
Correlation of Structure and Electrocatalytic Performance of Bulk Oxides for Water Electrolysis


Abstract
1. Introduction
2. The Introduction of Water Electrolysis
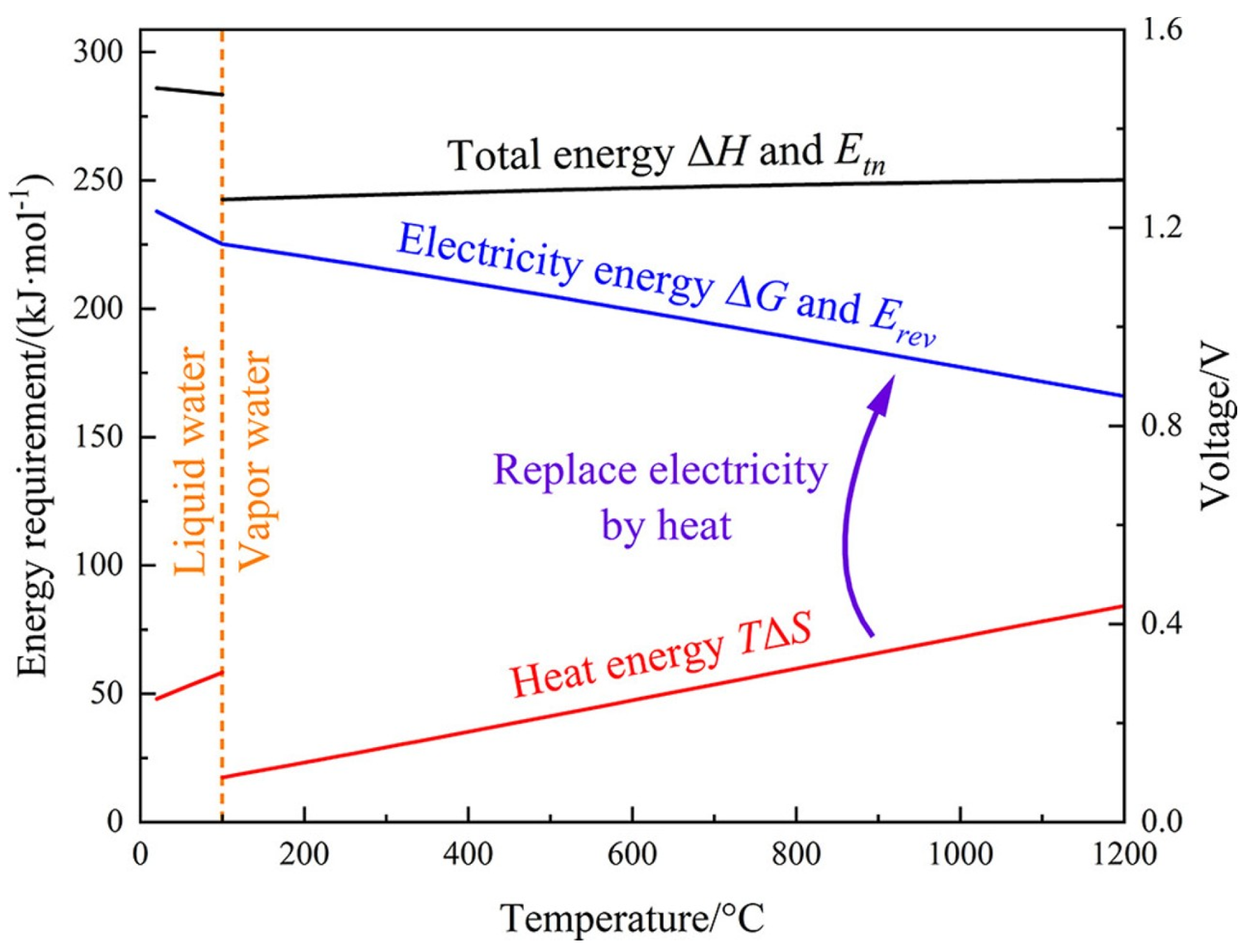
2.1. Fundamental Concepts of Water Electrolysis
2.2. Oxygen Evolution Reaction and Hydrogen Evolution Reaction
2.3. Performance Evaluation Parameters
- Overpotential (η). The overpotential for water electrolysis is the extra applied voltage relative to the theoretical voltage under standard conditions, which is the commonly used parameter to assess the activity of the given electrocatalyst. Typically, the overpotential at jgeo (the geometric current density normalized to the electrode surface area) = 10 mA/cm2 (corresponding to a solar-to-hydrogen efficiency of 12.3%) is used as a key parameter to evaluate the catalytic activity [33]. However, the overpotential of the electrocatalysts is strongly affected by the specific surface area and loading mass, especially for nanocatalysts with large specific surface areas, which hinders the use of the overpotential to reveal the intrinsic catalytic activity [34].
- Tafel Slope (b) and Exchange Current Density (j0). The two parameters can be obtained from the Tafel equation, η = a + blog j, where b is the Tafel slope, j is the current density, and η is the overpotential, respectively. Extrapolating the linear part of the Tafel plot to zero overpotential gives the exchange current density (j0), which reflects the intrinsic catalytic activity of the catalyst. Generally, an excellent electrocatalyst should have low b and high j0 values [35].
- Mass Activity (MA) and Specific Activity (SA). To further characterize the intrinsic activity of the catalyst, additional parameters, such as the mass activity (A/g) and specific activity, have been proposed based on the electrocatalyst mass loading, specific surface area, and electrochemically active surface area (ECSA). Mass activity is the current normalized by the current based on the catalyst mass loading. For electrocatalysts in equal mass, higher mass activity indicates greater catalytic efficiency, making it a useful parameter for assessing cost efficiency [36]. Specific activity is obtained by normalizing the current with the specific surface area or ECSA of the electrocatalysts, providing a more accurate reflection of intrinsic catalytic differences and facilitating the understanding of structure–activity relationships [37].
- Turnover Frequency (TOF), Faradaic Efficiency (FE), and Stability. Turnover frequency represents the number of product molecules generated per active site per unit of time, making it a key parameter for evaluating the intrinsic catalytic activity of the electrocatalysts [38]. Faradaic efficiency is defined as the ratio of the experimental to the theoretical product, indicating the efficiency and selectivity of the electrocatalysts [39]. In general, the FE of efficient electrocatalysts for water electrocatalysis is expected to be close to 100%. In addition to the above parameters, the stability of electrocatalysts, including both performance stability and structural stability, is the key indicator to assess the application potential of the electrocatalyst [40]. Performance stability is usually assessed by long-term operating electrocatalytic measurements, such as cyclic voltammetry (CV), chronopotentiometry (CP), and chronoamperometry (CA) tests. Structural stability requires in situ or post-reaction characterizations to evaluate the changes in the composition, structure, and morphology of the electrocatalysts.
