
Design, Synthesis, and Antitumor Biological Evaluation of Galaxamide and Its Analogs


Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Activity of Galaxamide and Its Analogs
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. Synthesis of 2
- N-Boc-Me-L-Leu-OH 2a. The 1a was synthesized according to the method of 2 from L-Leu (9.0 g, 38.9 mmol, 1.0 equiv), CH3I (25 g, 177.3 mmol, 4.5 equiv), and NaH (60% dispersion in mineral oil, 5.5 g, 137.5 mmol, 3.5 equiv). Following a wash with brine, the mixed EtOAc extracts were dried over Na2SO4, filtered, and evaporated to produce 2a (9.0 g, 36.7 mmol, 94% yield) as a white solid. [α]25D = −36.2 (c = 1.0, CHCl3). m.p. 55–56 °C. 1H NMR (600 MHz, CDCl3) δ 4.83 (t, J = 7.9 Hz, 1H, major), 4.71–4.52 (m, 1H, minor), 2.85 (s, 3H, major), 2.82 (s, 3H, minor), 1.75 (t, J = 7.8 Hz, 2H), 1.66–1.56 (m, 1H), 1.49 (s, 9H, major), 1.48 (s, 9H, minor), 0.97 (dd, J = 10.8, 6.6 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 178.0, 156.6 (major), 155.7 (minor), 57.0 (major), 56.2 (minor), 37.8 (major), 37.3 (minor), 30.7 (major), 30.6 (minor), 28.3, 24.9 (major), 24.7 (minor), 23.2 (major), 23.1 (minor), 21.3 (major), 21.1 (minor). HRMS (ESI+) calcd. for C12H23NO4 [M + Na]+: 268.1520; found: 268.1527.
- N-Boc-Me-D-Leu-OH 2b. The 1b was synthesized according to the method of 2 from D-Leu (9.0 g, 38.9 mmol, 1.0 equiv), CH3I (25 g, 177.3 mmol, 4.5 equiv), and NaH (60% dispersion in mineral oil, 5.5 g, 137.5 mmol, 3.5 equiv). Following a wash with brine, the mixed EtOAc extracts were dried over Na2SO4, filtered, and evaporated to produce 2b (8.8 g, 35.9 mmol, 91% yield) as a white solid. [α]25D = +36.2 (c = 1.0, CHCl3). m.p. 55–56 °C. 1H NMR (600 MHz, Chloroform-d) δ 4.87 (t, J = 7.7 Hz, 1H, major), 4.63 (dd, J = 11.2, 5.1 Hz, 1H, minor), 2.84 (s, 3H, major), 2.81 (s, 3H, minor), 1.79–1.64 (m, 2H), 1.64–1.53 (m, 1H), 1.48 (s, 9H, major), 1.47 (s, minor), 0.96 (dd, J = 11.4, 6.3 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 177.9 (major), 177.7 (minor), 156.5 (major), 155.7 (minor), 80.6 (major), 80.3 (minor), 57.1 (major), 56.1 (minor), 37.8 (major), 37.3 (minor), 30.6, 28.3 (major), 28.3 (minor), 24.9 (major), 24.6 (minor), 23.2 (major), 23.1 (minor), 21.2 (major), 21.1 (minor). HRMS (ESI+) calcd. for C12H23NO4 [M + Na]+: 268.1520; found: 268.1527.
3.1.2. Synthesis of Dipeptide 3
- N-Boc-Me-L-Leu-L-Leu-OBn 3a. The 3a was synthesized according to the method of 3 from 2a (10.0 g, 40.8 mmol, 1.0 equiv) and L-Leu-OBn·TosOH (9.9 g, 44.9 mmol, 1.1 equiv), HOBT (6.0 g, 44.9 mmol, 1.1 equiv), and EDCI (8.5 g, 44.9 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 90:10, v/v) to obtain 3a (15.0 g, 33.4 mmol, 82% yield) as a white solid. [α]25D = −272.8 (c = 1.0, CHCl3). m.p. 80–81 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.23 (d, J = 7.8 Hz, 1H), 7.41–7.29 (m, 5H), 5.14–5.01 (m, 2H), 4.64 (s, 1H, major), 4.47 (s, 1H, minor), 4.43–4.27 (m, 1H), 2.70 (s, 3H), 1.69–1.40 (m, 6H), 1.39 (s, 9H, major), 1.36 (s, 9H, minor)., 0.95–0.70 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 171.9, 136.4, 128.8, 128.4, 128.3, 79.3, 66.4, 50.8, 30.2, 28.5, 24.9, 24.7, 23.2, 21.6. HRMS (ESI) m/z calcd. for C25H40N2O5 [M + Na]+: 471.2830; found: 471.2838.
- N-Boc-Me-D-Leu-L-Leu-OBn 3b. The 3b was synthesized according to the method of 3 from 2b (10.0 g, 40.8 mmol, 1.0 equiv) and L-Leu-OBn·TosOH (9.9 g, 44.9 mmol, 1.1 equiv), HOBT (6.0 g, 44.9 mmol, 1.1 equiv), and EDCI (8.5 g, 44.9 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 90:10, v/v) to obtain 3b (14.7 g, 32.8 mmol, 80% yield) as a white solid. [α]25D = −245.8 (c = 1.0, CHCl3). m.p. 80–81 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.21 (d, J = 37.5 Hz, 1H), 7.44–7.18 (m, 5H), 5.12 (s, 2H), 4.74 (s, 1H, major), 4.50 (s, 1H, minor), 4.37 (s, 1H), 2.70 (s, 3H), 1.59 (m, 6H), 1.40 (s, 9H), 0.98–0.72 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.7, 171.6, 136.4, 128.8, 128.4, 128.3, 79.3, 66.4, 50.8, 38.0, 28.5, 24.8, 23.2, 21.6. HRMS (ESI) m/z calcd. for C25H40N2O5 [M + Na]+: 471.2830; found: 471.2838.
- N-Boc-Me-L-Leu-D-Leu-OBn 3c. The 3c was synthesized according to the method of 3 from 2a (10.0 g, 40.8 mmol, 1.0 equiv) and D-Leu-OBn·TosOH (9.9 g, 44.9 mmol, 1.1 equiv), HOBT (6.0 g, 44.9 mmol, 1.1 equiv), and EDCI (8.5 g, 44.9 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 90:10, v/v) to obtain 3c (15.2 g, 33.9 mmol, 82% yield) as a white solid. [α]25D = −427.9 (c = 1.0, CHCl3). m.p. 80–81 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.42–8.16 (m, 1H), 7.44–7.26 (m, 5H), 5.11 (d, J = 4.2 Hz, 2H), 4.71 (d, J = 8.5 Hz, 1H, major), 4.49 (d, J = 8.4 Hz, 1H, minor), 4.39–4.23 (m, 1H), 2.69 (s, 3H, major), 2.67 (s, 3H, minor), 1.73–1.41 (m, 6H), 1.40 (s, 9H, major), 1.38 (s, 9H, minor), 0.94–0.76 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.7, 171.7, 136.4, 128.9, 128.5, 128.3, 79.3, 66.4, 50.80, 38.0, 28.5, 24.8, 23.3, 21.6. HRMS (ESI) m/z calcd. for C25H40N2O5 [M + Na]+: 471.2830; found: 471.2838.
- N-Boc-Me-D-Leu-D-Leu-OBn 3d. The 3d was synthesized according to the method of 3 from 2b (10.0 g, 40.8 mmol, 1.0 equiv) and D-Leu-OBn·TosOH (9.9 g, 44.9 mmol, 1.1 equiv), HOBT (6.0 g, 44.9 mmol, 1.1 equiv), and EDCI (8.5 g, 44.9 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 90:10, v/v) to obtain 3d (14.8 g, 33.0 mmol, 79% yield) as a white solid. [α]25D = −215.6 (c = 1.0, CHCl3). m.p. 80–81 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.20 (d, J = 7.8 Hz, 1H), 7.54–7.21 (m, 5H), 5.11 (q, J = 12.6 Hz, 2H), 4.67 (s, 1H, major), 4.49 (s, 1H, minor), 4.37 (d, J = 15.0 Hz, 1H), 2.73 (s, 3H), 1.73–1.43 (m, 6H), 1.40 (s, 9H), 0.94–0.73 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 171.9, 136.4, 128.8, 128.5, 128.3, 79.3, 66.4, 50.8, 28.5, 24.9, 24.7, 23.2, 21.5. HRMS (ESI) m/z calcd. for C25H40N2O5 [M + Na]+: 471.2830; found: 471.2838.
3.1.3. Synthesis of 4
- N-Me-L-Leu-L-Leu-OBn 4a. The 4a was synthesized according to the method of 4 from 3a (15.0 g, 33.4 mmol, 1.0 equiv), TFA (10 mL), DCM (40 mL). After that excess TFA and DCM was removed in vacuo to produce 5a (10.0 g, 28.7 mmol, 86% yield) as a colorless oil. [α]25D = −272.8 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 8.20 (d, J = 8.2 Hz, 1H), 7.52–7.17 (m, 5H), 5.11 (d, J = 3.8 Hz, 2H), 4.42 (ddd, J = 10.5, 8.1, 4.5 Hz, 1H), 2.93 (t, J = 7.2 Hz, 1H), 2.15 (s, 3H), 1.64–1.59 (m, 2H), 1.55–1.48 (m, 1H), 1.27 (m, 2H), 1.04–0.69 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 175.2, 172.8, 136.4, 128.9, 128.5, 128.4, 66.44, 62.7, 50.3, 43.1, 34.6, 24.8, 24.7, 23.3, 23.3, 22.9, 21.5. HRMS (ESI) m/z calcd. for C20H32N2O3 [M + H]+: 349.2486; found: 349.2487.
- N-Me-D-Leu-L-Leu-OBn 4b. The 4b was synthesized according to the method of 4 from 3b (15.0 g, 33.4 mmol, 1.0 equiv), TFA (10 mL), DCM (40 mL). After that excess TFA and DCM was removed in vacuo to produce 5b (9.8 g, 28.1 mmol, 84% yield) as a colorless oil. [α]25D = −245.8 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 8.25 (d, J = 8.0 Hz, 1H), 7.44–7.23 (m, 5H), 5.12 (s, 2H), 4.40 (dd, J = 11.2, 6.7 Hz, 1H), 2.97 (t, J = 7.3 Hz, 1H), 2.19 (s, 3H), 1.65 (m, 3H), 1.55 (d, J = 10.8 Hz, 1H), 1.31 (dt, J = 15.8, 6.9 Hz, 2H), 0.98–0.72 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 175.4, 172.8, 136.4, 128.8, 128.5, 128.3, 66.4, 62.7, 50.6, 42.9, 34.7, 24.8, 24.8, 23.3, 23.1, 22.9, 21.4. HRMS (ESI) m/z calcd. for C20H32N2O3 [M + H]+: 349.2486; found: 349.2487.
- N-Me-L-Leu-D-Leu-OBn 4c. The 4c was synthesized according to the method of 4 from 3c (15.0 g, 33.4 mmol, 1.0 equiv), TFA (10 mL), DCM (40 mL). After that excess TFA and DCM was removed in vacuo to produce 5c (10.4 g, 29.8 mmol, 89% yield) as a colorless oil. [α]25D = −427.9 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 8.27 (d, J = 8.0 Hz, 1H), 7.44–7.25 (m, 5H), 5.11 (s, 2H), 4.36 (ddt, J = 7.5, 5.2, 2.8 Hz, 1H), 3.02–2.86 (m, 1H), 2.16 (d, J = 1.7 Hz, 3H), 1.70–1.55 (m, 3H), 1.53 (dt, J = 9.5, 3.6 Hz, 1H), 1.34–1.20 (m, 2H), 0.97–0.71 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 175.5, 172.9, 136.4, 128.9, 128.5, 128.4, 66.4, 62.6, 50.6, 42.9, 34.6, 24.8, 24.8, 23.3, 23.1, 23.0, 21.4. HRMS (ESI) m/z calcd. for C20H32N2O3 [M + H]+: 349.2486; found: 349.2487.
- N-Me-D-Leu-D-Leu-OBn 4d. The 4d was synthesized according to the method of 4 from 3d (15.0 g, 33.4 mmol, 1.0 equiv), TFA (10 mL), DCM (40 mL). After that excess TFA and DCM was removed in vacuo to produce 5d (9.5 g, 27.3 mmol, 81% yield) as a colorless oil. [α]25D = −215.6 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 8.17 (d, J = 8.2 Hz, 1H), 7.35 (q, J = 8.5 Hz, 5H), 5.12 (d, J = 3.3 Hz, 2H), 4.46 (q, J = 9.4, 7.5 Hz, 1H), 2.93 (t, J = 7.4 Hz, 1H), 2.17 (s, 3H), 1.64 (m, 3H), 1.55 (d, J = 11.8 Hz, 1H), 1.38–1.17 (m, 2H), 1.03–0.67 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 175.2, 172.8, 136.4, 128.9, 128.5, 128.4, 66.4, 62.8, 50.3, 43.1, 34.6, 24.8, 24.7, 23.3, 23.3, 22.9, 21.4. HRMS (ESI) m/z calcd. for C20H32N2O3 [M + H]+: 349.2486; found: 349.2487.
