
First-Principles Study of Rh Segregation in the Au–Rh(111) Alloy with Adsorbed NO, CO, or O2
 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Adsorption Behavior
3.2. Segregation Behavior
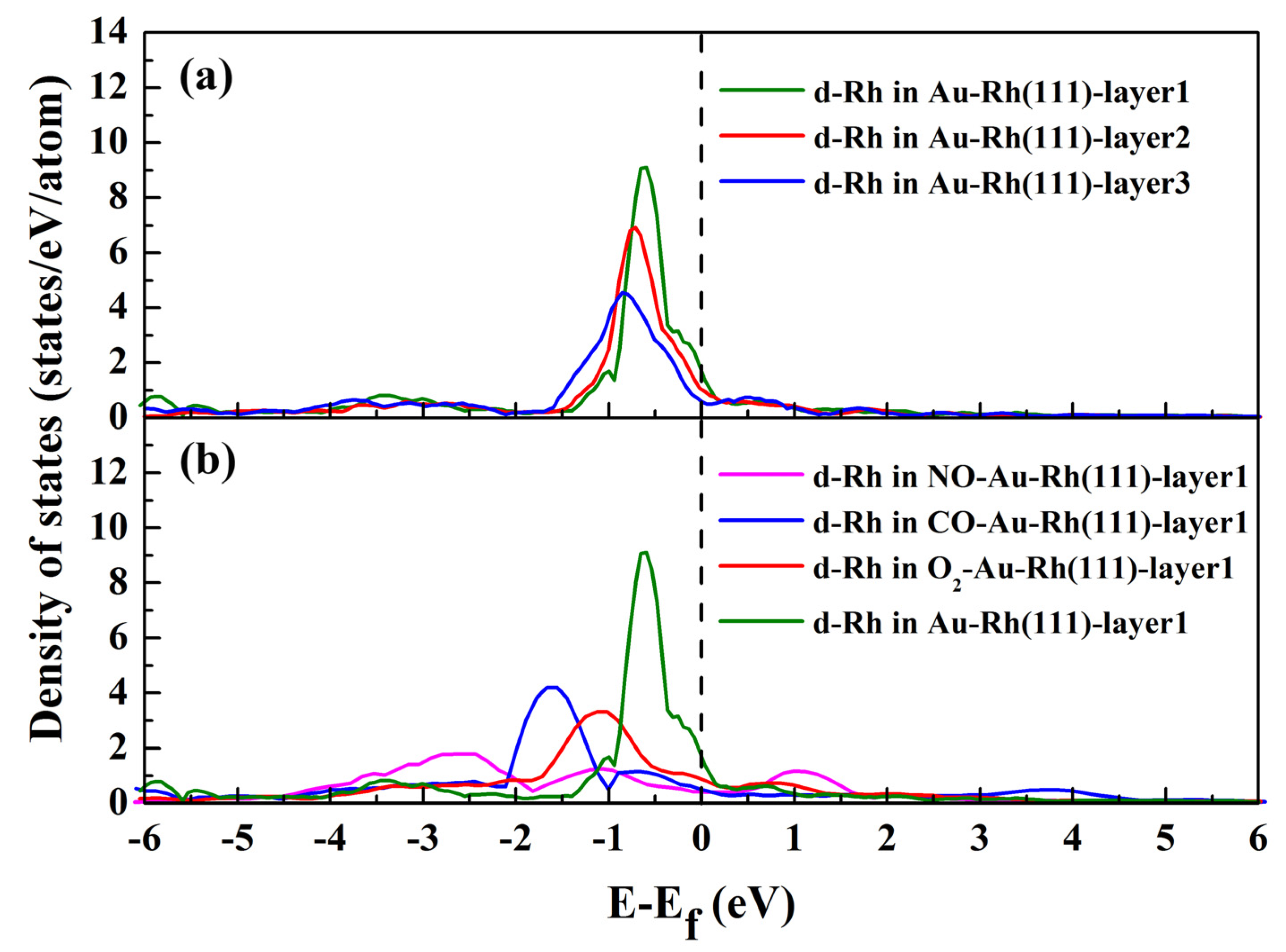
3.3. Electronic Structure Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, S.; Jelic, J.; Rein, D.; Najafishirtari, S.; Schmidt, F.P.; Girgsdies, F.; Kang, L.; Wandzilak, A.; Rabe, A.; Doronkin, D.E. Highly loaded bimetallic iron-cobalt catalysts for hydrogen release from ammonia. Nat. Commun. 2024, 15, 871. [Google Scholar] [CrossRef]
- Gholinejad, M.; Bashirimousavi, S.; Sansano, J.M. Novel magnetic bimetallic AuCu catalyst for reduction of nitroarenes and degradation of organic dyes. Sci. Rep. 2024, 14, 5852. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yang, F.; Chen, S.; Wu, H.; Yang, J.; Shen, F. Highly Efficient Catalytic Oxidation of Glucose to Formic Acid over Mn-Mo Doped Carbon Nanotube. Molecules 2025, 30, 1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Zhu, S.; Hitch, M. Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification. Molecules 2025, 30, 1471. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, Q.; Xiao, W.; Wang, J.; Wang, L. Design of highly efficient Ni-based water-electrolysis catalysts by a third transition metal addition into Ni3Mo. Intermetallics 2018, 94, 99–105. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, W.; Wang, J.; Wang, L. Understanding the surface segregation behavior of transition metals on Ni (111): A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 26616–26622. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Z.; Huang, W.; Zhou, S.; Hu, Z.; Wang, L. Density functional theory study of Ni segregation in CuNi (111) alloy with chemisorbed CO, O, or H. J. Phys. Chem. Solids 2022, 171, 111021. [Google Scholar] [CrossRef]
- Yu, Y.; Huang, W.; Liu, Z.; Hu, Z.; Wang, L. First-principles study of surface segregation in bimetallic Cu3M (1 1 1) (M= Au, Ag, and Zn) alloys in presence of adsorbed CO. Comput. Mater. Sci. 2022, 212, 111550. [Google Scholar] [CrossRef]
- Mashkovsky, I.; Bukhtiyarov, A.; Markov, P.; Bragina, G.; Baeva, G.; Smirnova, N.; Panafidin, M.; Chetyrin, I.; Gerasimov, E.Y.; Zubavichus, Y. Catalytic performance of a single atom Pd1Ag10/Al2O3 catalyst for the selective hydrogenation of acetylene: The role of CO-induced segregation. Appl. Surf. Sci. 2025, 681, 161516. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, H.Y.; Ham, H.C.; Yoo, S.J.; Kim, H.J.; Cho, E.; Manthiram, A.; Jang, J.H. Reversible surface segregation of Pt in a Pt3Au/C catalyst and its effect on the oxygen reduction reaction. J. Phys. Chem. C 2013, 117, 9164–9170. [Google Scholar] [CrossRef]