3. Overview of Bulk Oxides for Water Electrolysis
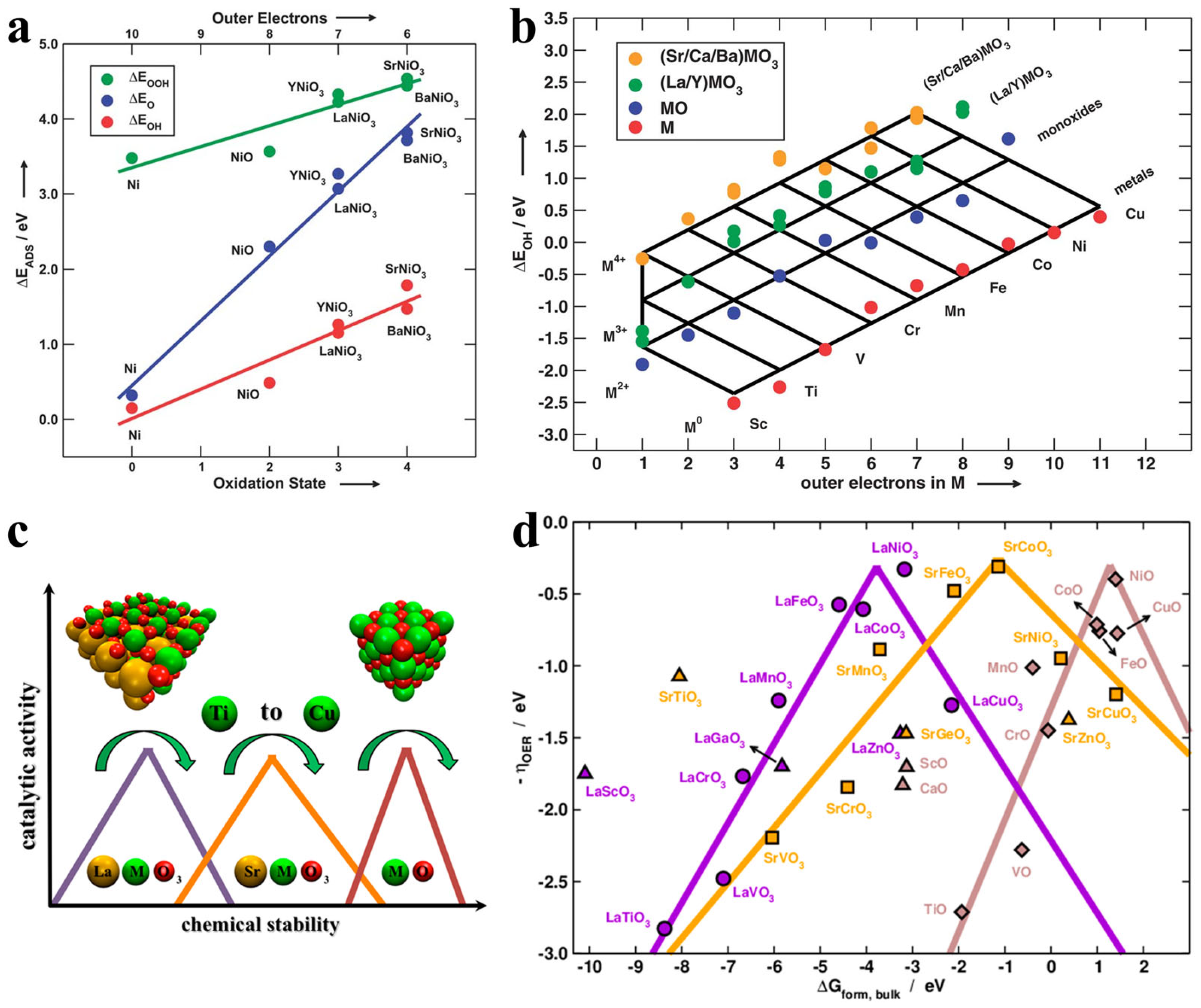
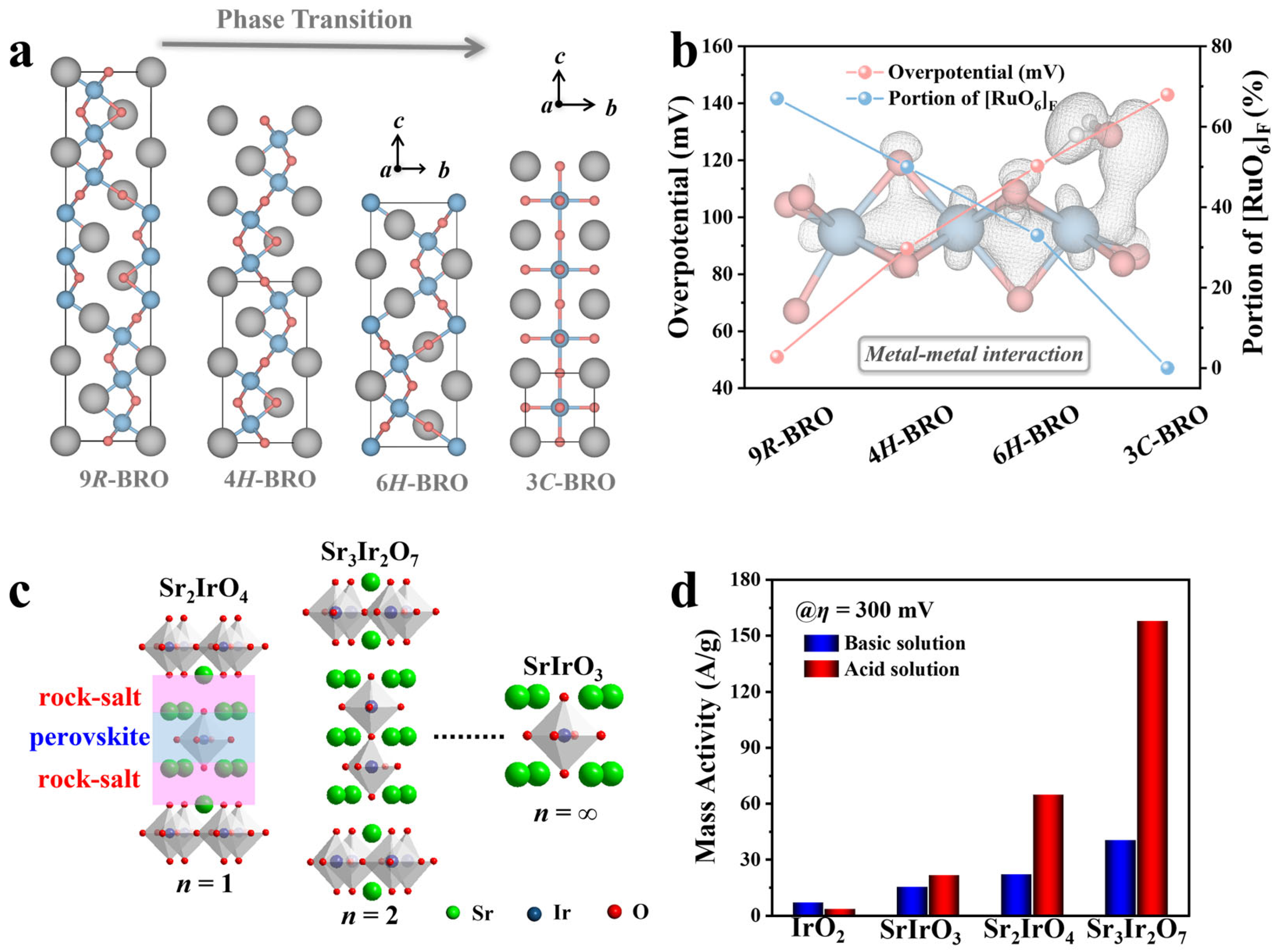
3.1. Overview of Bulk Oxide Electrocatalysts for Oxygen Evolution Reaction
3.2. Overview of Bulk Oxide Electrocatalysts for Hydrogen Evolution Reaction
4. Design Strategy for Bulk Oxides with High Water Electrolysis Performance
4.1. Crystal Structure Engineering
4.2. Heteroatom Doping
4.3. Defect Engineering
4.4. Morphology Engineering
5. Conclusions and Perspective
5.1. Developing New Electrocatalysts
5.2. Deepening the Understanding of Structure–Activity Correlations
5.3. Correlation of Atomic Properties of Transition Metals with Ligand Interactions
5.4. Practical Applications for Water Electrolysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lagadec, M.F.; Grimaud, A. Water electrolysers with closed and open electrochemical systems. Nat. Mater. 2020, 19, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Gunathilake, C.; Soliman, I.; Panthi, D.; Tandler, P.; Fatani, O.; Ghulamullah, N.A.; Marasinghe, D.; Farhath, M.; Madhujith, T.; Conrad, K.; et al. A comprehensive review on hydrogen production, storage, and applications. Chem. Soc. Rev. 2024, 53, 10900–10969. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Xu, Z.L.; Li, F.M.; Chen, F.Y.; Yu, J.Y.; Yan, Y.; Chen, Y.; Xia, B.Y. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 2023, 52, 5652–5683. [Google Scholar] [CrossRef]
- Wan, R.; Yuan, T.; Wang, L.; Li, B.; Liu, M.; Zhao, B. Earth-abundant electrocatalysts for acidic oxygen evolution. Nat. Catal. 2024, 7, 1288–1304. [Google Scholar] [CrossRef]
- Yang, Y.; Peltier, C.R.; Zeng, R.; Schimmenti, R.; Li, Q.; Huang, X.; Yan, Z.; Potsi, G.; Selhorst, R.; Lu, X.; et al. Electrocatalysis in Alkaline Media and Alkaline Membrane-Based Energy Technologies. Chem. Rev. 2022, 122, 6117–6321. [Google Scholar] [CrossRef]
- Song, Y.; Chen, H.; Wang, X.; Weng, C.; Zou, K.; Wang, C.; Yuan, Y.; Ma, Y.; Yang, X.; Lin, W. Engineering Ir-based catalysts for high current density applications in proton exchange membrane water electrolyzers. Energy Environ. Sci. 2025, 18, 130–154. [Google Scholar] [CrossRef]
- Zhu, C.; Tian, H.; Tan, P.; Huang, B.; Zhao, S.; Cai, G.; Yuan, C.; Zhao, M.-H.; Cao, M.; Zhao, J.; et al. Ruthenate perovskite with face-sharing motifs for alkaline hydrogen evolution. Chem Catal. 2024, 4, 101132. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Cava, R.J. Hexagonal Perovskites as Quantum Materials. Chem. Rev. 2021, 121, 2935–2965. [Google Scholar] [CrossRef]
- Zhu, Y.; Tang, Z.; Yuan, L.; Li, B.; Shao, Z.; Guo, W. Beyond conventional structures: Emerging complex metal oxides for efficient oxygen and hydrogen electrocatalysis. Chem. Soc. Rev. 2025, 54, 1027–1092. [Google Scholar] [CrossRef]
- Humayun, M.; Li, Z.; Israr, M.; Khan, A.; Luo, W.; Wang, C.; Shao, Z. Perovskite Type ABO3 Oxides in Photocatalysis, Electrocatalysis, and Solid Oxide Fuel Cells: State of the Art and Future Prospects. Chem. Rev. 2025, 125, 3165–3241. [Google Scholar] [CrossRef]
- Wang, K.; Han, C.; Shao, Z.; Qiu, J.; Wang, S.; Liu, S. Perovskite Oxide Catalysts for Advanced Oxidation Reactions. Adv. Funct. Mater. 2021, 31, 2102089. [Google Scholar] [CrossRef]
- Meharban, F.; Lin, C.; Wu, X.; Tan, L.; Wang, H.; Hu, W.; Zhou, D.; Li, X.; Luo, W. Scaling Up Stability: Navigating from Lab Insights to Robust Oxygen Evolution Electrocatalysts for Industrial Water Electrolysis. Adv. Energy Mater. 2024, 14, 2402886. [Google Scholar] [CrossRef]
- Du, Y.; Liu, J.; Chen, J.; Wang, S.; Tang, Y.; Wang, A.L.; Fu, G.; Lu, X.F. Design Principle and Regulation Strategy of Noble Metal-Based Materials for Practical Proton Exchange Membrane Water Electrolyzer. Adv. Energy Mater. 2024, 15, 2404113. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef]
- Zhao, Q.; Yan, Z.; Chen, C.; Chen, J. Spinels: Controlled Preparation, Oxygen Reduction/Evolution Reaction Application, and Beyond. Chem. Rev. 2017, 117, 10121–10211. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric Oxides as Low-Cost and Highly-Efficient Oxygen Reduction/Evolution Catalysts for Low-Temperature Electrochemical Devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, M.; Gu, X.; Shi, Y.; Deng, Z.; Cai, N. Water Electrolysis toward Elevated Temperature: Advances, Challenges and Frontiers. Chem. Rev. 2023, 123, 7119–7192. [Google Scholar] [CrossRef]
- Song, J.; Wei, C.; Huang, Z.-F.; Liu, C.; Zeng, L.; Wang, X.; Xu, Z.J. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 2020, 49, 2196–2214. [Google Scholar] [CrossRef]