3.1.4. Synthesis of 7
- N-Boc-Me-L-Leu-L-Leu-OH 7a. The 7a was synthesized according to the method of 7 from 3a (15.0 g, 33.4 mmol, 1.0 equiv), 10% Pd/C (5 g, 4.7 mmol, 0.1 equiv), H2 (1 atm), 2 h. The mixture was filtered and concentrated in vacuo to give 7a (10.0 g, 27.9 mmol, 84% yield) as a colorless oil. [α]25D = −272.8 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 12.48 (s, 1H), 7.99 (d, J = 12.9 Hz, 1H), 4.63 (t, J = 8.0 Hz, 1H, major), 4.47 (d, J = 10.4 Hz, 1H, minor), 4.31–4.00 (m, 1H), 2.71 (s, 3H), 1.78–1.47 (m, 5H), 1.45–1.27 (m, 9H), 1.10–0.57 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 174.4, 171.6, 152.2, 79.3, 50.5, 31.0, 28.5, 25.0, 24.8, 23.6, 23.4, 21.6. HRMS (ESI) m/z calcd. for C18H34N2O5 [M + Na]+: 381.2360; found: 381.2261.
- N-Boc-Me-D-Leu-L-Leu-OH 7b. The 7b was synthesized according to the method of 7 from 3b (15.0 g, 33.4 mmol, 1.0 equiv), 10% Pd/C (5 g, 4.7 mmol, 0.1 equiv), H2 (1 atm), 2 h. The mixture was filtered and concentrated in vacuo to give 7b (9.8 g, 27.3 mmol, 82% yield) as a colorless oil. [α]25D = −245.8 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 7.93 (dd, J = 25.2, 9.2 Hz, 1H), 4.63 (s, 1H, major), 4.47 (d, J = 10.6 Hz, 1H, minor), 4.35–4.10 (m, 1H), 2.72 (s, 3H), 1.68–1.47 (m, 5H), 1.40 (s, 9H), 1.00–0.64 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 174.5, 171.6, 79.3, 56.8, 50.7, 38.0, 37.6, 30.3, 28.5, 25.0, 24.8, 23.6, 23.4, 21.6. HRMS (ESI) m/z calcd. for C18H34N2O5 [M + Na]+: 381.2360; found: 381.2261.
- N-Boc-Me-L-Leu-D-Leu-OH 7c. The 7c was synthesized according to the method of 7 from 3c (15.0 g, 33.4 mmol, 1.0 equiv), 10% Pd/C (5 g, 4.7 mmol, 0.1 equiv), H2 (1 atm), 2 h. The mixture was filtered and concentrated in vacuo to give 7c (9.5 g, 26.5 mmol, 79% yield) as a colorless oil. [α]25D = −427.9 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 12.47 (s, 1H), 7.98 (d, J = 12.9 Hz, 1H), 4.62 (t, J = 8.0 Hz, 1H, major), 4.46 (d, J = 10.4 Hz, 1H, minor), 4.31–4.00 (m, 1H), 2.71 (s, 3H), 1.78–1.47 (m, 5H), 1.45–1.27 (m, 9H), 1.10–0.57 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 174.4, 171.6, 152.2, 79.3, 50.5, 31.0, 28.5, 25.0, 24.8, 23.6, 23.4, 21.6. HRMS (ESI) m/z calcd. for C18H34N2O5 [M + Na]+: 381.2360; found: 381.2261.
- N-Boc-Me-D-Leu-D-Leu-OH 7d. The 7d was synthesized according to the method of 7 from 3d (15.0 g, 33.4 mmol, 1.0 equiv), 10% Pd/C (5 g, 4.7 mmol, 0.1 equiv), H2 (1 atm), 2 h. The mixture was filtered and concentrated in vacuo to give 7d (10.3 g, 28.8 mmol, 86% yield) as a colorless oil. [α]25D = −215.6 (c = 1.0, CHCl3). 1H NMR (600 MHz, DMSO-d6) δ 7.92 (dd, J = 25.2, 9.2 Hz, 1H), 4.62 (s, 1H, major), 4.46 (d, J = 10.6 Hz, 1H, minor), 4.35–4.11 (m, 1H), 2.72 (s, 3H), 1.68–1.47 (m, 5H), 1.40 (s, 9H), 1.00–0.64 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 174.5, 171.6, 79.3, 56.8, 50.7, 38.0, 37.6, 30.3, 28.5, 25.0, 24.8, 23.6, 23.4, 21.6. HRMS (ESI) m/z calcd. for C18H34N2O5 [M + Na]+: 381.2360; found: 381.2261.
3.1.5. Synthesis of 5
- N-Boc-D-Leu-Me-D-Leu-D-Leu-OBn 5a. The 5a was synthesized according to the method of 5 from 4d (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5h (12.8 g, 22.8 mmol, 78% yield) as a white solid. [α]25D = −267.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, CDCl3) δ 7.28 (td, J = 13.7, 6.9 Hz, 5H), 6.45 (d, J = 8.3 Hz, 1H), 5.33–5.18 (m, 1H), 5.15–4.99 (m, 2H), 4.71–4.43 (m, 2H), 2.88 (s, 3H, major), 2.67 (s, 3H, minor), 1.77–1.49 (m, 9H), 1.40 (s, 9H, major), 1.35 (s, 9H, minor), 1.06–0.73 (m, 18H). 13C NMR (151 MHz, CDCl3) δ 174.6, 172.4, 170.5, 155.7, 135.6, 128.5, 128.5, 128.3, 128.1, 128.1, 66.8, 54.3, 50.7, 49.2, 41.6, 40.9, 35.6, 30.1, 28.3, 28.2, 24.8, 24.7, 24.6, 23.4, 23.0, 22.8, 21.9, 21.7, 21.6. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-D-Leu-Me-L-Leu-L-Leu-OBn 5b. The 5b was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5b (13.8 g, 24.6 mmol, 85% yield) as a white solid. [α]25D = −388.8 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.44 (d, J = 7.5 Hz, 1H, minor), 7.73 (d, J = 7.6 Hz, 1H, major), 7.45–7.17 (m, 5H), 7.09 (d, J = 7.0 Hz, 1H, major), 6.76 (d, J = 8.6 Hz, 1H, minor), 5.22–4.95 (m, 3H), 4.57–4.20 (m, 2H), 2.90 (s, 3H, major), 2.76 (s, 3H, minor), 1.76–1.39 (m, 7H), 1.34 (s, 9H), 1.26 (q, J = 7.0, 5.0 Hz, 2H), 1.00–0.65 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 173.9, 172.5, 171.3, 156.4, 136.4, 128.9, 128.4, 128.2, 78.6, 66.3, 54.3, 51.1, 49.6, 36.9, 30.9, 28.6, 28.6, 24.9, 24.7, 24.7, 23.7, 23.5, 23.2, 22.0, 21.6, 21.5. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-L-Leu-Me-D-Leu-L-Leu-OBn 5c. The 5c was synthesized according to the method of 5 from 4b (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-L-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5c (14.2 g, 25.3 mmol, 87% yield) as a white solid. [α]25D = −375.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.34 (d, J = 7.5 Hz, 1H, minor), 7.73 (d, J = 7.6 Hz, 1H, major), 7.50–7.17 (m, 5H), 7.17–6.67 (m, 1H), 5.27–4.83 (m, 3H), 4.64–4.16 (m, 2H), 2.90 (s, 3H, major), 2.76 (s, 3H, minor), 1.81–1.38 (m, 7H), 1.34 (s, 9H), 1.26 (q, J = 6.1, 5.0 Hz, 2H), 1.01–0.70 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 173.8, 172.4, 171.2, 156.3, 136.3, 128.8, 128.3, 128.1, 78.6, 66.2, 54.19, 51.0, 49.5, 36.8, 30.8, 28.5, 28.5, 24.8, 24.6, 24.6, 23.6, 23.4, 23.1, 21.9, 21.5, 21.4. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-D-Leu-Me-D-Leu-L-Leu-OBn 5d. The 5d was synthesized according to the method of 5 from 4b (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5d (13.5 g, 24.1 mmol, 83% yield) as a white solid. [α]25D = −255.8.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.21 (d, J = 7.7 Hz, 1H, minor), 8.12 (d, J = 7.6 Hz, 1H, major), 7.34 (m, 5H), 6.96 (d, J = 8.2 Hz, 1H), 5.14 (m, 1H), 5.10 (s, 2H), 4.69–4.10 (m, 2H), 2.94 (s, 3H, major), 2.55 (s, 3H, minor), 1.78–1.44 (m, 7H), 1.36 (s, 9H), 1.31–1.15 (m, 2H), 0.83 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 173.8, 172.6, 171.5, 156.0, 136.4, 128.9, 128.4, 128.2, 78.3, 66.3, 53.9, 50.8, 49.4, 37.6, 30.8, 28.6, 28.5, 24.7, 24.7, 24.6, 23.7, 23.6, 23.2, 21.9, 21.6. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-L-Leu-Me-L-Leu-D-Leu-OBn 5e. The 5e was synthesized according to the method of 5 from 4c (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-L-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5e (13.6 g, 24.2 mmol, 84% yield) as a white solid. [α]25D = −365.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.42 (dd, J = 14.4, 7.5 Hz, 1H, minor), 7.78 (d, J = 7.8 Hz, 1H, major), 7.34 (m, 5H), 7.05 (d, J = 7.0 Hz, 1H, major), 6.78 (d, J = 8.6 Hz, 1H, minor), 5.21–5.00 (m, 3H), 4.35 (d, J = 7.5 Hz, 2H), 2.83 (s, 3H, major), 2.73 (s, 3H, minor), 1.72–1.43 (m, 7H), 1.36 (s, 9H), 1.31 (s, 2H), 0.84 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 173.8, 172.5, 171.2, 156.3, 136.3, 128.9, 128.5, 128.4, 128.4, 78.60, 66.4, 54.1, 50.9, 49.7, 37.1, 30.8, 28.6, 24.9, 24.7, 24.7, 23.7, 23.6, 23.0, 21.9, 21.8, 21.6. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-D-Leu-Me-L-Leu-D-Leu-OBn 5f. The 5f was synthesized according to the method of 5 from 4c (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5f (13.0 g, 23.2 mmol, 80% yield) as a white solid. [α]25D = −369.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.43 (dd, J = 14.6, 7.4 Hz, 1H, minor), 7.79 (d, J = 7.7 Hz, 1H, major), 7.35 (q, J = 9.1, 8.7 Hz, 5H), 7.06 (d, J = 7.0 Hz, 1H, major), 6.79 (d, J = 8.6 Hz, 1H, minor), 5.12 (dd, J = 14.7, 5.5 Hz, 3H), 4.59–4.14 (m, 2H), 2.84 (s, 3H, major), 2.74 (s, 3H, minor), 1.83–1.43 (m, 7H), 1.37 (s, 9H), 1.22 (s, 2H), 0.85 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 172.4, 128.8, 78.6, 54.1, 50.9, 49.6, 37.1, 30.7, 28.6, 24.9, 24.7, 24.6, 23.7, 23.6, 23.0, 21.9, 21.8, 21.5. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-L-Leu-Me-D-Leu-D-Leu-OBn 5g. The 5g was synthesized according to the method of 5 from 4d (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-L-Leu-OH (7.2 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5g (13.2 g, 23.5 mmol, 81% yield) as a white solid. [α]25D = −323.6 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, CDCl3) δ 7.37–7.17 (m, 5H), 6.74 (d, J = 8.1 Hz, 1H), 5.16 (d, J = 15.8 Hz, 1H), 5.15–5.03 (m, 3H), 4.52 (td, J = 9.1, 4.6 Hz, 2H), 2.93 (s, 3H), 1.81–1.43 (m, 9H), 1.36 (s, 9H), 1.00–0.75 (m, 18H). 13C NMR (151 MHz, CDCl3) δ 174.5, 172.3, 170.5, 156.0, 135.6, 128.5, 128.1, 128.0, 79.7, 66.7, 54.7, 51.0, 49.3, 41.7, 40.4, 36.0, 30.4, 28.3, 24.9, 24.8, 24.7, 23.2, 23.2, 22.8, 21.8, 21.6, 21.5. HRMS (ESI) m/z calcd. for C31H51N3O6 [M + Na]+: 584.3670; found: 584.3672.
- N-Boc-D-Pro-Me-L-Leu-L-Leu-OBn 5h. The 5h was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-L-Pro-OH (6.7 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5h (13.0 g, 23.8 mmol, 83% yield) as a white solid. [α]25D = −175.6 (c = 1.0, CHCl3). m.p. 158–159 °C. 1H NMR (600 MHz, CDCl3) δ7.39–7.26 (m, 5H, major), 7.17 (d, J = 7.9 Hz, 5H, minor), 5.40 (dd, J = 11.0, 4.5 Hz, 1H), 5.12 (q, J = 12.6 Hz, 2H), 4.64–4.49 (m, 2H), 3.65–3.50 (m, 1H), 3.47 (dd, J = 5.0, 2.1 Hz, 1H), 3.03 (s, 3H, minor)2.94 (s, 3H, major), 2.22–2.03 (m, 2H), 1.98–1.84 (m, 2H), 1.84–1.53 (m, 4H), 1.39 (s, 9H), 1.01–0.79 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 174.0, 172.3, 171.2, 154.8, 136.0, 128.4, 127.9, 127.9, 66.4, 55.8, 55.0, 51.4, 47.0, 39.8, 35.7, 30.8, 29.5, 28.4, 25.0, 24.9, 23.4, 22.9, 21.6, 21.4. HRMS (ESI) m/z calcd. for C30H47N3O6 [M + Na]+: 586.3357; found: 586.3357.
