to predict cocrystal formation. Int. J. Pharm. 2011, 407, 63–71. [Google Scholar] [CrossRef]
- Sun, Y.; Tao, J.; Zhang GG, Z.; Yu, L. Solubilities of Crystalline Drugs in Polymers: An Improved Analytical Method and Comparison of Solubilities of Indomethacin and Nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci. 2010, 99, 4023–4031. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Polymer | δd/MPa0.5 | δp/MPa0.5 | δh/MPa0.5 | δt/MPa0.5 | Δδt (D, P) |
|---|---|---|---|---|---|
| QUR | 22.37 | 6.85 | 1.1 | 23.42 | N/A |
| P188 | 20.78 | 0.77 | 0.1 | 20.8 | 2.62 |
| PEG8000 | 21.03 | 0.83 | 0.1 | 21.05 | 2.37 |
| Drug | Concentration (μg/mL) |
|---|---|
| QUR | 0.45 ± 0.05 |
| P188 (0.3 mg/mL) | 0.56 ± 0.08 |
| P188 (1 mg/mL) | 0.97 ± 0.05 |
| P188 (3 mg/mL) | 1.63 ± 0.03 |
| PEG8000 (0.3 mg/mL) | 0.51 ± 0.05 |
| PEG8000 (1 mg/mL) | 0.86 ± 0.03 |
| PEG8000 (3 mg/mL) | 0.90 ± 0.07 |
| Parameters | QUR | QUR/P188-CSD | QUR/PEG8000-CSD |
|---|---|---|---|
| AUC(0–24) (μg/mL/h) | 8.46 ± 1.38 | 29.73 ± 9.95 | 211.69 ± 22.48 |
| AUC(0–∞) (μg/mL/h) | 19.08 ± 7.51 | 74.85 ± 80.42 | 563.09 ± 267.75 |
| Cmax (μg/mL) | 0.71 ± 0.19 | 3.89 ± 1.65 | 16.03 ± 0.95 |
| Tmax (h) | 0.90 ± 0.60 | 0.40 ± 0.22 | 0.92 ± 0.13 |
| t1/2 (h) | 28.46 ± 20.83 | 27.67 ± 24.19 | 35.84 ± 20.05 |
| CLz/F (L/h/kg) | 9.00 ± 3.67 | 3.33 ± 1.73 | 0.30 ± 0.10 |
| MRT(0–24) (h) | 10.94 ± 1.37 | 9.16 ± 1.03 | 10.48 ± 0.36 |
| MRT(0–∞) (h) | 46.73 ± 28.62 | 36.86 ± 35.74 | 51.05 ± 29.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yan, Q.; Hu, C. Enhancing Oral Absorption of Quercetin Through Multifactorial Synergies in Crystal Dispersion Systems. Molecules 2025, 30, 2390. https://doi.org/10.3390/molecules30112390
Liu Y, Yan Q, Hu C. Enhancing Oral Absorption of Quercetin Through Multifactorial Synergies in Crystal Dispersion Systems. Molecules. 2025; 30(11):2390. https://doi.org/10.3390/molecules30112390
Chicago/Turabian StyleLiu, Yao, Qiuli Yan, and Chunhui Hu. 2025. "Enhancing Oral Absorption of Quercetin Through Multifactorial Synergies in Crystal Dispersion Systems" Molecules 30, no. 11: 2390. https://doi.org/10.3390/molecules30112390
APA StyleLiu, Y., Yan, Q., & Hu, C. (2025). Enhancing Oral Absorption of Quercetin Through Multifactorial Synergies in Crystal Dispersion Systems. Molecules, 30(11), 2390. https://doi.org/10.3390/molecules30112390