- Xu, X.; Pan, Y.; Zhong, Y.; Shi, C.; Guan, D.; Ge, L.; Hu, Z.; Chin, Y.Y.; Lin, H.J.; Chen, C.T.; et al. New Undisputed Evidence and Strategy for Enhanced Lattice-Oxygen Participation of Perovskite Electrocatalyst Through Cation Deficiency Manipulation. Adv. Sci. 2022, 9, e2200530. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, X.; Zhong, Y.; Ge, L.; Chen, Y.; Veder, J.M.; Guan, D.; O’Hayre, R.; Li, M.; Wang, G.; et al. Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation. Nat. Commun. 2020, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.H. Recent Development of Oxygen Evolution Electrocatalysts in Acidic Environment. Adv. Mater. 2021, 33, e2006328. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Chen, H.; Gao, R.; Shi, L.; Yang, L.; Zou, X. Iridium-containing water-oxidation catalysts in acidic electrolyte. Chin. J. Catal. 2021, 42, 1054–1077. [Google Scholar] [CrossRef]
- Wang, X.; Xi, S.; Huang, P.; Du, Y.; Zhong, H.; Wang, Q.; Borgna, A.; Zhang, Y.W.; Wang, Z.; Wang, H.; et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 2022, 611, 702–708. [Google Scholar] [CrossRef]
- Lin, C.; Li, J.-L.; Li, X.; Yang, S.; Luo, W.; Zhang, Y.; Kim, S.-H.; Kim, D.-H.; Shinde, S.S.; Li, Y.-F.; et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 2021, 4, 1012–1023. [Google Scholar] [CrossRef]
- Fang, M.; Dong, G.; Wei, R.; Ho, J.C. Hierarchical Nanostructures: Design for Sustainable Water Splitting. Adv. Energy Mater. 2017, 7, 1700559. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef]
- Li, L.; Wang, P.; Shao, Q.; Huang, X. Metallic nanostructures with low dimensionality for electrochemical water splitting. Chem. Soc. Rev. 2020, 49, 3072–3106. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Y.; Sheng, W.; Xu, Z.J.; Jaroniec, M.; Qiao, S.-Z. Strategies for design of electrocatalysts for hydrogen evolution under alkaline conditions. Mater. Today 2020, 36, 125–138. [Google Scholar] [CrossRef]
- Ahsan, M.A.; He, T.; Noveron, J.C.; Reuter, K.; Puente-Santiago, A.R.; Luque, R. Low-dimensional heterostructures for advanced electrocatalysis: An experimental and computational perspective. Chem. Soc. Rev. 2022, 51, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Xu, Z.J. The Comprehensive Understanding of 10 mA cm geo−2 as an Evaluation Parameter for Electrochemical Water Splitting. Small Methods 2018, 2, 1800168. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Norskov, J.K. Materials for solar fuels and chemicals. Nat. Mater. 2016, 16, 70–81. [Google Scholar] [CrossRef]
- Zhu, C.; Cai, G.-H.; Yuan, C.; Huang, B.; Li, G.; Croft, M.; Greenblatt, M.; Li, M.-R. Intersite Charge Transfer Enhanced Oxygen Evolution Reactivity on A2IrO3 (A = Li, Na, Cu) Delafossite Electrocatalysts. J. Electrochem. Soc. 2022, 169, 056523. [Google Scholar] [CrossRef]
- Nong, H.N.; Reier, T.; Oh, H.-S.; Gliech, M.; Paciok, P.; Vu, T.H.T.; Teschner, D.; Heggen, M.; Petkov, V.; Schlögl, R.; et al. A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOx core–shell electrocatalysts. Nat. Catal. 2018, 1, 841–851. [Google Scholar] [CrossRef]
- Wei, C.; Sun, S.; Mandler, D.; Wang, X.; Qiao, S.Z.; Xu, Z.J. Approaches for measuring the surface areas of metal oxide electrocatalysts for determining their intrinsic electrocatalytic activity. Chem. Soc. Rev. 2019, 48, 2518–2534. [Google Scholar] [CrossRef]
- Lettenmeier, P.; Wang, L.; Golla-Schindler, U.; Gazdzicki, P.; Canas, N.A.; Handl, M.; Hiesgen, R.; Hosseiny, S.S.; Gago, A.S.; Friedrich, K.A. Nanosized IrOx-Ir Catalyst with Relevant Activity for Anodes of Proton Exchange Membrane Electrolysis Produced by a Cost-Effective Procedure. Angew. Chem. Int. Ed. 2016, 55, 742–746. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Gu, L.; Zhang, Y.; Li, G.D.; Zou, X.; Chen, J.S. Corrosion engineering towards efficient oxygen evolution electrodes with stable catalytic activity for over 6000 hours. Nat. Commun. 2018, 9, 2609. [Google Scholar] [CrossRef]
- Geiger, S.; Kasian, O.; Ledendecker, M.; Pizzutilo, E.; Mingers, A.M.; Fu, W.T.; Diaz-Morales, O.; Li, Z.; Oellers, T.; Fruchter, L.; et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 2018, 1, 508–515. [Google Scholar] [CrossRef]
- Xu, X.; Wang, W.; Zhou, W.; Shao, Z. Recent Advances in Novel Nanostructuring Methods of Perovskite Electrocatalysts for Energy-Related Applications. Small Methods 2018, 2, 1800071. [Google Scholar] [CrossRef]