- Wang, J.W.; Wang, Y.F.; Zhang, J.G.; Yu, Y.L.; Zhou, G.G. Optimization of electrocatalytic properties of NiMoCo foam electrode for water electrolysis by post-treatment processing. Rare Met. 2015, 34, 802–807. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- McCrory, C.C.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Lu, L.; Peng, L.; Li, L.; Li, J.; Huang, X.; Wei, Z. Improved hydrogen oxidation reaction under alkaline conditions by Au–Pt alloy nanoparticles. J. Energy Chem. 2020, 40, 52–56. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, P.; Huang, X.; Hu, C.; Wen, Z.; Lu, Z.H. Anchoring PdAu nanoclusters inside aminated metal-organic framework for fast dehydrogenation of formic acid. Fuel 2024, 359, 130459. [Google Scholar] [CrossRef]
- Weng, X.; Liu, Y.; Wang, K.K.; Feng, J.J.; Yuan, J.; Wang, A.J.; Xu, Q.Q. Single-step aqueous synthesis of AuPt alloy nanodendrites with superior electrocatalytic activity for oxygen reduction and hydrogen evolution reaction. Int. J. Hydrogen Energy 2016, 41, 18193–18202. [Google Scholar] [CrossRef]
- Wei, H.; Wei, X.; Yang, X.; Yin, G.; Wang, A.; Liu, X.; Huang, Y.; Zhang, T. Supported Au-Ni nano-alloy catalysts for the chemoselective hydrogenation of nitroarenes. Chin. J. Catal. 2015, 36, 160–167. [Google Scholar] [CrossRef]
- Luo, J.; Njoki, P.N.; Lin, Y.; Wang, L.; Zhong, C.J. Activity-composition correlation of AuPt alloy nanoparticle catalysts in electrocatalytic reduction of oxygen. Electrochem. Commun. 2006, 8, 581–587. [Google Scholar] [CrossRef]
- Chin, Y.; King, D.L.; Roh, H.S.; Wang, Y.; Heald, S.M. Structure and reactivity investigations on supported bimetallic AuNi catalysts used for hydrocarbon steam reforming. J. Catal. 2006, 244, 153–162. [Google Scholar] [CrossRef]
- Barakat, T.; Rooke, J.C.; Genty, E.; Cousin, R.; Siffert, S.; Su, B.L. Gold catalysts in environmental remediation and water-gas shift technologies. Energy Environ. Sci. 2013, 6, 371–391. [Google Scholar] [CrossRef]
- Mancilla, A.; Mendoza-Cruz, R.; Portales, B.; Zanella, R. Catalytic oxidation of carbon monoxide at low temperature using AuRh/TiO2 bimetallic catalysts: Effect of the synthesis method. Mater. Today Chem. 2024, 38, 102092. [Google Scholar] [CrossRef]
- Wang, X.; Lu, G.; Guo, Y.; Zhang, Z.; Guo, Y. Role of Rh promoter on increasing stability of Au/Al2O3 catalyst for CO oxidation at low temperature. Environ. Chem. Lett. 2011, 9, 185–189. [Google Scholar] [CrossRef]
- Van Delft, F.; Siera, J.; Nieuwenhuys, B. The transient behaviour of Pt-Rh (410) alloy surfaces upon interaction with O2, NO and CO. Surf. Sci. 1989, 208, 365–382. [Google Scholar] [CrossRef]