- N-Boc-D-Phe-Me-L-Leu-L-Leu-OBn 5i. The 5i was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Phe-OH (8.3 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5i (15.0 g, 25.2 mmol, 88% yield) as a white solid. [α]25D = −333.0 (c = 1.0, CHCl3). m.p. 119–120 °C. 1H NMR (600 MHz, Chloroform-d) δ 7.45–7.11 (m, 10H), 6.77 (d, J = 8.1 Hz, 1H), 5.25 (d, J = 7.6 Hz, 1H), 5.19–5.02 (m, 3H), 4.78 (d, J = 7.6 Hz, 1H), 4.54 (d, J = 7.4 Hz, 1H), 2.98 (d, J = 7.4 Hz, 2H), 2.66 (s, 3H), 1.80–1.53 (m, 4H), 1.40 (d, J = 2.7 Hz, 9H), 0.97–0.75 (m, 12H). 13C NMR (151 MHz, Chloroform-d) δ 173.2, 172.2, 170.4, 155.5, 135.9, 135.7, 129.3, 128.6, 128.5, 128.2, 128.0, 127.1, 66.7, 54.9, 52.0, 51.0, 40.4, 39.2, 35.9, 30.5, 28.3, 24.9, 24.4, 23.1, 22.8, 21.9, 21.5. HRMS (ESI) m/z calcd. for C34H49N3O6 [M + Na]+: 618.3514; found: 618.3514.
- N-Boc-Gly-Me-L-Leu-L-Leu-OBn 5j. The 5j was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-Gly-OH (4.9 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5j (11.5 g, 24.7 mmol, 79% yield) as a white solid. [α]25D = −205.3 (c = 1.0, CHCl3). m.p. 117–118 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.20 (d, J = 7.6 Hz, 1H), 7.52–7.04 (m, 5H), 6.70 (t, J = 5.8 Hz, 1H), 5.11 (t, J = 13.2 Hz, 2H), 5.06 (s, 1H), 4.30 (ddd, J = 10.3, 7.5, 4.9 Hz, 1H), 3.93–3.65 (m, 2H), 2.82 (s, 3H, major), 2.77 (s, 3H, minor), 1.70–1.44 (m, 5H), 1.38 (s, 9H), 1.33 (d, J = 3.9 Hz, 1H), 0.96–0.64 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 171.6, 170.2, 156.2, 136.4, 128.9, 128.5, 128.4, 128.3, 78.5, 78.4, 66.5, 66.4, 54.2, 51.0, 42.5, 37.5, 30.1, 28.7, 24.7, 23.6, 23.2, 21.9, 21.6. HRMS (ESI) m/z calcd. for C27H43N3O6 [M + Na]+: 528.3044; found: 528.3043.
- N-Boc-D-Ala-Me-L-Leu-L-Leu-OBn 5k. The 5k was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Ala-OH (5.9 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5k (12.5 g, 24.3 mmol, 84% yield) as a white solid. [α]25D = −555.6 (c = 1.0, CHCl3). m.p. 105–106 °C. 1H NMR (600 MHz, Chloroform-d) δ 7.32 (dp, J = 14.9, 7.1, 6.5 Hz, 5H), 6.64 (d, J = 8.1 Hz, 1H), 5.34 (d, J = 7.4 Hz, 1H), 5.23–5.05 (m, 3H), 4.55 (t, J = 7.3 Hz, 2H), 2.93 (s, 3H), 1.74 (d, J = 8.1 Hz, 1H), 1.69–1.53 (m, 4H), 1.40 (s, 9H), 1.29 (d, J = 6.8 Hz, 3H), 1.01–0.75 (m, 12H). 13C NMR (151 MHz, Chloroform-d) δ 174.5, 172.3, 170.4, 155.6, 135.6, 128.5, 128.2, 128.1, 79.7, 66.8, 54.9, 51.0, 46.8, 40.6, 35.9, 30.5, 28.3, 24.9, 24.9, 23.1, 22.7, 21.7, 21.6, 18.0. HRMS (ESI) m/z calcd. for C28H45N3O6 [M + Na]+: 542.3201; found: 542.3209.
- N-Boc-O-Me-D-Ser-Me-L-Leu-L-Leu-OBn 5l. The 5l was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-O-Me-D-Ser-OH (6.9 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5l (14.0 g, 25.5 mmol, 89% yield) as a white solid. [α]25D = −275.6 (c = 1.0, CHCl3). m.p. 121–122 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.72 (d, J = 7.7 Hz, 1H), 7.46–7.24 (m, 5H), 7.12 (d, J = 6.7 Hz, 1H), 5.15–5.08 (m, 2H), 5.06 (d, J = 12.6 Hz, 1H), 4.58 (d, J = 6.8 Hz, 1H), 4.41–4.24 (m, 1H), 3.44 (t, J = 6.8 Hz, 2H), 3.22 (s, 3H), 2.91 (s, 3H, major), 2.77 (s, 3H, minor), 1.77–1.38 (m, 6H), 1.34 (s, 9H), 1.03–0.60 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.5, 172.0, 171.3, 156.0, 136.4, 128.9, 128.4, 128.2, 78.9, 72.1, 66.3, 58.9, 54.5, 51.1, 50.7, 39.5, 36.9, 31.1, 28.5, 24.7, 24.5, 23.8, 23.2, 21.7, 21.5. HRMS (ESI) m/z calcd. for C29H47N3O7 [M + Na]+: 572.3306; found: 572.3307.
- N-Boc-O-Me-D-Tyr-Me-L-Leu-L-Leu-OBn 5m. The 5m was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-O-Me-D-Tyr-OH (9.3 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5m (17.0 g, 27.2 mmol, 95% yield) as a white solid. [α]25D = −157.3 (c = 1.0, CHCl3). m.p. 120–121 °C. 1H NMR (600 MHz, Chloroform-d) δ 7.34 (ddd, J = 12.7, 7.5, 3.6 Hz, 5H), 7.12 (d, J = 8.2 Hz, 2H), 6.82 (d, J = 8.4 Hz, 3H), 5.29–5.00 (m, 4H), 4.74 (d, J = 7.4 Hz, 1H), 4.54 (td, J = 8.2, 6.1 Hz, 1H), 3.79 (s, 3H), 2.93 (d, J = 7.5 Hz, 2H), 2.68 (s, 3H), 1.84–1.52 (m, 4H), 1.40 (s, 9H), 1.35–1.24 (m, 2H), 0.95–0.76 (m, 12H). 13C NMR (151 MHz, Chloroform-d) δ 173.3, 172.2, 170.4, 158.8, 155.5, 130.3, 128.5, 128.0, 114.0, 66.7, 55.2, 54.9, 52.1, 51.0, 40.4, 38.3, 35.9, 30.5, 28.3, 24.9, 24.4, 23.1, 22.8, 22.0, 21.5. HRMS (ESI) m/z calcd. for C35H51N3O7 [M + Na]+: 648.3620; found: 648.3621.
- N-Boc-Methyl ester-D-Asp-Me-L-Leu-L-Leu-OBn 5n. The 5n was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-Methyl ester-D-Asp-OH (7.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5n (14.0 g, 24.3 mmol, 84% yield) as a white solid. [α]25D = −269.7 (c = 1.0, CHCl3). m.p. 124–125 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.80 (d, J = 7.7 Hz, 1H, minor), 7.74 (d, J = 7.8 Hz, 1H, major), 7.34 (m, 6H), 5.18–4.95 (m, 3H), 4.80–4.52 (m, 1H), 4.47–4.21 (m, 1H), 3.54 (d, J = 14.7 Hz, 3H), 3.00 (s, 3H, major), 2.91 (s, 3H, minor), 2.89–2.68 (m, 1H), 2.65–2.52 (m, 1H), 1.60 (m, 5H), 1.36 (s, 9H), 0.98–0.64 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.50, 172.03, 171.22, 171.02, 155.39, 136.40, 128.85, 128.46, 128.19, 78.77, 66.34, 60.19, 51.93, 51.84, 51.07, 50.94, 47.07, 36.94, 36.64, 36.32, 30.95, 30.77, 28.49, 24.70, 23.74, 23.22, 21.82, 21.55. HRMS (ESI) m/z calcd. for C30H47N3O8 [M + Na]+: 600.3256; found: 600.3265.
- N-Boc-3-Flu-D-Phe-Me-L-Leu-L-Leu-OBn 5o. The 5o was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-3-Flu-D-Phe-OH (9.3 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5o (15.5 g, 25.2 mmol, 88% yield) as a white solid. [α]25D = −555.6 (c = 1.0, CHCl3). m.p. 121–122 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.66 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 6.2 Hz, 1H), 7.38–7.24 (m, 5H), 7.20–6.88 (m, 3H), 5.16–5.06 (m, 2H), 5.04 (d, J = 12.4 Hz, 1H), 4.66 (q, J = 7.2 Hz, 1H), 4.31 (ddd, J = 12.0, 7.7, 5.0 Hz, 1H), 2.95–2.83 (m, 2H), 2.73 (s, 3H), 1.80–1.41 (m, 4H), 1.31 (s, 9H), 1.26–1.10 (m, 2H), 0.97–0.52 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 172.5, 171.2, 163.3, 161.7, 156.2, 136.4, 130.5, 130.4, 128.9, 128.5, 128.4, 128.3, 128.1, 126.1, 116.7, 116.6, 113.8, 113.7, 78.9, 66.2, 60.2, 54.4, 52.0, 51.1, 37.3, 36.7, 30.9, 28.5, 24.7, 24.1, 23.7, 23.2, 21.7, 21.4, 21.2. HRMS (ESI) m/z calcd. for C34H48FN3O6 [M + Na]+: 636.3420; found: 636.3421.
- N-Boc-2-Me-D-Phe-Me-L-Leu-L-Leu-OBn 5p. The 5p was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-2-Me-D-Ser-OH (8.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5p (15.0 g, 24.6 mmol, 86% yield) as a white solid. [α]25D = −505.6 (c = 1.0, CHCl3). m.p. 142–143 °C. 1H NMR (600 MHz, Chloroform-d) δ 7.41–7.26 (m, 5H), 7.09 (s, 4H), 6.79 (d, J = 8.0 Hz, 1H), 5.21 (d, J = 7.5 Hz, 1H), 5.19–5.04 (m, 3H), 4.75 (d, J = 7.5 Hz, 1H), 4.54 (d, J = 7.4 Hz, 1H), 3.02–2.84 (m, 2H), 2.67 (s, 3H), 2.32 (s, 3H), 1.73–1.51 (m, 4H), 1.39 (s, 9H), 1.31 (td, J = 11.2, 10.3, 5.9 Hz, 1H), 1.05 (s, 1H), 1.01–0.70 (m, 12H). 13C NMR (151 MHz, Chloroform-d) δ 173.2, 172.2, 170.5, 155.6, 136.7, 135.7, 132.8, 129.2, 129.2, 128.5, 128.1, 128.0, 66.7, 54.9, 52.0, 51.0, 40.3, 38.7, 35.9, 30.6, 28.3, 24.9, 24.4, 23.1, 22.8, 21.9, 21.5, 21.0. HRMS (ESI) m/z calcd. for C35H51N3O6 [M + Na]+: 632.3420; found: 632.3421.
- N-Boc-4-Chl-D-Phe-Me-L-Leu-L-Leu-OBn 5q. The 5q was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-3-Chl-D-Phe-OH (9.4 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5q (16.0 g, 25.4 mmol, 89% yield) as a white solid. [α]25D = −399.6 (c = 1.0, CHCl3). m.p. 151–152 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.64 (d, J = 7.7 Hz, 1H), 7.42 (d, J = 6.1 Hz, 1H), 7.39–7.20 (m, 8H), 5.21–5.04 (m, 2H), 5.02 (d, J = 9.1 Hz, 1H), 4.66 (d, J = 7.2 Hz, 1H), 4.54–4.18 (m, 1H), 2.97–2.80 (m, 2H), 2.74 (s, 3H), 1.81–1.43 (m, 4H), 1.30 (s, 9H), 1.25 (m, 2H), 0.93–0.61 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 172.4, 171.2, 156.2, 136.4, 131.8, 131.8, 128.9, 128.5, 128.4, 128.1, 78.9, 66.2, 54.4, 52.0, 51.1, 36.9, 36.7, 31.0, 28.5, 24.7, 24.1, 23.7, 23.2, 21.8, 21.4. HRMS (ESI) m/z calcd. for C34H48ClN3O6 [M + Na]+: 652.3124; found: 652.3132.
- N-Boc-4-Bro-D-Phe-Me-L-Leu-L-Leu-OBn 5r. The 5r was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-4-Bro-D-Phe-OH (10.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5r (17.0 g, 25.2 mmol, 88% yield) as a white solid. [α]25D = −375.6 (c = 1.0, CHCl3). m.p. 165–166 °C. 1H NMR (600 MHz, CDCl3) δ 7.48–7.22 (m, 6H), 7.08 (d, J = 7.9 Hz, 2H), 6.75 (d, J = 8.1 Hz, 1H), 5.28 (d, J = 7.7 Hz, 1H), 5.20–5.01 (m, 3H), 4.77 (q, J = 7.6 Hz, 1H), 4.53 (td, J = 8.5, 5.6 Hz, 1H), 2.93 (dd, J = 7.4, 3.9 Hz, 2H), 2.73 (s, 3H), 1.73–1.46 (m, 4H), 1.38 (s, 9H), 1.01–0.67 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 172.8, 172.3, 170.3, 155.4, 135.6, 135.0, 131.7, 131.2, 131.1, 128.6, 128.5, 128.3, 128.2, 128.1, 121.1, 80.2, 66.8, 54.9, 51.7, 51.0, 40.4, 38.5, 36.0, 30.7, 28.3, 24.9, 24.5, 23.1, 22.8, 21.9, 21.5. HRMS (ESI) m/z calcd. for C34H48BrN3O6 [M + Na]+: 696.2619; found: 696.2614.