- Zhou, D.; Li, P.; Lin, X.; McKinley, A.; Kuang, Y.; Liu, W.; Lin, W.F.; Sun, X.; Duan, X. Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: Identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly. Chem. Soc. Rev. 2021, 50, 8790–8817. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Vojvodic, A.; Norskov, J.K. Chemistry. Optimizing perovskites for the water-splitting reaction. Science 2011, 334, 1355–1356. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Inoglu, N.G.; Su, H.-Y.; Martínez, J.I.; Man, I.C.; Koper, M.T.M.; Kitchin, J.R.; Rossmeisl, J. Number of outer electrons as descriptor for adsorption processes on transition metals and their oxides. Chem. Sci. 2013, 4, 1245–1249. [Google Scholar] [CrossRef]
- Xu, X.; Pan, Y.; Zhong, Y.; Ran, R.; Shao, Z. Ruddlesden–Popper perovskites in electrocatalysis. Mater. Horiz. 2020, 7, 2519–2565. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Díaz-Morales, O.A.; Kolb, M.J.; Koper, M.T.M. Why Is Bulk Thermochemistry a Good Descriptor for the Electrocatalytic Activity of Transition Metal Oxides? ACS Catal. 2015, 5, 869–873. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef]
- Miao, X.; Zhang, L.; Wu, L.; Hu, Z.; Shi, L.; Zhou, S. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 2019, 10, 3809. [Google Scholar] [CrossRef]
- Yagi, S.; Yamada, I.; Tsukasaki, H.; Seno, A.; Murakami, M.; Fujii, H.; Chen, H.; Umezawa, N.; Abe, H.; Nishiyama, N.; et al. Covalency-reinforced oxygen evolution reaction catalyst. Nat. Commun. 2015, 6, 8249. [Google Scholar] [CrossRef]
- Kim, J.; Yin, X.; Tsao, K.C.; Fang, S.; Yang, H. Ca2Mn2O5 as oxygen-deficient perovskite electrocatalyst for oxygen evolution reaction. J. Am. Chem. Soc. 2014, 136, 14646–14649. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Bhosale, R.R.; Almomani, F.; Malik, S.S.; Suslov, S.; Tarlochan, F. Combustion synthesis of bifunctional LaMO3 (M = Cr, Mn, Fe, Co, Ni) perovskites for oxygen reduction and oxygen evolution reaction in alkaline media. J. Electroanal. Chem. 2018, 809, 22–30. [Google Scholar] [CrossRef]
- Hong, W.T.; Stoerzinger, K.A.; Lee, Y.-L.; Giordano, L.; Grimaud, A.; Johnson, A.M.; Hwang, J.; Crumlin, E.J.; Yang, W.; Shao-Horn, Y. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 2017, 10, 2190–2200. [Google Scholar] [CrossRef]
- Sun, Y.; Liao, H.; Wang, J.; Chen, B.; Sun, S.; Ong, S.J.H.; Xi, S.; Diao, C.; Du, Y.; Wang, J.-O.; et al. Covalency competition dominates the water oxidation structure–activity relationship on spinel oxides. Nat. Catal. 2020, 3, 554–563. [Google Scholar] [CrossRef]
- May, K.J.; Carlton, C.E.; Stoerzinger, K.A.; Risch, M.; Suntivich, J.; Lee, Y.-L.; Grimaud, A.; Shao-Horn, Y. Influence of Oxygen Evolution during Water Oxidation on the Surface of Perovskite Oxide Catalysts. J. Phys. Chem. Lett. 2012, 3, 3264–3270. [Google Scholar] [CrossRef]
- Risch, M.; Grimaud, A.; May, K.J.; Stoerzinger, K.A.; Chen, T.J.; Mansour, A.N.; Shao-Horn, Y. Structural Changes of Cobalt-Based Perovskites upon Water Oxidation Investigated by EXAFS. J. Phys. Chem. C 2013, 117, 8628–8635. [Google Scholar] [CrossRef]
- Duan, Y.; Sun, S.; Xi, S.; Ren, X.; Zhou, Y.; Zhang, G.; Yang, H.; Du, Y.; Xu, Z.J. Tailoring the Co 3d-O 2p Covalency in LaCoO3 by Fe Substitution to Promote Oxygen Evolution Reaction. Chem. Mater. 2017, 29, 10534–10541. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, S.; Song, J.; Xi, S.; Chen, B.; Du, Y.; Fisher, A.C.; Cheng, F.; Wang, X.; Zhang, H.; et al. Enlarged CoO Covalency in Octahedral Sites Leading to Highly Efficient Spinel Oxides for Oxygen Evolution Reaction. Adv. Mater. 2018, 30, e1802912. [Google Scholar] [CrossRef]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chueh, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6, 6097. [Google Scholar] [CrossRef]
- Suntivich, J.; Hong, W.T.; Lee, Y.-L.; Rondinelli, J.M.; Yang, W.; Goodenough, J.B.; Dabrowski, B.; Freeland, J.W.; Shao-Horn, Y. Estimating Hybridization of Transition Metal and Oxygen States in Perovskites from O K-Edge X-Ray Absorption Spectroscopy. J. Phys. Chem. C 2014, 118, 1856–1863. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Cui, Z.; Li, Y.; Xin, S.; Zhou, J.; Long, Y.; Jin, C.; Goodenough, J.B. Exceptional oxygen evolution reactivities on CaCoO3 and SrCoO3. Sci. Adv. 2019, 5, eaav6262. [Google Scholar] [CrossRef]
- Ye, X.; Song, S.; Li, L.; Chang, Y.-C.; Qin, S.; Liu, Z.; Huang, Y.-C.; Zhou, J.; Zhang, L.-j.; Dong, C.-L.; et al. A’–B Intersite Cooperation-Enhanced Water Splitting in Quadruple Perovskite Oxide CaCu3Ir4O12. Chem. Mater. 2021, 33, 9295–9305. [Google Scholar] [CrossRef]