- Piccolo, L.; Li, Z.; Demiroglu, I.; Moyon, F.; Konuspayeva, Z.; Berhault, G.; Afanasiev, P.; Lefebvre, W.; Yuan, J.; Johnston, R.L. Understanding and controlling the structure and segregation behaviour of AuRh nanocatalysts. Sci. Rep. 2016, 6, 35226. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1993, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 2665–2668. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B Condens. Matter 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Hendrik, J.; James, D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dhifallah, M.; Dhouib, A.; Aldulaijan, S.; Di Renzo, F.; Guesmi, H. First-principles study of Au–Cu alloy surface changes induced by gas adsorption of CO, NO, or O2. J. Chem. Phys. 2016, 145, 024701. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dai, C.; Fisher, A.; Shen, Y.; Cheng, D. A full understanding of oxygen reduction reaction mechanism on Au (1 1 1) surface. J. Phys.-Condens. Matter 2017, 29, 365201. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, Y. Triangular nanoplates of silver: Synthesis, characterization, and use as sacrificial templates for generating triangular nanorings of gold. Adv. Mater. 2003, 15, 695–699. [Google Scholar] [CrossRef]
- Dhouib, A.; Guesmi, H. DFT study of the M segregation on MAu alloys (M=Ni, Pd, Pt) in presence of adsorbed oxygen O and O2. Chem. Phys. Lett. 2012, 521, 98–103. [Google Scholar] [CrossRef]
- Sansa, M.; Dhouib, A.; Guesmi, H. Density functional theory study of CO-induced segregation in gold-based alloys. J. Chem. Phys. 2014, 141, 064709. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, W.; Wang, J.; Wang, L. First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen. Materials 2016, 9, 5. [Google Scholar] [CrossRef]
- Ishikawa, A.; Tateyama, Y. First-principles microkinetic analysis of NO+ CO reactions on Rh (111) surface toward understanding NOx reduction pathways. J. Phys. Chem. C 2018, 122, 17378–17388. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Stoltze, P.; Nørskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Gottfried, J.; Schmidt, K.; Schroeder, S.; Christmann, K. Spontaneous and electron-induced adsorption of oxygen on Au (110)-(1×2). Surf. Sci. 2002, 511, 65–82. [Google Scholar] [CrossRef]
- Wang, G.; Hove, M.A.V.; Ross, P.N.; Baskes, M.I. Quantitative prediction of surface segregation in bimetallic Pt–M alloy nanoparticles (M=Ni, Re, Mo). Prog. Surf. Sci. 2005, 79, 28–45. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, Z.; Xiao, C.; Wang, G. Density functional theory calculation of platinum surface segregation energy in Pt3Ni (111) surface doped with a third transition metal. Surf. Sci. 2011, 605, 1577–1582. [Google Scholar] [CrossRef]
- DeBoer, F.R.; Boom, R.; Miedema, A.R. Cohesion in Metals, 2nd ed.; North-Holland Physics Publishing: Amsterdam, The Netherlands, 1989; pp. 657–660. [Google Scholar]