- N-Boc-D-Cyclohexyl-Gly-Me-L-Leu-L-Leu-OBn 5s. The 5s was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc D-Cyclohexyl-Gly-OH (8.1 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5s (15.0 g, 25.5 mmol, 89% yield) as a white solid. [α]25D = −275.6 (c = 1.0, CHCl3). m.p. 122–123 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.62 (d, J = 7.5 Hz, 1H, minor), 7.75 (d, J = 7.6 Hz, 1H, major), 7.34 (m, 5H), 7.01 (d, J = 7.0 Hz, 1H), 5.11 (dd, J = 14.4, 6.5 Hz, 2H), 5.05 (d, J = 12.7 Hz, 1H), 4.48–4.21 (m, 1H), 4.18 (t, J = 7.6 Hz, 1H), 2.92 (s, 3H, major), 2.83 (s, 3H, minor), 1.75–1.48 (m, 10H), 1.34 (d, J = 18.2 Hz, 10H), 1.25–0.93 (m, 5H), 0.84 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 173.2, 172.5, 171.4, 156.5, 136.4, 128.9, 128.9, 128.4, 128.1, 78.7, 66.3, 55.8, 54.3, 51.1, 37.1, 31.3, 29.2, 28.9, 28.6, 26.2, 26.2, 26.0, 24.8, 24.7, 23.8, 23.2, 21.5, 21.4. HRMS (ESI) m/z calcd. for C33H53N3O6 [M + Na]+: 610.3827; found: 610.3833.
- N-Boc-D-Phenyl-Gly-Me-L-Leu-L-Leu-OBn 5t. The 5t was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Phenyl-Gly-OH (7.9 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5t (15.0 g, 25.8 mmol, 90% yield) as a white solid. [α]25D = −335.6 (c = 1.0, CHCl3). m.p. 121–122 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.05 (d, J = 7.6 Hz, 1H), 7.57–6.88 (m, 10H), 5.52 (d, J = 7.4 Hz, 1H), 5.11 (dd, J = 17.0, 8.8 Hz, 2H), 5.07 (s, 1H), 4.33 (dd, J = 7.6, 3.5 Hz, 1H), 2.87–2.65 (m, 3H), 1.75–1.38 (m, 5H), 1.36 (d, J = 7.6 Hz, 9H), 1.02–0.49 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 171.5, 171.3, 136.4, 128.9, 128.8, 128.6, 128.5, 128.4, 128.3, 78.9, 66.4, 55.9, 54.4, 51.0, 37.2, 30.9, 28.6, 24.7, 24.5, 23.6, 23.3, 21.6, 21.5. HRMS (ESI) m/z calcd. for C33H47N3O6 [M + Na]+: 604.3357; found: 604.3367.
- N-Boc-D-Val-Me-L-Leu-L-Leu-OBn 5u. The 5u was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Val-OH (6.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5u (13.5 g, 24.7 mmol, 86% yield) as a white solid. [α]25D = −275.6 (c = 1.0, CHCl3). m.p. 108–109 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.23 (d, J = 7.7 Hz, 1H), 7.44–7.18 (m, 5H), 6.90 (d, J = 8.4 Hz, 1H), 5.16 (dd, J = 10.7, 4.5 Hz, 1H), 5.13–5.04 (m, 2H), 4.38–4.21 (m, 1H), 4.09 (t, J = 8.7 Hz, 1H), 2.99 (s, 3H, major), 2.67 (s, 3H, minor), 2.00–1.79 (m, 1H), 1.69–1.37 (m, 6H), 1.36 (s, 9H), 0.95–0.66 (m, 18H). 13C NMR (151 MHz, DMSO-d6) δ 173.4, 172.6, 171.5, 156.1, 136.4, 128.9, 128.5, 128.3, 78.4, 66.4, 56.3, 53.6, 50.8, 37.5, 31.1, 30.1, 28.6, 28.6, 24.6, 24.5, 23.7, 23.3, 22.0, 21.4, 19.2. HRMS (ESI) m/z calcd. for C30H49N3O6 [M + Na]+: 570.3514; found: 570.3524.
- N-Boc-D-Met-Me-L-Leu-L-Leu-OBn 5v. The 5v was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-D-Met-OH (7.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5v (14.0 g, 23.6 mmol, 82% yield) as a white solid. [α]25D = −275.6 (c = 1.0, CHCl3). m.p. 95–96 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.69 (d, J = 7.7 Hz, 1H), 7.35 (m, 5H), 7.22 (d, J = 6.7 Hz, 1H), 5.12 (d, J = 12.6 Hz, 2H), 5.06 (d, J = 12.7 Hz, 1H), 4.49 (d, J = 7.1 Hz, 1H), 4.32 (d, J = 10.3 Hz, 1H), 2.93 (s, 3H, major), 2.78 (s, 3H, minor), 2.47 (d, J = 7.4 Hz, 2H), 2.01 (s, 3H), 1.86–1.73 (m, 2H), 1.62 (td, J = 35.0, 31.7, 19.2 Hz, 6H), 1.36 (d, J = 14.4 Hz, 9H), 0.85 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 173.2, 172.5, 171.2, 156.3, 136.4, 128.9, 128.4, 128.1, 78.8, 66.3, 54.4, 51.1, 50.2, 36.8, 31.1, 30.9, 30.0, 28.6, 28.6, 24.9, 24.7, 23.7, 23.2, 21.7, 21.5, 15.1. HRMS (ESI) m/z calcd. for C30H49N3O6S [M + Na]+: 602.3234; found: 602.3244.
- N-Boc-3-Me-D-Phe-Me-L-Leu-L-Leu-OBn 5w. The 5w was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-3-Me-D-Phe-OH (8.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5w (15.0 g, 24.6 mmol, 86% yield) as a white solid. [α]25D = −475.4 (c = 1.0, CHCl3). m.p. 121–122 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.64 (d, J = 7.7 Hz, 1H), 7.49–7.22 (m, 5H), 7.24–6.82 (m, 4H), 5.10 (t, J = 14.0 Hz, 2H), 5.04 (d, J = 12.5 Hz, 1H), 4.65 (t, J = 7.1 Hz, 1H), 4.42–4.15 (m, 1H), 2.97–2.73 (m, 2H), 2.67 (s, 3H), 2.25 (s, 3H), 1.82–1.44 (m, 4H), 1.30 (d, J = 21.2 Hz, 9H), 1.22 (m, 2H), 0.99–0.56 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 173.1, 172.5, 171.2, 156.3, 137.6, 137.2, 136.4, 130.4, 128.9, 128.49, 128.4, 128.1, 127.6, 126.9, 78.9, 66.1, 54.4, 52.2, 51.1, 37.6, 36.7, 30.9, 28.5, 24.7, 24.0, 23.8, 23.3, 21.8, 21.5, 21.4. HRMS (ESI) m/z calcd. for C35H51N3O6 [M + Na]+: 632.3671; found: 632.3678.
- N-Boc-4-Me-D-Phe-Me-L-Leu-L-Leu-OBn 5x. The 5x was synthesized according to the method of 5 from 4a (10.0 g, 28.7 mmol, 1.0 equiv) and N-Boc-2-Me-D-Phe-OH (8.8 g, 31.5 mmol, 1.1 equiv), HOBT (4.2 g, 31.5 mmol, 1.1 equiv), and EDCI (6.0 g, 31.5 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 80:20, v/v) to obtain 5x (14.5 g, 23.8 mmol, 83% yield) as a white solid. [α]25D = −356.7 (c = 1.0, CHCl3). m.p. 161–162 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.61 (d, J = 7.6 Hz, 1H), 7.43 (d, J = 5.7 Hz, 1H), 7.40–7.23 (m, 4H), 7.09 (m, 4H), 5.28–4.93 (m, 3H), 4.62 (q, J = 6.9 Hz, 1H), 4.41–4.24 (m, 1H), 2.97–2.73 (m, 2H), 2.66 (s, 3H), 2.25 (s, 3H), 1.76–1.38 (m, 4H), 1.37–1.07 (m, 9H), 0.97–0.43 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 173.1, 172.4, 171.2, 156.3, 136.4, 136.0, 134.1, 129.7, 129.2, 128.9, 128.4, 128.1, 78.9, 66.2, 54.4, 52.2, 51.1, 39.3, 37.2, 36.6, 30.9, 28.5, 24.7, 23.9, 23.7, 23.2, 21.8, 21.4, 21.1. HRMS (ESI) m/z calcd. for C35H51N3O6 [M + Na]+: 632.3671; found: 632.3678.
3.1.6. Synthesis of 6
3.1.7. Synthesis of 8
- N-Boc-Me-D-Leu-D-Leu-D-Leu-Me-D-Leu-D-Leu-OBn 8a. The 8a was synthesized according to the method of 8 from 7d (10.0 g, 27.9 mmol, 1.0 equiv) and 6a (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8a (19.4 g, 24.2 mmol, 84% yield) as a white solid. [α]25D = −175.6 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.24 (d, J = 8.0 Hz, 1H), 8.21–8.02 (m, 1H), 7.83 (m, 1H), 7.34 (m, 5H), 5.10 (d, J = 8.0 Hz, 3H), 4.82–4.43 (m, 2H), 4.43–4.06 (m, 2H), 2.92 (s, 3H, major), 2.67 (d, J = 18.0 Hz, 3H), 2.57 (s, 3H, minor), 1.79–1.19 (m, 25H), 0.84 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 172.6, 172.1, 171.3, 136.4, 128.9, 128.5, 128.2, 79.4, 66.3, 53.95, 50.8, 47.3, 41.6, 37.8, 37.7, 30.9, 28.5, 24.8, 24.7, 24.5, 23.6, 23.5, 23.4, 23.3, 22.1, 22.0, 22.0, 21.6. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-L-Leu-L-Leu-D-Leu-Me-L-Leu-L-Leu-OBn 8b. The 8b was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6b (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8b (19.5 g, 24.3 mmol, 87% yield) as a white solid. [α]25D = −164.7 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.13 (m, 1H), 8.01–7.84 (m, 1H), 7.68 (m, 1H), 7.35 (m, 5H), 5.16–5.06 (m, 2H), 4.87 (d, J = 198.8 Hz, 1H), 4.68–4.61 (m, 1H), 4.58–4.42 (m, 1H), 4.42–4.23 (m, 2H), 2.96 (s, 3H, major), 2.80 (s, 3H, minor), 2.69 (d, J = 5.1 Hz, 3H), 1.82–1.43 (m, 11H), 1.43–1.28 (m, 13H), 0.85 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.6, 172.0, 171.3, 171.0, 155.4, 136.4, 128.8, 128.5, 128.3, 79.4, 66.4, 55.3, 53.8, 50.8, 47.3, 41.3, 37.4, 30.7, 30.1, 28.4, 24.9, 24.7, 24.6, 24.6, 24.5, 23.6, 23.5, 23.3, 22.2, 22.1, 22.0, 21.6, 21.4. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-D-Leu-L-Leu-L-Leu-Me-D-Leu-L-Leu-OBn 8c. The 8c was synthesized according to the method of 8 from 7b (10.0 g, 27.9 mmol, 1.0 equiv) and 6c (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8c (18.7 g, 23.3 mmol, 83% yield) as a white solid. [α]25D = −154.2 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.46 (d, J = 7.6 Hz, 1H, minor), 8.28–7.98 (m, 1H, major), 7.98–7.50 (m, 2H), 7.34 (m, 5H), 5.10 (d, J = 6.8 Hz, 3H), 4.71–4.43 (m, 2H), 4.43–4.25 (m, 2H), 2.84 (s, 3H, major), 2.74 (s, 3H, minor), 2.70 (d, J = 15.3 Hz, 3H), 1.84–1.11 (m, 24H), 0.84 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 173.0, 172.6, 172.5, 171.1, 136.3, 128.9, 128.5, 128.4, 128.4, 79.41, 66.4, 54.2, 51.0, 50.9, 48.2, 37.8, 30.9, 28.4, 25.0, 24.9, 24.7, 24.6, 23.7, 23.6, 23.5, 23.3, 22.0, 21.9, 21.6, 21.5. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-D-Leu-L-Leu-D-Leu-Me-D-Leu-L-Leu-OBn 8d. The 8d was synthesized according to the method of 8 from 7b (10.0 g, 27.9 mmol, 1.0 equiv) and 6d (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8d (17.9 g, 22.3 mmol, 79% yield) as a white solid. [α]25D = −145.6 (c = 1.0, CHCl3). m.p. 138–139 °C 1H NMR (600 MHz, DMSO-d6) δ 8.54–7.95 (m, 1H), 7.90–7.55 (m, 2H), 7.34 (m, 5H), 5.11 (t, J = 9.7 Hz, 3H), 4.77–4.42 (m, 2H), 4.42–4.22 (m, 2H), 2.79 (d, J = 64.4 Hz, 3H), 2.70 (d, J = 15.3 Hz, 3H), 1.79–1.44 (m, 11H), 1.38 (s, 13H), 0.99–0.66 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.9, 172.6, 172.5, 171.1, 136.3, 128.9, 128.5, 128.4, 128.4, 79.4, 66.4, 54.2, 50.9, 50.9, 48.2, 39.6, 37.8, 30.9, 28.4, 25.0, 24.9, 24.7, 24.6, 23.7, 23.6, 23.5, 23.3, 22.0, 21.9, 21.6, 21.5. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-L-Leu-D-Leu-L-Leu-Me-L-Leu-D-Leu-OBn 8e. The 8e was synthesized according to the method of 8 from 7c (10.0 g, 27.9 mmol, 1.0 equiv) and 6e (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8e (19.1 g, 23.8 mmol, 84% yield) as a white solid. [α]25D = −134.6 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.24 (d, J = 8.0 Hz, 1H), 8.19–8.01 (m, 1H), 7.83 (m, 1H), 7.34 (m, 5H), 5.10 (d, J = 8.0 Hz, 3H), 4.72 (q, J = 7.9 Hz, 1H), 4.68–4.44 (m, 1H), 4.44–4.32 (m, 1H), 4.32–4.23 (m, 1H), 2.92 (s, 3H), 2.69–2.54 (m, 3H), 1.70–1.42 (m, 11H), 1.42–1.24 (m, 12H), 0.84 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 172.6, 172.1, 171.3, 136.4, 128.9, 128.5, 128.2, 79.4, 66.3, 53.9, 50.8, 47.3, 41.6, 37.8, 37.7, 30.9, 28.5, 24.8, 24.7, 24.6, 23.6, 23.5, 23.4, 23.3, 22.1, 22.0, 22.0, 21.6. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-L-Leu-D-Leu-D-Leu-Me-L-Leu-D-Leu-OBn 8f. The 8f was synthesized according to the method of 8 from 4c (10.0 g, 27.9 mmol, 1.0 equiv) and 6f (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8f (19.7 g, 24.6 mmol, 86% yield) as a white solid. [α]25D = −165.6 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.56–8.01 (m, 1H), 7.97–7.53 (m, 2H), 7.35 (dp, J = 15.6, 7.2 Hz, 5H), 5.11 (t, J = 9.7 Hz, 3H), 4.78–4.43 (m, 2H), 4.43–4.26 (m, 2H), 2.84 (s, 3H, major), 2.74 (s, 3H, minor), 2.70 (d, J = 15.3 Hz, 3H), 1.79–1.45 (m, 11H), 1.39 (s, 14H), 0.84 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 172.6, 172.1, 171.3, 136.4, 128.9, 128.5, 128.2, 79.4, 66.3, 53.95, 50.8, 47.3, 41.6, 37.8, 37.7, 30.9, 28.5, 24.8, 24.7, 24.5, 23.6, 23.5, 23.4, 23.3, 22.1, 22.0, 22.0, 21.6. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-D-Leu-D-Leu-L-Leu-Me-D-Leu-D-Leu-OBn 8g. The 8g was synthesized according to the method of 8 from 7d (10.0 g, 27.9 mmol, 1.0 equiv) and 6g (14.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8g (18.6 g, 23.2 mmol, 81% yield) as a white solid. [α]25D = −154.8 (c = 1.0, CHCl3). m.p. 138–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.23 (d, J = 7.7 Hz, 1H), 8.11–7.90 (m, 1H), 7.76 (m, 8.4 Hz, 1H), 7.35 (m, 5H), 5.27–4.95 (m, 3H), 4.73 (m, 1H), 4.68–4.40 (m, 1H), 4.40–4.20 (m, 2H), 2.91 (s, 3H), 2.75–2.58 (m, 3H), 1.71–1.43 (m, 11H), 1.42–1.23 (m, 14H), 0.98–0.67 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.6, 172.0, 171.3, 171.0, 155.4, 136.4, 128.8, 128.5, 128.3, 79.4, 66.4, 55.3, 53.8, 50.8, 47.3, 41.3, 37.4, 30.7, 30.1, 28.4, 24.9, 24.7, 24.6, 24.6, 24.5, 23.6, 23.5, 23.3, 22.2, 22.1, 22.0, 21.6, 21.4. HRMS (ESI) m/z calcd. for C44H75N5O8 [M + Na]+: 824.5508; found: 824.5507.
- N-Boc-Me-L-Leu-L-Leu-D-Pro-Me-L-Leu-L-Leu-OBn 8h. The 8h was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6h (13.7 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8h (19.0 g, 24.2 mmol, 88% yield) as a white solid. [α]25D = −178.9 (c = 1.0, CHCl3). m.p. 169–170 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.19 (d, J = 7.6 Hz, 1H), 7.90 (m, 1H), 7.39–7.29 (m, 5H), 5.18–5.06 (m, 2H), 5.05–4.81 (m, 1H), 4.77–4.58 (m, 1H), 4.58–4.42 (m, 2H), 4.37–4.20 (m, 1H), 3.71 (d, J = 8.3 Hz, 1H), 3.57–3.38 (m, 1H), 2.91 (s, 3H, major), 2.69 (d, J = 7.7 Hz, 3H), 2.60 (d, J = 5.5 Hz, 1H, minor), 2.19–2.07 (m, 1H), 2.04–1.95 (m, 1H), 1.93–1.82 (m, 1H), 1.75–1.53 (m, 5H), 1.52–1.39 (m, 9H), 1.34–1.21 (m, 10H), 1.02–0.62 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.2, 171.5, 136.4, 128.9, 128.5, 128.3, 66.4, 56.9, 50.8, 47.1, 37.3, 35.0, 34.8, 31.6, 30.3, 28.5, 24.8, 24.7, 24.6, 23.7, 23.7, 23.3, 22.0, 21.5. HRMS (ESI) m/z calcd. for C43H71N5O8 [M + Na]+: 808.5195; found: 808.5203.
- N-Boc-Me-L-Leu-L-Leu-D-Phe-Me-L-Leu-L-Leu-OBn 8i. The 8i was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6i (15.2 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8i (20.0 g, 23.9 mmol, 86% yield) as a white solid. [α]25D = −155.6 (c = 1.0, CHCl3). m.p. 141–142 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.37–8.22 (m, 1H), 8.19 (d, J = 7.6 Hz, 1H), 7.63 (m 1H), 7.35 (m, 5H), 7.27–7.09 (m, 5H), 5.12 (dd, J = 15.7, 11.2 Hz, 2H), 5.08 (d, J = 7.2 Hz, 1H), 4.89 (q, J = 7.3 Hz, 1H), 4.66–4.38 (m, 1H), 4.38–4.17 (m, 2H), 2.98 (dd, J = 13.9, 5.9 Hz, 1H), 2.84 (s, 3H, major), 2.83–2.78 (m, 1H), 2.69 (d, J = 12.3 Hz, 3H), 2.55(s, 3H, minor), 1.67–1.44 (m, 7H), 1.44–1.17 (m, 14H), 0.96–0.65 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.0, 171.4, 155.4, 137.9, 136.4, 129.9, 129.7, 128.9, 128.8, 128.5, 128.3, 128.2, 126.7, 79.4, 66.4, 60.2, 53.9, 50.9, 41.6, 37.6, 30.8, 30.2, 28.4, 24.9, 24.8, 24.6, 23.7, 23.2, 22.0, 21.7, 21.6, 21.2. HRMS (ESI) m/z calcd. for C47H73N5O8 [M + Na]+: 858.5351; found: 858.5359.
- N-Boc-Me-L-Leu-L-Leu-Gly-Me-L-Leu-L-Leu-OBn 8j. The 8j was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6j (12.4 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8j (17.5 g, 23.5 mmol, 84% yield) as a white solid. [α]25D = −167.5 (c = 1.0, CHCl3). m.p. 137–138 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.60 (d, J = 7.6 Hz, 1H, minor), 8.23 (d, J = 7.6 Hz, 1H, major), 8.03–7.73 (m, 2H), 7.51–7.16 (m, 5H), 5.17–5.09 (m, 2H), 5.07 (t, J = 5.4 Hz, 1H), 4.64 (s, 1H, major), 4.50 (d, J = 6.4 Hz, 1H, minor), 4.41–4.24 (m, 2H), 3.97 (d, J = 5.1 Hz, 2H), 2.82 (s, 3H, major), 2.78 (s, 3H, minor), 2.71 (s, 3H), 1.57 (m, 10H), 1.40 (d, J = 9.9 Hz, 11H), 1.02–0.61 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.5, 171.5, 170.50, 169.5, 136.4, 128.9, 128.5, 128.4, 128.3, 66.4, 54.3, 51.0, 41.4, 41.1, 37.5, 30.2, 28.5, 25.0, 24.8, 24.7, 24.7, 23.7, 23.6, 23.2, 23.2, 23.0, 22.6, 21.9, 21.7, 21.6. HRMS (ESI) m/z calcd. for C40H67N5O8 [M + Na]+: 768.4882; found: 768.4892.
- N-Boc-Me-L-Leu-L-Leu-D-Ala-Me-L-Leu-L-Leu-OBn 8k. The 8k was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6k (12.9 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8k (18.0 g, 23.7 mmol, 85% yield) as a white solid. [α]25D = −187.5 (c = 1.0, CHCl3). m.p. 128–129 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.67–8.04 (m, 1H), 7.99 (dd, J = 29.8, 7.5 Hz, 1H), 7.72 (dd, J = 79.4, 8.9 Hz, 1H), 7.48–7.23 (m, J = 6.6 Hz, 5H), 5.16–5.08 (m, 2H), 5.06 (d, J = 7.9 Hz, 1H), 4.77–4.42 (m, 2H), 4.32 (dp, J = 17.5, 6.0, 5.5 Hz, 2H), 2.91 (s, 3H, major), 2.83 (s, 3H, minor), 2.70 (s, 3H), 1.83–1.47 (m, 9H), 1.40 (d, J = 6.9 Hz, 13H), 1.16 (d, J = 6.7 Hz, 3H), 0.98–0.54 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 173.3, 173.1, 172.6, 171.3, 155.9, 136.4, 128.9, 128.5, 128.4, 128.2, 79.5, 66.5, 66.3, 54.4, 51.5, 51.1, 45.7, 44.7, 37.2, 31.1, 30.2, 28.4, 25.0, 24.9, 24.7, 24.7, 23.7, 23.6, 23.5, 23.4, 23.2, 22.2, 21.8, 21.6, 21.6, 21.3. HRMS (ESI) m/z calcd. for C41H69N5O8 [M + Na]+: 782.5039; found: 782.5032.
- N-Boc-Me-L-Leu-L-Leu-O-Me-D-Ser-Me-L-Leu-L-Leu-OBn 8l. The 8l was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6l (13.8 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8l (19.1 g, 24.1 mmol, 86% yield) as a white solid. [α]25D = −198.6 (c = 1.0, CHCl3). m.p. 139–140 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.14 (m, 1H), 7.97 (m, 1H), 7.74 (m, 1H), 7.47–7.16 (m, 5H), 5.11 (m, 2H), 5.06 (d, J = 9.6 Hz, 1H), 4.97–4.74 (m, 1H), 4.66–4.45 (m, 1H), 4.42–4.18 (m, 2H), 3.45 (d, J = 6.3 Hz, 2H), 3.22 (s, 3H, major), 3.17 (s, 3H, minor), 2.94 (s, 3H, major), 2.80 (s, 3H, minor), 2.70 (d, J = 5.2 Hz, 3H), 1.82–1.46 (m, 9H), 1.40 (d, J = 7.1 Hz, 13H), 1.00–0.63 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.4, 171.3, 171.0, 170.5, 156.0, 136.4, 128.8, 128.4, 128.4, 128.2, 79.4, 72.7, 72.2, 66.5, 66.3, 58.9, 54.6, 51.5, 51.1, 41.4, 38.9, 37.2, 31.3, 28.4, 24.9, 24.7, 24.6, 23.8, 23.5, 23.2, 21.8, 21.6, 21.5. HRMS (ESI) m/z calcd. for C42H71N5O9 [M + Na]+: 812.5144; found: 812.5152.
- N-Boc-Me-L-Leu-L-Leu-O-Me-D-Tyr-Me-L-Leu-L-Leu-OBn 8m. The 8m was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6m (16.1 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8m (23.2 g, 26.6 mmol, 95% yield) as a white solid. [α]25D = −156.6 (c = 1.0, CHCl3). m.p. 141–142 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.60–8.19 (m, 1H), 8.16 (d, J = 7.5 Hz, 1H), 7.65 (mm, 1H), 7.35 (m, 5H), 7.12 (d, J = 8.1 Hz, 2H), 6.76 (d, J = 8.0 Hz, 2H), 5.11 (q, J = 12.9 Hz, 3H), 4.83 (t, J = 7.4 Hz, 1H), 4.65–4.42 (m, 1H), 4.32 (m, 2H), 3.70 (d, J = 8.9 Hz, 3H), 2.91 (dd, J = 14.1, 5.8 Hz, 1H), 2.85 (s, 3H, major), 2.79–2.73 (m, 1H), 2.69 (d, J = 13.1 Hz, 3H), 2.56 (s, 3H, minor), 1.72–1.13 (m, 21H), 1.03–0.57 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.1, 171.4, 158.3, 136.4, 130.7, 129.7, 128.9, 128.5, 128.3, 128.2, 113.9, 79.4, 66.4, 55.3, 53.9, 50.9, 37.6, 30.9, 30.16, 28.43, 24.92, 24.78, 24.54, 23.67, 23.22, 21.96, 21.64. HRMS (ESI) m/z calcd. for C48H75N5O9 [M + H]+: 888.5457; found: 888.5456.