- Zhu, Y.; Tahini, H.A.; Hu, Z.; Chen, Z.G.; Zhou, W.; Komarek, A.C.; Lin, Q.; Lin, H.J.; Chen, C.T.; Zhong, Y.; et al. Boosting Oxygen Evolution Reaction by Creating Both Metal Ion and Lattice-Oxygen Active Sites in a Complex Oxide. Adv. Mater. 2020, 32, e1905025. [Google Scholar] [CrossRef]
- Diaz-Morales, O.; Raaijman, S.; Kortlever, R.; Kooyman, P.J.; Wezendonk, T.; Gascon, J.; Fu, W.T.; Koper, M.T. Iridium-based double perovskites for efficient water oxidation in acid media. Nat. Commun. 2016, 7, 12363. [Google Scholar] [CrossRef]
- Hubert, M.A.; Patel, A.M.; Gallo, A.; Liu, Y.; Valle, E.; Ben-Naim, M.; Sanchez, J.; Sokaras, D.; Sinclair, R.; Nørskov, J.K.; et al. Acidic Oxygen Evolution Reaction Activity–Stability Relationships in Ru-Based Pyrochlores. ACS Catal. 2020, 10, 12182–12196. [Google Scholar] [CrossRef]
- Lebedev, D.; Povia, M.; Waltar, K.; Abdala, P.M.; Castelli, I.E.; Fabbri, E.; Blanco, M.V.; Fedorov, A.; Copéret, C.; Marzari, N.; et al. Highly Active and Stable Iridium Pyrochlores for Oxygen Evolution Reaction. Chem. Mater. 2017, 29, 5182–5191. [Google Scholar] [CrossRef]
- Yang, L.; Yu, G.; Ai, X.; Yan, W.; Duan, H.; Chen, W.; Li, X.; Wang, T.; Zhang, C.; Huang, X.; et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers. Nat. Commun. 2018, 9, 5236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liang, X.; Chen, H.; Yan, W.; Shi, L.; Liu, Y.; Li, J.; Zou, X. Identifying Key Structural Subunits and Their Synergism in Low-Iridium Triple Perovskites for Oxygen Evolution in Acidic Media. Chem. Mater. 2020, 32, 3904–3910. [Google Scholar] [CrossRef]
- Retuerto, M.; Pascual, L.; Piqué, O.; Kayser, P.; Salam, M.A.; Mokhtar, M.; Alonso, J.A.; Peña, M.; Calle-Vallejo, F.; Rojas, S. How oxidation state and lattice distortion influence the oxygen evolution activity in acid of iridium double perovskites. J. Mater. Chem. A 2021, 9, 2980–2990. [Google Scholar] [CrossRef]
- Zhu, C.; Tian, H.; Huang, B.; Cai, G.; Yuan, C.; Zhang, Y.; Li, Y.; Li, G.; Xu, H.; Li, M.-R. Boosting oxygen evolution reaction by enhanced intrinsic activity in Ruddlesden−Popper iridate oxides. Chem. Eng. J. 2021, 423, 130185. [Google Scholar] [CrossRef]
- Wang, L.; Shi, L.; Liu, Q.; Huang, Y.; Yan, W.; Liang, X.; Zhao, X.; Chen, H.; Zou, X. Structurally Robust Honeycomb Layered Strontium Iridate as an Oxygen Evolution Electrocatalyst in Acid. ACS Catal. 2023, 13, 7322–7330. [Google Scholar] [CrossRef]
- Rodríguez-García, I.; Gómez de la Fuente, J.L.; Galyamin, D.; Tolosana-Moranchel, Á.; Kayser, P.; Salam, M.A.; Alonso, J.A.; Calle-Vallejo, F.; Rojas, S.; Retuerto, M. Dy2NiRuO6 perovskite with high activity and durability for the oxygen evolution reaction in acidic electrolyte. J. Mater. Chem. A 2024, 12, 16854–16862. [Google Scholar] [CrossRef]
- Zhong, X.; Sui, L.; Yang, M.; Koketsu, T.; Klingenhof, M.; Selve, S.; Reeves, K.G.; Ge, C.; Zhuang, L.; Kan, W.H.; et al. Stabilization of layered lithium-rich manganese oxide for anion exchange membrane fuel cells and water electrolysers. Nat. Catal. 2024, 7, 546–559. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, Q.; Zhong, Y.; Tahini, H.A.; Shao, Z.; Wang, H. Metal oxide-based materials as an emerging family of hydrogen evolution electrocatalysts. Energy Environ. Sci. 2020, 13, 3361–3392. [Google Scholar] [CrossRef]
- Anantharaj, S.; Noda, S.; Jothi, V.R.; Yi, S.; Driess, M.; Menezes, P.W. Strategies and Perspectives to Catch the Missing Pieces in Energy-Efficient Hydrogen Evolution Reaction in Alkaline Media. Angew. Chem. Int. Ed. 2021, 60, 18981–19006. [Google Scholar] [CrossRef]
- Parsons, R. The rate of electrolytic hydrogen evolution and the heat of adsorption of hydrogen. Trans. Faraday Soc. 1958, 54, 1053–1063. [Google Scholar] [CrossRef]
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Nilsson, A.; Petterssonb, L.G.M.; Hammer, B.; Bligaard, T.; Christensen, C.H.; Norskov, J.K. The electronic structure effect in heterogeneous catalysis. Catal. Lett. 2005, 100, 111–114. [Google Scholar] [CrossRef]
- Greeley, J.; Norskov, J.K.; Kibler, L.A.; El-Aziz, A.M.; Kolb, D.M. Hydrogen evolution over bimetallic systems: Understanding the trends. Chemphyschem 2006, 7, 1032–1035. [Google Scholar] [CrossRef]
- Hansen, J.N.; Prats, H.; Toudahl, K.K.; Morch Secher, N.; Chan, K.; Kibsgaard, J.; Chorkendorff, I. Is There Anything Better than Pt for HER? ACS Energy Lett. 2021, 6, 1175–1180. [Google Scholar] [CrossRef]