- Allinger, N.L.; Zhou, X.; Bergsma, J. Molecular mechanics parameters. J. Mol. Struct. THEOCHEM 1994, 312, 69–83. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L. Calculated surface segregation in transition metal alloys. Comput. Mater. Sci. 1999, 15, 119–143. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L.; Norskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990–16000. [Google Scholar] [CrossRef]
- Krebs, E.; Silvi, B.; Raybaud, P. Mixed sites and promoter segregation: A DFT study of the manifestation of Le Chatelier’s principle for the Co (Ni) MoS active phase in reaction conditions. Catal. Today 2008, 130, 160–169. [Google Scholar] [CrossRef]
- Ma, Y.; Balbuena, P.B. Surface segregation in bimetallic Pt3M (M = Fe, Co, Ni) alloys with adsorbed oxygen. Surf. Sci. 2009, 603, 349–353. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Wang, L.G.; Tsymbal, E.Y.; Jaswal, S.S. Structural and magnetic properties of clean and methylthiolate-adsorbed Co(0001) surfaces: A first-principles study. J. Magn. Magn. Mater. 2005, 286, 119–123. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top | Bridge | fcc | hcp | ||
|---|---|---|---|---|---|
| Au(111) | NO | −0.28 | −0.18 | −0.10 | −0.07 |
| CO | −0.35 | −0.26 | −0.18 | −0.17 | |
| O2 | No adsorption | ||||
| Rh(111) | NO | −1.85 | −2.17 | −2.37 | −2.37 |
| CO | −1.80 | −1.79 | −1.85 | −1.86 | |
| O2 | No adsorption | −1.28 | −1.32 | −1.31 | |
| Position of the Rh Atom | NO | CO | O2 |
|---|---|---|---|
| 1st layer | −2.44 | −2.14 | −0.79 |
| 2nd layer | −0.37 | −0.46 | No adsorption |
| 3rd layer | −0.31 | −0.37 | No adsorption |
| 4th layer | −0.31 | −0.38 | No adsorption |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, Y.; Yu, Y.; Gu, H.; Kang, Y.; Zhao, G.; Li, Y.; Huang, Q. First-Principles Study of Rh Segregation in the Au–Rh(111) Alloy with Adsorbed NO, CO, or O2. Molecules 2025, 30, 2389. https://doi.org/10.3390/molecules30112389
Wen Y, Yu Y, Gu H, Kang Y, Zhao G, Li Y, Huang Q. First-Principles Study of Rh Segregation in the Au–Rh(111) Alloy with Adsorbed NO, CO, or O2. Molecules. 2025; 30(11):2389. https://doi.org/10.3390/molecules30112389
Chicago/Turabian StyleWen, Yufeng, Yanlin Yu, Huaizhang Gu, Yuexin Kang, Guoqi Zhao, Yuanxun Li, and Qiuling Huang. 2025. "First-Principles Study of Rh Segregation in the Au–Rh(111) Alloy with Adsorbed NO, CO, or O2" Molecules 30, no. 11: 2389. https://doi.org/10.3390/molecules30112389
APA StyleWen, Y., Yu, Y., Gu, H., Kang, Y., Zhao, G., Li, Y., & Huang, Q. (2025). First-Principles Study of Rh Segregation in the Au–Rh(111) Alloy with Adsorbed NO, CO, or O2. Molecules, 30(11), 2389. https://doi.org/10.3390/molecules30112389