- N-Boc-Me-L-Leu-L-Leu-Methylester-D-Asp-Me-L-Leu-L-Leu-OBn 8n. The 8n was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6n (14.6 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8n (20.0 g, 24.5 mmol, 88% yield) as a white solid. [α]25D = −139.8 (c = 1.0, CHCl3). m.p. 139–140 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.70–8.27 (m, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.49–7.18 (m, 5H), 5.10 (d, J = 6.0 Hz, 2H), 5.07 (d, J = 9.1 Hz, 2H), 4.69–4.42 (m, 1H), 4.39–4.23 (m, 2H), 3.54 (s, 3H), 2.95 (d, J = 8.8 Hz, 3H), 2.91–2.85 (m, 1H), 2.75–2.63 (m, 3H), 2.61–2.52 (m, 1H), 1.73–1.17 (m, 21H), 1.05–0.55 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.5, 171.8, 171.1, 136.4, 128.9, 128.5, 128.2, 66.4, 54.2, 51.9, 51.0, 41.5, 37.0, 36.6, 30.8, 28.4, 24.9, 24.7, 24.6, 24.4, 23.8, 23.6, 23.2, 21.9, 21.7, 21.5. HRMS (ESI) m/z calcd. for C43H71N5O10 [M + H]+: 840.5093; found: 840.5101.
- N-Boc-Me-L-Leu-L-Leu-3-Flu-D-Phe-Me-L-Leu-L-Leu-OBn 8o. The 8o was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6o (15.7 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8o (21.2 g, 24.6 mmol, 88% yield) as a white solid. [α]25D = −145.6 (c = 1.0, CHCl3). m.p. 140–141 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.47 (d, J = 6.8 Hz, 1H, major), 8.38 (d, J = 7.6 Hz, 1H, minor), 7.96 (d, J = 7.5 Hz, 1H, major), 7.86 (d, J = 7.5 Hz, 1H, minor), 7.63 (d, J = 8.7 Hz, 1H, major), 7.46 (d, J = 8.9 Hz, 1H, minor), 7.43–7.27 (m, 5H), 7.07 (m, 3H), 5.20–4.98 (m, 3H), 4.99–4.71 (m, 1H), 4.67–4.39 (m, 1H), 4.31 (m, 2H), 2.96 (dd, J = 13.4, 7.1 Hz, 1H), 2.88 (t, J = 15.5 Hz, 3H), 2.82–2.70 (m, 1H), 2.73–2.58 (m, 3H), 1.89–1.00 (m, 21H), 1.00–0.53 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.1, 171.2, 163.3, 161.7, 136.4, 136.3, 128.8, 128.8, 128.6, 128.4, 128.2, 126.0, 116.8, 116.6, 113.8, 113.7, 79.4, 66.6, 66.3, 60.2, 51.3, 51.1, 41.7, 37.7, 37.2, 36.9, 31.2, 30.2, 28.4, 24.9, 24.7, 24.6, 24.5, 23.7, 23.6, 23.4, 23.4, 23.2, 23.0, 21.9, 21.7, 21.6, 21.5. HRMS (ESI) m/z calcd. for C47H72FN5O8 [M + Na]+: 876.5257; found: 876.5254.
- N-Boc-Me-L-Leu-L-Leu-2-Me-D-Phe-Me-L-Leu-L-Leu-OBn 8q. The 8q was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6q (15.6 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8q (21.1 g, 24.6 mmol, 88% yield) as a white solid. [α]25D = −156.6 (c = 1.0, CHCl3). m.p. 161–162 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.59–8.15 (m, 1H), 7.98–7.63 (m, 1H), 7.57–7.25 (m, 5H), 7.22 (d, J = 7.5 Hz, 1H), 7.08 (m, 3H), 5.17–5.03 (m, 2H), 5.01 (dt, J = 11.2, 5.6 Hz, 1H), 4.92 (d, J = 7.5 Hz, 1H), 4.68–4.42 (m, 1H), 4.36 (dt, J = 14.1, 6.6 Hz, 1H), 4.32–4.19 (m, 1H), 3.02 (d, J = 11.0 Hz, 1H), 2.80 (dd, J = 15.1, 8.7 Hz, 1H), 2.75 (s, 1H), 2.68 (d, J = 10.4 Hz, 4H), 2.31 (s, 3H, major), 2.23 (s, 3H, minor), 1.80–1.44 (m, 6H), 1.45–1.20 (m, 13H), 0.96–0.59 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.5, 172.5, 171.2, 156.0, 136.4, 135.9, 134.0, 129.7, 129.1, 128.8, 128.8, 128.4, 128.4, 128.1, 79.4, 66.5, 66.2, 51.3, 51.1, 41.8, 41.5, 37.2, 36.9, 31.1, 30.1, 30.0, 28.4, 24.9, 24.7, 24.6, 23.7, 23.6, 23.4, 23.2, 23.1, 21.7, 21.5, 21.1. HRMS (ESI) m/z calcd. for C48H75N5O8 [M + Na]+: 872.5508; found: 872.5514.
- N-Boc-Me-L-Leu-L-Leu-3-Chl-D-Phe-Me-L-Leu-L-Leu-OBn 8p. The 8p was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6p (16.2 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8p (22.2 g, 25.3 mmol, 90% yield) as a white solid. [α]25D = −116.6 (c = 1.0, CHCl3). m.p. 161–162 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.45 (d, J = 6.7 Hz, 1H, major), 8.35 (d, J = 7.5 Hz, 1H, minor), 7.94 (d, J = 7.5 Hz, 1H, major), 7.85 (d, J = 7.5 Hz, 1H, minor), 7.64 (d, J = 8.5 Hz, 1H, major), 7.48 (d, J = 9.0 Hz, 1H, minor), 7.41–7.11 (m, 8H), 5.10 (m, 2H), 4.99 (m, 1H), 4.91 (d, J = 7.4 Hz, 1H), 4.67–4.40 (m, 1H), 4.41–4.19 (m, 2H), 3.00–2.69 (m, 5H), 2.68 (d, J = 9.9 Hz, 3H), 1.86–1.02 (m, 20H), 1.02–0.17 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.5, 172.2, 170.6, 155.3, 136.4, 131.7, 128.9, 128.8, 128.5, 128.4, 128.2, 79.5, 79.4, 66.6, 66.3, 51.3, 51.1, 50.8, 41.7, 37.7, 37.2, 37.0, 31.2, 30.2, 28.4, 24.9, 24.7, 24.5, 24.3, 23.7, 23.5, 23.4, 23.2, 23.1, 22.0, 22.0, 21.7, 21.5, 21.1. HRMS (ESI) m/z calcd. for C47H72ClN5O8 [M + Na]+: 892.4962; found: 892.4966.
- N-Boc-Me-L-Leu-L-Leu-3-Bro-D-Phe-Me-L-Leu-L-Leu-OBn 8r. The 8r was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6r (17.6 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8r (24.0 g, 26.3 mmol, 94% yield) as a white solid. [α]25D = −175.6 (c = 1.0, CHCl3). m.p. 171–172 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.44 (d, J = 6.8 Hz, 1H, major), 8.34 (d, J = 7.6 Hz, 1H, minor), 7.95 (d, J = 7.5 Hz, 1H, major), 7.86 (d, J = 7.4 Hz, 1H, minor), 7.64 (d, J = 8.6 Hz, 1H, major), 7.48 (d, J = 8.8 Hz, 1H, minor), 7.45 (m, 2H), 7.35 (m, 5H), 7.19 (m, 2H), 5.24–5.01 (m, 2H), 5.00–4.68 (m, 2H), 4.64–4.38 (m, 1H), 4.38–4.07 (m, 2H), 2.86 (m, 4H), 2.77 (d, J = 13.3 Hz, 1H), 2.67 (d, J = 8.7 Hz, 3H), 1.82–1.00 (m, 20H), 1.01–0.49 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 173.0, 172.6, 171.1, 170.6, 156.0, 136.4, 132.1, 131.4, 131.0, 128.8, 128.8, 128.5, 128.4, 128.4, 128.1, 120.3, 79.4, 66.6, 66.2, 60.2, 51.4, 51.1, 50.8, 41.8, 41.5, 37.2, 36.9, 31.2, 28.4, 24.9, 24.7, 24.6, 23.7, 23.5, 23.4, 23.2, 23.1, 22.1, 22.0, 21.7, 21.7, 21.5, 21.1. HRMS (ESI) m/z calcd. for C47H72BrN5O8 [M + H]+: 936.4456; found: 936.4454.
- N-Boc-Me-L-Leu-L-Leu-D-Cyclohexyl-Gly-Me-L-Leu-L-Leu-OBn 8s. The 8s was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6s (14.9 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8s (21.1 g, 25.4 mmol, 91% yield) as a white solid. [α]25D = −145.6 (c = 1.0, CHCl3). m.p. 140–141 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.09 (d, J = 7.0 Hz, 1H, major), 7.98 (d, J = 7.5 Hz, 1H, minor), 7.90 (d, J = 8.8 Hz, 1H, major), 7.84 (dd, J = 14.0, 8.4 Hz, 1H, minor), 7.75–7.65 (m, 1H), 7.35 (m, 5H), 5.18–5.08 (m, 2H), 5.06 (d, J = 12.9 Hz, 1H), 4.62 (d, J = 8.1 Hz, 1H), 4.52–4.20 (m, 3H), 2.97 (s, 3H, major), 2.86 (s, 3H, minor), 2.69 (d, J = 7.4 Hz, 3H), 1.89–0.94 (m, 32H), 0.95–0.58 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.7, 172.6, 172.5, 172.1, 170.7, 155.9, 136.4, 136.3, 128.9, 128.9, 128.6, 128.5, 128.2, 79.4, 66.5, 66.2, 54.5, 51.1, 41.4, 38.0, 37.1, 31.5, 29.2, 28.4, 26.2, 26.0, 26.0, 25.0, 24.9, 24.8, 24.6, 23.9, 23.5, 23.3, 23.3, 22.3, 21.6, 21.5, 21.4, 21.3. HRMS (ESI) m/z calcd. for C46H77N5O8 [M + Na]+: 850.5670; found:850.5675.
- N-Boc-Me-L-Leu-L-Leu-D-Phenyl-Gly-Me-L-Leu-L-Leu-OBn 8t. The 8t was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6t (14.8 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8t (20.0 g, 24.3 mmol, 87% yield) as a white solid. [α]25D = −167.5 (c = 1.0, CHCl3). m.p. 140–141 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.41 (d, J = 7.3 Hz, 1H), 8.19 (d, J = 7.5 Hz, 1H), 8.09–7.76 (m, 1H), 7.46–7.14 (m, 9H), 5.77 (q, J = 7.4, 6.8 Hz, 1H), 5.13 (d, J = 12.2 Hz, 2H), 5.08 (d, J = 12.7 Hz, 1H), 4.75–4.43 (m, 1H), 4.43–4.13 (m, 2H), 2.88 (s, 3H, minor), 2.75 (s, 3H, major), 2.69 (s, 3H), 1.84–1.07 (m, 20H), 1.09–0.24 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 171.7, 171.4, 171.0, 136.4, 128.9, 128.8, 128.5, 128.4, 128.3, 79.5, 66.4, 54.4, 51.5, 51.0, 40.8, 37.4, 31.1, 30.2, 28.4, 25.0, 24.7, 24.6, 24.4, 23.6, 23.5, 23.2, 22.2, 21.9, 21.6, 21.3. HRMS (ESI) m/z calcd. for C46H71N5O8 [M + Na]+: 844.5195; found: 844.5204.
- N-Boc-Me-L-Leu-L-Leu-D-Val-Me-L-Leu-L-Leu-OBn 8u. The 8u was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6u (13.7 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8u (19.1 g, 24.1 mmol, 86% yield) as a white solid. [α]25D = −175.6 (c = 1.0, CHCl3). m.p. 128–129 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.26 (d, J = 7.8 Hz, 1H), 7.99 (m, 1H), 7.85–7.63 (m, 1H), 7.49–7.26 (m, 5H), 5.14 (d, J = 6.1 Hz, 1H), 5.13–5.05 (m, 2H), 4.49 (t, J = 8.7 Hz, 2H), 4.31 (m, 2H), 2.96 (s, 3H), 2.70 (d, J = 4.2 Hz, 3H), 1.97 (q, J = 7.2 Hz, 1H), 1.68–1.43 (m, 9H), 1.43–1.36 (m, 10H), 1.35–1.22 (m, 3H), 0.85 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 172.5, 172.4, 172.2, 171.3, 136.4, 128.8, 128.5, 128.3, 79.4, 66.4, 54.3, 53.6, 50.8, 41.3, 37.5, 31.0, 30.2, 28.5, 24.9, 24.7, 24.6, 23.5, 23.3, 22.1, 21.9, 21.4, 19.4. HRMS (ESI) m/z calcd. for C43H73N5O8 [M + Na]+: 810.5351; found: 810.5359.