- Podjaski, F.; Weber, D.; Zhang, S.; Diehl, L.; Eger, R.; Duppel, V.; Alarcón-Lladó, E.; Richter, G.; Haase, F.; Fontcuberta i Morral, A.; et al. Rational strain engineering in delafossite oxides for highly efficient hydrogen evolution catalysis in acidic media. Nat. Catal. 2019, 3, 55–63. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M (Ni, Co, Fe, Mn) hydr (oxy) oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- Zheng, J.; Yan, Y.; Xu, B. Correcting the Hydrogen Diffusion Limitation in Rotating Disk Electrode Measurements of Hydrogen Evolution Reaction Kinetics. J. Electrochem. Soc. 2015, 162, F1470–F1481. [Google Scholar] [CrossRef]
- Liu, E.; Li, J.; Jiao, L.; Doan, H.T.T.; Liu, Z.; Zhao, Z.; Huang, Y.; Abraham, K.M.; Mukerjee, S.; Jia, Q. Unifying the Hydrogen Evolution and Oxidation Reactions Kinetics in Base by Identifying the Catalytic Roles of Hydroxyl-Water-Cation Adducts. J. Am. Chem. Soc. 2019, 141, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Zhuang, Z.; Gao, M.; Zheng, J.; Chen, J.G.; Yan, Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 6848. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Sheng, W.; Zhuang, Z.; Xu, B.; Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2016, 2, e1501602. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; van der Vliet, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef]
- Mao, B.; Sun, P.; Jiang, Y.; Meng, T.; Guo, D.; Qin, J.; Cao, M. Identifying the Transfer Kinetics of Adsorbed Hydroxyl as a Descriptor of Alkaline Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 2020, 59, 15232–15237. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, H.; Sun, C.; Xiao, D.; Wang, H.T.; Xiao, Y.; Zhao, S.; Chen, K.H.; Lin, W.X.; Shao, Y.C.; et al. Bimetallic nanoalloys planted on super-hydrophilic carbon nanocages featuring tip-intensified hydrogen evolution electrocatalysis. Nat. Commun. 2024, 15, 7179. [Google Scholar] [CrossRef]
- Chen, X.H.; Li, X.L.; Li, T.; Jia, J.H.; Lei, J.L.; Li, N.B.; Luo, H.Q. Enhancing neutral hydrogen production by disrupting the rigid hydrogen bond network on Ru nanoclusters through Nb2O5-mediated water reorientation. Energy Environ. Sci. 2024, 17, 5091–5101. [Google Scholar] [CrossRef]
- Ledezma-Yanez, I.; Wallace, W.D.Z.; Sebastián-Pascual, P.; Climent, V.; Feliu, J.M.; Koper, M.T.M. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat. Energy 2017, 2, 17031. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, J.; Hu, Z.; Zhou, W.; Xu, X.; Zhong, Y.; Liu, B.; Chen, Y.; Xu, M.; Lin, H.J.; et al. Searching General Sufficient and Necessary Conditions for Ultrafast Hydrogen Evolving Electrocatalysis. Adv. Funct. Mater. 2019, 29, 1900704. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, J.; Huang, Y.C.; Dong, C.L.; Wang, J.Q.; Zhou, W.; Shao, Z. Screening highly active perovskites for hydrogen-evolving reaction via unifying ionic electronegativity descriptor. Nat. Commun. 2019, 10, 3755. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Yang, X.; Sun, J.; Wang, X.; Zhu, J.; Fu, Y. Competitive Adsorption Mechanism of Defect-Induced d-Orbital Single Electrons in SrRuO3 for Alkaline Hydrogen Evolution Reaction. Adv. Energy Mater. 2023, 13, 2301779. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, C.; Yang, J.; Han, T.; Liang, M.; Zhao, M.-H.; Li, G.; Li, M.-R. Boost alkaline hydrogen evolution reaction by intermetallic charge transfer in perovskite ruthenate encapsulated in ZIF-67 derived cobalt sulfide nanoparticles. Appl. Surf. Sci. 2024, 663, 160128. [Google Scholar] [CrossRef]
- Zhu, Y.; Tahini, H.A.; Hu, Z.; Dai, J.; Chen, Y.; Sun, H.; Zhou, W.; Liu, M.; Smith, S.C.; Wang, H.; et al. Unusual synergistic effect in layered Ruddlesden-Popper oxide enables ultrafast hydrogen evolution. Nat. Commun. 2019, 10, 149. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Zhou, W.; Zhu, Z.; Su, C.; Liu, M.; Shao, Z. A Perovskite Electrocatalyst for Efficient Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6442–6448. [Google Scholar] [CrossRef]
- Sun, Q.; Dai, Z.; Zhang, Z.; Chen, Z.; Lin, H.; Gao, Y.; Chen, D. Double perovskite PrBaCo2O5.5: An efficient and stable electrocatalyst for hydrogen evolution reaction. J. Power Sources 2019, 427, 194–200. [Google Scholar] [CrossRef]
- Zhang, F.; Hao, S.; Zheng, G.; Lei, L.; Zhang, X. Enhanced hydrogen evolution from the face-sharing [RuO6] octahedral motif. J. Energy Chem. 2021, 56, 276–282. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, Y.; Tahini, H.A.; Lin, Q.; Chen, Y.; Guan, D.; Zhou, C.; Hu, Z.; Lin, H.J.; Chan, T.S.; et al. Single-phase perovskite oxide with super-exchange induced atomic-scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 2020, 11, 5657. [Google Scholar] [CrossRef]
- Xu, X.; Pan, Y.; Zhong, Y.; Ge, L.; Jiang, S.P.; Shao, Z. From scheelite BaMoO4 to perovskite BaMoO3: Enhanced electrocatalysis toward the hydrogen evolution in alkaline media. Compos. Part B Eng. 2020, 198, 108214. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, Y.; Chen, Y.; Wen, X.; Long, M.; Wu, X.; Hu, Z.; Guan, D.; Wang, X.; Zhou, C.; et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 2022, 13, 1189. [Google Scholar] [CrossRef]
- Voiry, D.; Mohite, A.; Chhowalla, M. Phase engineering of transition metal dichalcogenides. Chem. Soc. Rev. 2015, 44, 2702–2712. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xia, Y. Crystal-phase and surface-structure engineering of ruthenium nanocrystals. Nat. Rev. Mater. 2020, 5, 440–459. [Google Scholar] [CrossRef]
- Chen, Y.; Lai, Z.; Zhang, X.; Fan, Z.; He, Q.; Tan, C.; Zhang, H. Phase engineering of nanomaterials. Nat. Rev. Chem. 2020, 4, 243–256. [Google Scholar] [CrossRef]
- Jung, K.N.; Jung, J.H.; Im, W.B.; Yoon, S.; Shin, K.H.; Lee, J.W. Doped lanthanum nickelates with a layered perovskite structure as bifunctional cathode catalysts for rechargeable metal-air batteries. ACS Appl. Mater. Interfaces 2013, 5, 9902–9907. [Google Scholar] [CrossRef]
- Takeguchi, T.; Yamanaka, T.; Takahashi, H.; Watanabe, H.; Kuroki, T.; Nakanishi, H.; Orikasa, Y.; Uchimoto, Y.; Takano, H.; Ohguri, N.; et al. Layered perovskite oxide: A reversible air electrode for oxygen evolution/reduction in rechargeable metal-air batteries. J. Am. Chem. Soc. 2013, 135, 11125–11130. [Google Scholar] [CrossRef]
- Cao, C.; Shang, C.; Li, X.; Wang, Y.; Liu, C.; Wang, X.; Zhou, S.; Zeng, J. Dimensionality Control of Electrocatalytic Activity in Perovskite Nickelates. Nano Lett. 2020, 20, 2837–2842. [Google Scholar] [CrossRef]
- Bartel, C.J.; Sutton, C.; Goldsmith, B.R.; Ouyang, R.; Musgrave, C.B.; Ghiringhelli, L.M.; Scheffler, M. New tolerance factor to predict the stability of perovskite oxides and halides. Sci. Adv. 2019, 5, aav0693. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, H.; Xue, L.; Sun, J.; Wang, X.; Xiong, P.; Zhu, J. Recent advances in the heteroatom doping of perovskite oxides for efficient electrocatalytic reactions. Nanoscale 2021, 13, 19840–19856. [Google Scholar] [CrossRef]
- Kuznetsov, D.A.; Naeem, M.A.; Kumar, P.V.; Abdala, P.M.; Fedorov, A.; Muller, C.R. Tailoring Lattice Oxygen Binding in Ruthenium Pyrochlores to Enhance Oxygen Evolution Activity. J. Am. Chem. Soc. 2020, 142, 7883–7888. [Google Scholar] [CrossRef]
- Retuerto, M.; Pascual, L.; Calle-Vallejo, F.; Ferrer, P.; Gianolio, D.; Pereira, A.G.; Garcia, A.; Torrero, J.; Fernandez-Diaz, M.T.; Bencok, P.; et al. Na-doped ruthenium perovskite electrocatalysts with improved oxygen evolution activity and durability in acidic media. Nat. Commun. 2019, 10, 2041. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Wu, Q.; Liu, H.; Gu, W.; Wang, X.; Cheng, Z.; Fu, Z.; Lu, Y. Optimized Electronic Configuration to Improve the Surface Absorption and Bulk Conductivity for Enhanced Oxygen Evolution Reaction. J. Am. Chem. Soc. 2019, 141, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Shi, L.; Liu, Y.; Chen, H.; Si, R.; Yan, W.; Zhang, Q.; Li, G.D.; Yang, L.; Zou, X. Activating Inert, Nonprecious Perovskites with Iridium Dopants for Efficient Oxygen Evolution Reaction under Acidic Conditions. Angew. Chem. Int. Ed. 2019, 58, 7631–7635. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Shi, L.; Cao, R.; Wan, G.; Yan, W.; Chen, H.; Liu, Y.; Zou, X. Perovskite-Type Solid Solution Nano-Electrocatalysts Enable Simultaneously Enhanced Activity and Stability for Oxygen Evolution. Adv. Mater. 2020, 32, e2001430. [Google Scholar] [CrossRef]
- Hua, B.; Li, M.; Pang, W.; Tang, W.; Zhao, S.; Jin, Z.; Zeng, Y.; Shalchi Amirkhiz, B.; Luo, J.-L. Activating p-Blocking Centers in Perovskite for Efficient Water Splitting. Chem 2018, 12, 2902–2916. [Google Scholar] [CrossRef]
- Su, C.; Wang, W.; Shao, Z. Cation-Deficient Perovskites for Clean Energy Conversion. Acc. Mater. Res. 2021, 2, 477–488. [Google Scholar] [CrossRef]