- N-Boc-Me-L-Leu-L-Leu-D-Met-Me-L-Leu-L-Leu-OBn 8v. The 8v was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6v (14.7 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8v (22.0 g, 26.9 mmol, 96% yield) as a white solid. [α]25D = −139.8 (c = 1.0, CHCl3). m.p. 118–119 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.21 (d, J = 7.2 Hz, 1H, major), 8.17 (d, J = 7.4 Hz, 1H, minor), 7.99 (d, J = 7.3 Hz, 1H, major), 7.93 (d, J = 7.6 Hz, 1H, minor), 7.75 (d, J = 8.5 Hz, 1H, major), 7.63 (d, J = 8.7 Hz, 1H, minor), 7.48–6.87 (m, 5H), 5.17–5.07 (m, 2H), 5.07–5.00 (m, 1H), 4.79 (m, 1H), 4.66–4.39 (m, 1H), 4.39–4.17 (m, 2H), 2.96 (s, 3H, major), 2.81 (s, 3H, minor), 2.70 (d, J = 4.6 Hz, 3H), 2.42 (q, J = 6.2, 4.6 Hz, 2H), 2.04 (s, 3H, major), 1.98 (s, 3H, minor), 1.81–1.14 (m, 22H), 1.10–0.56 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.6, 172.4, 171.3, 170.8, 136.4, 128.9, 128.5, 128.4, 128.2, 79.5, 66.5, 66.3, 54.5, 51.7, 51.1, 41.5, 37.9, 37.2, 31.2, 30.1, 29.7, 28.4, 24.9, 24.7, 23.7, 23.5, 23.4, 23.2, 22.2, 21.8, 21.6, 21.6, 21.5, 15.1. HRMS (ESI) m/z calcd. for C43H73N5O8S [M + Na]+: 842.5072; found: 842.5082.
- N-Boc-Me-L-Leu-L-Leu-3-Me-D-Phe-Me-L-Leu-L-Leu-OBn 8w. The 8w was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6w (15.6 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8w (20.2 g, 23.6 mmol, 85% yield) as a white solid. [α]25D = −178.6 (c = 1.0, CHCl3). m.p. 122–123 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.47 (d, J = 6.6 Hz, 1H, major), 8.36 (d, J = 7.3 Hz, 1H, minor), 7.95 (d, J = 7.4 Hz, 1H, major), 7.86 (d, J = 7.5 Hz, 1H, minor), 7.65 (d, J = 8.7 Hz, 1H, major), 7.47 (d, J = 9.2 Hz, 1H, minor), 7.35 (m, 5H), 7.24–6.88 (m, 4H), 5.23–5.04 (m, 2H), 5.01 (dd, J = 11.3, 4.8 Hz, 1H), 4.95–4.68 (m, 1H), 4.68–4.41 (m, 1H), 4.41–4.12 (m, 2H), 3.02–2.56 (m, 8H), 2.25 (d, J = 8.3 Hz, 3H), 1.81–1.56 (m, 3H), 1.45 (m, 17H), 1.19–0.95 (m, 1H), 0.96–0.45 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.9, 172.6, 172.4, 171.9, 170.6, 156.0, 137.6, 137.1, 136.4, 130.4, 128.9, 128.9, 128.5, 128.2, 127.6, 126.9, 79.4, 66.6, 66.2, 57.0, 55.6, 54.4, 51.1, 41.8, 41.6, 37.7, 36.9, 31.1, 30.2, 30.1, 28.4, 24.9, 24.7, 24.6, 24.2, 23.7, 23.6, 23.5, 23.2, 23.1, 22.0, 21.9, 21.7, 21.7, 21.5, 21.5, 21.2. HRMS (ESI) m/z calcd. for C48H75N5O8 [M + Na]+: 872.5508; found: 872.5517.
- N-Boc-Me-L-Leu-L-Leu-4-Me-D-Phe-Me-L-Leu-L-Leu-OBn 8x. The 8x was synthesized according to the method of 8 from 7a (10.0 g, 27.9 mmol, 1.0 equiv) and 6x (15.6 g, 30.7 mmol, 1.1 equiv), HOBT (4.1 g, 30.7 mmol, 1.1 equiv), and EDCI (5.9 g, 30.7 mmol, 1.1 equiv). The crude product was purified by silica gel chromatography (hexane/EtOAc = 70:30, v/v) to obtain 8x (21.0 g, 24.6 mmol, 88% yield) as a white solid. [α]25D = −156.7 (c = 1.0, CHCl3). m.p. 142–143 °C. 1H NMR (600 MHz, DMSO-d6) δ8.47 (d, J = 6.6 Hz, 1H, major), 8.36 (d, J = 7.3 Hz, 1H, minor), 7.95 (d, J = 7.4 Hz, 1H, major), 7.86 (d, J = 7.5 Hz, 1H, minor), 7.65 (d, J = 8.7 Hz, 1H, major), 7.47 (d, J = 9.2 Hz, 1H, minor), 7.35 (m, 5H), 7.24–6.88 (m, 4H), 5.23–5.04 (m, 2H), 5.01 (dd, J = 11.3, 4.8 Hz, 1H), 4.95–4.68 (m, 1H), 4.68–4.41 (m, 1H), 4.41–4.12 (m, 2H), 3.02–2.56 (m, 8H), 2.25 (d, J = 8.3 Hz, 3H), 1.81–1.56 (m, 3H), 1.45 (m, 17H), 1.19–0.95 (m, 1H), 0.96–0.45 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 172.9, 172.6, 172.4, 171.9, 170.6, 156.0, 137.6, 137.1, 136.4, 130.38, 128.9, 128.9, 128.5, 128.2, 127.6, 126.9, 79.4, 66.6, 66.3, 57.0, 55.6, 54.4, 51.1, 41.8, 41.6, 37.7, 36.9, 31.1, 30.2, 30.1, 28.4, 24.9, 24.7, 24.6, 24.2, 23.7, 23.6, 23.5, 23.3, 23.1, 22.0, 21.9, 21.7, 21.7, 21.5, 21.5, 21.2. HRMS (ESI) m/z calcd. for C48H75N5O8 [M + Na]+: 872.5508; found: 872.5517.
3.1.8. Synthesis of 9
3.1.9. Synthesis of 10
3.1.10. Synthesis of Gala
- Cyclo(Me-D-Leu-D-Leu-D-Leu-Me-D-Leu-D-Leu) Gala01. The product 10a (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala01 (62 mg, 0.10 mmol, 48% yield) as a white solid. [α]25D = −5.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.62 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), 5.10 (t, J = 7.6 Hz, 1H), 4.74 (dq, J = 35.7, 7.5 Hz, 2H), 4.48 (dd, J = 10.1, 5.2 Hz, 1H), 4.15 (dt, J = 13.2, 6.6 Hz, 1H), 2.98 (s, 3H), 2.72 (s, 3H), 1.69–1.14 (m, 15H), 0.87 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 174.5, 172.2, 171.3, 170.8, 170.5, 57.9, 53.6, 51.3, 48.3, 47.7, 41.34, 41.7, 38.7, 37.2, 35.1, 31.3, 29.9, 25.2, 24.9, 24.8, 24.8, 24.7, 23.5, 23.4, 23.1, 23.0, 23.0, 22.0, 22.0, 21.9, 21.7. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-L-Leu-L-Leu-D-Leu-Me-L-Leu-L-Leu) Gala02. The product 10b (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala02 (60 mg, 0.10 mmol, 45% yield) as a white solid. [α]25D = −13.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.63 (d, J = 8.1 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 9.5, 6.2 Hz, 1H), 4.77 (td, J = 8.3, 5.6 Hz, 1H), 4.71 (q, J = 7.6 Hz, 1H), 4.49 (dd, J = 10.4, 5.2 Hz, 1H), 4.15 (ddd, J = 10.4, 8.1, 5.2 Hz, 1H), 2.98 (s, 3H), 2.73 (s, 3H), 1.75–1.32 (m, 15H), 1.02–0.61 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 174.5, 172.3, 171.4, 170.8, 170.6, 57.9, 53.7, 51.4, 48.3, 47.7, 41.4, 41.1, 38.7, 37.2, 35.2, 31.3, 29.9, 25.3, 24.9, 24.9, 24.8, 24.8, 23.5, 23.4, 23.2, 23.0, 23.0, 22.1, 22.0, 21.9, 21.7. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-D-Leu-L-Leu-L-Leu-Me-D-Leu-L-Leu) Gala03. The product 10c (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala03 (63 mg, 0.11 mmol, 48% yield) as a white solid. [α]25D = −75.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.29 (d, J = 8.7 Hz, 1H), 7.45 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 9.4 Hz, 1H), 5.06 (t, J = 7.8 Hz, 1H), 4.80 (td, J = 9.1, 5.5 Hz, 1H), 4.73–4.50 (m, 2H), 4.16 (td, J = 9.5, 5.1 Hz, 1H), 2.95 (s, 3H), 2.54 (s, 3H), 1.75–1.31 (m, 15H), 1.09–0.55 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 173.7, 171.7, 171.5, 171.2, 169.7, 55.9, 53.5, 52.3, 47.6, 47.5, 41.6, 41.3, 40.9, 37.1, 34.9, 30.9, 29.3, 25.0, 25.0, 24.8, 24.8, 24.7, 23.4, 23.3, 23.2, 23.0, 22.9, 22.7, 22.3, 22.1, 21.4. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-D-Leu-L-Leu-D-Leu-Me-D-Leu-L-Leu) Gala04. The product 10d (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala04 (59 mg, 0.11 mmol, 0.45% yield) as a white solid. [α]25D = −55.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.28 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.00 (d, J = 9.4 Hz, 1H), 5.05 (t, J = 7.8 Hz, 1H), 4.79 (q, J = 3.6 Hz, 1H), 4.73–4.45 (m, 2H), 4.15 (q, J = 4.3 Hz, 1H), 2.94 (s, 3H), 2.53 (s, 3H), 1.76–1.26 (m, 15H), 1.02–0.65 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 173.7, 171.7, 171.5, 171.2, 169.7, 55.9, 53.5, 52.3, 47.6, 47.5, 41.6, 41.3, 40.9, 37.1, 34.9, 30.9, 29.3, 25.0, 24.9, 24.8, 24.7, 24.6, 23.4, 23.3, 23.2, 23.0, 22.9, 22.7, 22.3, 22.1, 21.4. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-L-Leu-D-Leu-L-Leu-Me-L-Leu-D-Leu) Gala05. The product 10e (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala05 (60 mg, 0.10 mmol, 45% yield) as a white solid. [α]25D = −75.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.30 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 9.4 Hz, 1H), 5.06 (t, J = 7.7 Hz, 1H), 4.79 (dd, J = 9.2, 5.8 Hz, 1H), 4.64 (t, J = 8.4 Hz, 2H), 4.15 (dt, J = 9.5, 4.7 Hz, 1H), 2.95 (s, 3H), 2.54 (s, 3H), 1.88–1.15 (m, 15H), 1.15–0.41 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 173.7, 171.7, 171.5, 171.1, 169.7, 55.9, 53.5, 52.3, 47.6, 47.51, 41.7, 41.2, 40.9, 37.1, 34.9, 30.9, 29.3, 25.0, 24.9, 24.8, 24.7, 24.7, 23.4, 23.4, 23.2, 23.0, 22.9, 22.7, 22.3, 22.1, 21.4. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-L-Leu-D-Leu-D-Leu-Me-L-Leu-D-Leu) Gala06. The product 10f (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala06 (69 mg, 0.12 mmol, 54% yield) as a white solid. [α]25D = −85.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.29 (d, J = 8.7 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 9.4 Hz, 1H), 5.05 (t, J = 7.7 Hz, 1H), 4.79 (td, J = 9.0, 5.5 Hz, 1H), 4.63 (t, J = 8.4 Hz, 2H), 4.15 (td, J = 9.5, 5.2 Hz, 1H), 2.94 (s, 3H), 2.53 (s, 3H), 1.76–1.14 (m, 16H), 0.97–0.58 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 173.7, 171.7, 171.5, 171.2, 169.7, 55.9, 53.5, 52.3, 47.6, 47.5, 41.6, 41.3, 40.9, 37.1, 34.9, 30.9, 29.3, 25.0, 24.9, 24.8, 24.8, 24.7, 23.4, 23.4, 23.2, 23.0, 22.9, 22.8, 22.3, 22.1, 21.4. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-D-Leu-D-Leu-L-Leu-Me-D-Leu-D-Leu) Gala07. The product 10g (134 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala07 (65 mg, 0.11 mmol, 50% yield) as a white solid. [α]25D = −10.6 (c = 1.0, CHCl3). m.p. 199–200 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.63 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), 5.11 (t, J = 7.6 Hz, 1H), 4.77 (d, J = 7.4 Hz, 1H), 4.71 (d, J = 7.8 Hz, 1H), 4.49 (dd, J = 10.1, 5.2 Hz, 1H), 4.27–3.96 (m, 1H), 2.99 (s, 3H), 2.73 (s, 3H), 1.73–1.26 (m, 15H), 0.87 (m, 30H). 13C NMR (151 MHz, DMSO-d6) δ 174.53, 172.27, 171.37, 170.84, 170.57, 57.94, 53.64, 51.37, 48.34, 47.69, 41.40, 41.09, 38.70, 37.23, 35.17, 31.34, 29.88, 25.26, 24.92, 24.87, 24.81, 24.77, 23.50, 23.40, 23.15, 23.02, 22.98, 22.06, 21.97, 21.91, 21.70. HRMS (ESI) m/z calcd. for C32H59N5O5 [M + Na]+: 616.4408; found: 616.4415.