- Arandiyan, H.; Mofarah, S.S.; Sorrell, C.C.; Doustkhah, E.; Sajjadi, B.; Hao, D.; Wang, Y.; Sun, H.; Ni, B.J.; Rezaei, M.; et al. Defect engineering of oxide perovskites for catalysis and energy storage: Synthesis of chemistry and materials science. Chem. Soc. Rev. 2021, 50, 10116–10211. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Chen, D.; Liu, J.; Zhang, Z.; Shao, Z.; Ciucci, F. Water Splitting with an Enhanced Bifunctional Double Perovskite. ACS Catal. 2017, 8, 364–371. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, P.; Bai, H.; Ai, H.; Du, X.; Chen, M.; Liu, D.; Ip, W.F.; Lo, K.H.; Kwok, C.T.; et al. Development of Perovskite Oxide-Based Electrocatalysts for Oxygen Evolution Reaction. Small 2021, 17, e2101605. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Yu, J.; Chen, Y.; Liu, M.; Shao, Z. Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions. Chem. Mater. 2016, 28, 1691–1697. [Google Scholar] [CrossRef]
- Da, Y.; Zeng, L.; Wang, C.; Gong, C.; Cui, L. A simple approach to tailor OER activity of SrxCo0.8Fe0.2O3 perovskite catalysts. Electrochim. Acta 2019, 300, 85–92. [Google Scholar] [CrossRef]
- Chen, G.; Zhu, Y.; Chen, H.M.; Hu, Z.; Hung, S.F.; Ma, N.; Dai, J.; Lin, H.J.; Chen, C.T.; Zhou, W.; et al. An Amorphous Nickel-Iron-Based Electrocatalyst with Unusual Local Structures for Ultrafast Oxygen Evolution Reaction. Adv. Mater. 2019, 31, e1900883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guan, D.; Gao, X.; Yu, J.; Chen, G.; Zhou, W.; Shao, Z. Morphology, crystal structure and electronic state one-step co-tuning strategy towards developing superior perovskite electrocatalysts for water oxidation. J. Mater. Chem. A 2019, 7, 19228–19233. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, Y.; Zhong, Y.; Miao, J.; Lin, B.; Zhou, W.; Shao, Z. Enabling High and Stable Electrocatalytic Activity of Iron-Based Perovskite Oxides for Water Splitting by Combined Bulk Doping and Morphology Designing. Adv. Mater. Interfaces 2018, 6, 1801317. [Google Scholar] [CrossRef]
- Chen, H.; Shi, L.; Liang, X.; Wang, L.; Asefa, T.; Zou, X. Optimization of Active Sites via Crystal Phase, Composition, and Morphology for Efficient Low-Iridium Oxygen Evolution Catalysts. Angew. Chem. Int. Ed. 2020, 59, 19654–19658. [Google Scholar] [CrossRef]
- Kim, M.; Ju, H.; Kim, J. Dihydrogen phosphate ion functionalized nanocrystalline thallium ruthenium oxide pyrochlore as a bifunctional electrocatalyst for aqueous Na-air batteries. Appl. Catal. B Environ. 2019, 245, 29–39. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Electrolyte | η at 10 mA cm−2 (mV) | Tafel Slope (mV dec−1) | Reference |
|---|---|---|---|---|
| Ba2MIrO6 (M = Y, La, Ce, Pr, Nd, Tb) | 0.1 M HClO4 | >370 | 60–120 | [69] |
| Y2Ir2O7 | 0.1 M HClO4 | 262 | 50 | [71] |
| 6H-SrIrO3 | 0.5 M H2SO4 | 248 | / | [72] |
| CaCu3Ru4O12 | 0.5 M H2SO4 | 171 | 40 | [54] |
| Ba3TiIr2O9 | 0.1 M HClO4 | 275 | 45.7 | [73] |
| Ba4Sr4(Co0.8Fe0.2)4O15 | 0.1 M KOH | 340 | 47 | [68] |
| Sr2MIrO6 (M = Ni, Co, Sc, Fe) | 0.1 M HClO4 | 295–420 | 48–90 | [74] |
| CaCu3Ir4O12 | 1 M KOH | 252 | 47 | [67] |
| Sr3Ir2O7 | 0.5 M H2SO4 | 259 | 50 | [75] |
| Cu2IrO3 | 1 M KOH | 361 | 51 | [35] |
| SrIr2O6 | 0.1 M HClO4 | 303 | 44.2 | [76] |
| Dy2NiRuO6 | 0.1 M HClO4 | 277 | 58 | [77] |
| Li2Mn0.85Ru0.15O3 | 0.1 M KOH | 260 | 49.6 | [78] |
| Catalysts | Electrolyte | η at 10 mA cm−2 (mV) | Tafel Slope (mV dec−1) | Reference |
|---|---|---|---|---|
| Pr0.5(Ba0.5Sr0.5)0.5Co0.8Fe0.2O3−δ | 1 M KOH | 237 | 45 | [105] |
| (Gd0.5La0.5)BaCo2O5.5+δ | 1 M KOH | 210 | 27.6 | [101] |
| PrBaCo2O5.5+δ | 0.1 M KOH | 291 | 89 | [106] |
| SrRuO3 | 1 M KOH | 101 | 67 | [104] |
| Sr2RuO4 | 1 M KOH | 61 | 51 | [104] |
| SrRu0.9Co0.1O3−x | 1 M KOH | 57.8 | 35 | [102] |
| Sr4Ru2O9 | 1 M KOH | 28 | 55 | [107] |
| SrTi0.7Ru0.3O3−x | 1 M KOH | 46 | 40 | [108] |
| BaMoO3 | 1 M KOH | 336 | 110 | [109] |
| 9R-BaRuO3 | 1 M KOH | 51 | 30 | [7] |
| La2Sr2PtO7+δ | 0.5 M H2SO4 | 13 | 22 | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Zhao, C.; Tian, H.; Tong, S.-Y. Correlation of Structure and Electrocatalytic Performance of Bulk Oxides for Water Electrolysis. Molecules 2025, 30, 2391. https://doi.org/10.3390/molecules30112391
Zhu C, Zhao C, Tian H, Tong S-Y. Correlation of Structure and Electrocatalytic Performance of Bulk Oxides for Water Electrolysis. Molecules. 2025; 30(11):2391. https://doi.org/10.3390/molecules30112391
Chicago/Turabian StyleZhu, Chuanhui, Changming Zhao, Hao Tian, and Shuk-Yin Tong. 2025. "Correlation of Structure and Electrocatalytic Performance of Bulk Oxides for Water Electrolysis" Molecules 30, no. 11: 2391. https://doi.org/10.3390/molecules30112391
APA StyleZhu, C., Zhao, C., Tian, H., & Tong, S.-Y. (2025). Correlation of Structure and Electrocatalytic Performance of Bulk Oxides for Water Electrolysis. Molecules, 30(11), 2391. https://doi.org/10.3390/molecules30112391