- Cyclo(Me-L-Leu-L-Leu-D-Pro-Me-L-Leu-L-Leu) Gala08. The product 10h (131 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala08 (65 mg, 0.11 mmol, 51% yield) as a white solid. [α]25D = −75.6 (c = 1.0, CHCl3). m.p. 230–231 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.37 (d, J = 7.3 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 4.91 (t, J = 7.8 Hz, 1H), 4.75 (td, J = 8.5, 8.1, 5.3 Hz, 2H), 4.60 (dd, J = 10.5, 5.8 Hz, 1H), 4.33 (ddd, J = 11.6, 7.5, 4.2 Hz, 1H), 3.51 (t, J = 8.5 Hz, 1H), 3.45 (q, J = 5.3, 3.8 Hz, 1H), 3.02 (d, J = 11.7 Hz, 6H), 2.17 (q, J = 6.4 Hz, 1H), 2.04–1.91 (m, 1H), 1.88 (dd, J = 12.5, 6.6 Hz, 1H), 1.81–1.74 (m, 1H), 1.74–1.66 (m, 1H), 1.65–1.29 (m, 11H), 0.99–0.69 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 173.9, 173.0, 171.3, 170.2, 169.5, 57.6, 56.0, 53.8, 50.2, 47.9, 46.4, 42.3, 36.8, 36.3, 31.0, 30.8, 28.5, 25.4, 25.2, 25.1, 24.9, 24.7, 23.7, 23.4, 23.3, 23.2, 22.7, 22.4, 22.0, 21.8. HRMS (ESI) m/z calcd. for C31H55N5O5 [M + Na]+: 600.4095; found: 600.4103.
- Cyclo(Me-L-Leu-L-Leu-D-Phe-Me-L-Leu-L-Leu) (Gala09). The product 10i (142 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala09 (70 mg, 0.11 mmol, 51% yield) as a white solid. [α]25D = −55.6 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.60 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.35–7.04 (m, 5H), 5.06 (dd, J = 9.8, 6.0 Hz, 1H), 4.87 (q, J = 7.9 Hz, 1H), 4.76 (q, J = 7.5 Hz, 1H), 4.58 (dd, J = 10.1, 5.6 Hz, 1H), 4.12 (td, J = 8.8, 5.7 Hz, 1H), 3.05 (dd, J = 13.1, 8.4 Hz, 1H), 2.97 (s, 3H), 2.76 (dd, J = 13.2, 6.2 Hz, 1H), 2.63 (s, 3H), 1.86–1.45 (m, 7H), 1.40 (q, J = 8.2, 7.3 Hz, 4H), 1.24–1.07 (m, 1H), 0.97–0.59 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.8, 171.3, 170.9, 170.4, 138.2, 129.7, 128.4, 126.6, 57.7, 53.7, 51.6, 51.2, 48.2, 41.4, 38.7, 37.8, 37.2, 35.2, 31.2, 29.8, 25.2, 24.8, 24.8, 24.7, 23.5, 23.4, 23.2, 22.2, 22.0, 21.9. HRMS (ESI) m/z calcd. for C35H57N5O5 [M + Na]+: 650.4252; found: 650.4259.
- Cyclo(Me-L-Leu-L-Leu-Gly-Me-L-Leu-L-Leu) Gala10. The product 10j (122 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala10 (60 mg, 0.11 mmol, 51% yield) as a white solid. [α]25D = −65.5 (c = 1.0, CHCl3). m.p. 200–201 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.34–7.84 (m, 1H), 7.76–7.38 (m, 1H), 7.22 (d, J = 4.3 Hz, 1H), 5.04 (t, J = 7.7 Hz, 1H), 4.83 (td, J = 9.0, 4.6 Hz, 1H), 4.60 (dd, J = 13.8, 9.1 Hz, 1H), 4.45 (dd, J = 9.7, 5.3 Hz, 1H), 4.09 (ddd, J = 10.8, 8.1, 4.8 Hz, 1H), 3.25 (d, J = 13.7 Hz, 1H), 3.01 (s, 3H), 2.64 (s, 3H), 1.78–1.30 (m, 12H), 1.03–0.62 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.7, 171.7, 171.3, 170.3, 170.2, 58.1, 53.8, 51.8, 48.3, 43.12, 41.5, 38.3, 37.4, 35.1, 31.6, 31.5, 30.4, 25.3, 24.8, 24.8, 24.8, 23.6, 23.5, 23.4, 23.1, 22.3, 22.2, 22.0, 21.4. HRMS (ESI) m/z calcd. for C28H51N5O5 [M + Na]+: 560.3782; found: 560.3786.
- Cyclo(Me-L-Leu-L-Leu-D-Ala-Me-L-Leu-L-Leu) Gala11. The product 10k (125 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala11 (59 mg, 0.11 mmol, 49% yield) as a white solid. [α]25D = −54.6 (c = 1.0, CHCl3). m.p. 191–192 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.78 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 5.13 (t, J = 7.6 Hz, 1H), 4.83 (td, J = 8.7, 5.0 Hz, 1H), 4.74 (dd, J = 8.7, 6.1 Hz, 1H), 4.40 (dd, J = 10.1, 5.1 Hz, 1H), 4.12 (ddd, J = 12.1, 8.2, 4.3 Hz, 1H), 3.01 (s, 3H), 2.63 (s, 3H), 1.80–1.65 (m, 2H), 1.65–1.29 (m, 10H), 1.12 (d, J = 6.3 Hz, 3H), 1.04–0.60 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.88, 172.24, 171.62, 170.87, 170.38, 58.31, 53.56, 51.46, 48.43, 45.25, 41.50, 38.45, 37.38, 35.00, 31.48, 29.57, 25.30, 24.84, 24.81, 23.68, 23.55, 23.31, 23.06, 22.28, 22.07, 21.92, 21.31, 17.85. HRMS (ESI) m/z calcd. for C29H53N5O5 [M + Na]+: 574.3939; found: 574.3943.
- Cyclo(Me-L-Leu-L-Leu-O-Me-D-Ser-Me-L-Leu-L-Leu) Gala12. The product 10l (132 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala12 (67 mg, 0.12 mmol, 52% yield) as a white solid. [α]25D = −75.6 (c = 1.0, CHCl3). m.p. 203–204 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.80 (d, J = 8.2 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 9.0 Hz, 1H), 5.10 (t, J = 7.7 Hz, 1H), 4.82 (td, J = 8.2, 4.1 Hz, 2H), 4.45 (dd, J = 10.1, 4.9 Hz, 1H), 4.13 (ddd, J = 10.8, 8.2, 4.6 Hz, 1H), 3.63 (t, J = 8.6 Hz, 1H), 3.28 (dd, J = 9.4, 5.7 Hz, 1H), 3.23 (s, 3H), 2.99 (s, 3H), 2.67 (s, 3H), 1.78–1.65 (m, 2H), 1.64–1.31 (m, 10H), 1.05–0.57 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.7, 171.5, 171.3, 170.7, 170.3, 71.7, 58.8, 58.0, 53.6, 51.4, 48.6, 48.4, 41.4, 38.4, 37.3, 35.1, 31.4, 29.7, 25.3, 25.0, 24.8, 23.6, 23.5, 23.3, 23.1, 22.2, 22.0, 22.0, 21.5. HRMS (ESI) m/z calcd. for C30H55N5O6 [M + Na]+: 604.4045; found: 604.4051.
- Cyclo(Me-L-Leu-L-Leu-O-Tyr-Me-L-Leu-L-Leu) Gala13. The product 10m (148 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala13 (75 mg, 0.11 mmol, 52% yield) as a white solid. [α]25D = −34.6 (c = 1.0, CHCl3). m.p. 205–206 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.57 (t, J = 7.9 Hz, 2H), 7.45 (d, J = 8.2 Hz, 1H), 7.25–6.99 (m, 2H), 6.91–6.66 (m, 2H), 5.04 (dd, J = 9.9, 6.0 Hz, 1H), 4.78 (dtd, J = 39.0, 8.5, 6.1 Hz, 2H), 4.57 (dd, J = 10.2, 5.6 Hz, 1H), 4.11 (ddd, J = 9.6, 7.9, 5.6 Hz, 1H), 3.70 (s, 3H), 2.96 (s, 3H), 2.94 (s, 1H), 2.69 (dd, J = 13.1, 6.4 Hz, 1H), 2.62 (s, 3H), 1.78–1.44 (m, 7H), 1.39 (m, 5H), 0.96–0.69 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.1, 172.0, 171.3, 170.9, 170.4, 158.3, 130.7, 130.0, 113.9, 57.7, 55.4, 53.6, 51.5, 51.3, 48.2, 41.4, 38.8, 37.2, 36.9, 35.3, 31.2, 29.9, 25.2, 24.9, 24.8, 24.6, 23.5, 23.4, 23.4, 23.2, 22.2, 22.0, 21.9. HRMS (ESI) m/z calcd. for C36H59N5O6 [M + Na]+: 680.4358; found: 680.4365.
- Cyclo(Me-L-Leu-L-Leu-Methylester-D-Asp-Me-L-Leu-L-Leu) Gala14. The product 10n (138 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala14 (70 mg, 0.11 mmol, 52% yield) as a white solid. [α]25D = −45.9 (c = 1.0, CHCl3). m.p. 204–205 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.88 (d, J = 8.0 Hz, 1H), 7.28 (dd, J = 8.7, 6.4 Hz, 2H), 5.09 (dd, J = 9.6, 6.0 Hz, 1H), 4.99 (td, J = 9.6, 4.3 Hz, 1H), 4.80 (td, J = 8.9, 4.8 Hz, 1H), 4.47 (dd, J = 9.9, 5.0 Hz, 1H), 4.05 (ddd, J = 11.4, 8.0, 4.4 Hz, 1H), 3.55 (s, 3H), 3.00 (s, 3H), 2.78 (dd, J = 16.1, 10.1 Hz, 1H), 2.67 (s, 3H), 2.47 (dd, J = 16.1, 4.3 Hz, 1H), 1.78–1.28 (m, 13H), 0.88 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.66, 171.57, 171.15, 171.05, 170.84, 170.33, 57.80, 53.77, 51.81, 51.64, 48.37, 46.57, 41.30, 38.24, 37.35, 36.25, 34.91, 31.40, 29.62, 25.25, 24.80, 24.78, 23.64, 23.49, 23.43, 23.11, 22.07, 22.01, 21.94, 21.45. HRMS (ESI) m/z calcd. for C31H55N5O7 [M + Na]+: 632.3994; found: 632.4000.
- Cyclo(Me-L-Leu-L-Leu-3-Flu-D-Phe-Me-L-Leu-L-Leu) Gala15. The product 10o (145 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala15 (75 mg, 0.12 mmol, 53% yield) as a white solid. [α]25D = −54.8 (c = 1.0, CHCl3). m.p. 205–206 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.59 (t, J = 9.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.27 (q, J = 7.4 Hz, 1H), 7.10–6.83 (m, 3H), 5.07 (dd, J = 10.0, 5.9 Hz, 1H), 4.89 (q, J = 7.8 Hz, 1H), 4.75 (q, J = 7.8 Hz, 1H), 4.58 (dd, J = 10.1, 5.6 Hz, 1H), 4.09 (td, J = 8.7, 5.6 Hz, 1H), 3.05 (dd, J = 13.2, 7.8 Hz, 1H), 2.97 (s, 3H), 2.79 (dd, J = 13.2, 6.8 Hz, 1H), 2.66 (s, 3H), 1.71–1.15 (m, 13H), 1.00–0.58 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.7, 171.3, 170.8, 170.4, 163.3, 141.2, 141.2, 130.3, 130.2, 125.9, 116.5, 116.4, 60.2, 57.7, 53.7, 51.6, 50.9, 48.3, 41.3, 38.7, 37.4, 37.2, 35.3, 31.2, 29.9, 25.2, 24.9, 24.8, 24.7, 23.5, 23.4, 23.3, 23.2, 22.2, 22.0, 21.9, 21.8. HRMS (ESI) m/z calcd. for C35H56FN5O5 [M + Na]+: 668.4158; found: 668.4164.
- Cyclo(Me-L-Leu-L-Leu-2-Me-D-Phe-Me-L-Leu-L-Leu) Gala16. The product 10p (145 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala16 (75 mg, 0.12 mmol, 53% yield) as a white solid. [α]25D = −35.6 (c = 1.0, CHCl3). m.p. 235–236 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.66 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.18–6.85 (m, 4H), 5.04 (dd, J = 9.9, 6.1 Hz, 1H), 4.90 (td, J = 8.8, 5.6 Hz, 1H), 4.76 (q, J = 7.6 Hz, 1H), 4.57 (dd, J = 10.3, 5.4 Hz, 1H), 4.12 (td, J = 8.8, 5.3 Hz, 1H), 3.08 (dd, J = 13.5, 9.0 Hz, 1H), 2.96 (s, 3H), 2.75 (dd, J = 13.5, 5.6 Hz, 1H), 2.59 (s, 3H), 2.32 (s, 3H), 1.77–1.29 (m, 11H), 1.18–1.04 (m, 1H), 1.00–0.53 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.7, 171.3, 171.0, 170.3, 136.7, 136.1, 130.4, 130.1, 126.7, 125.9, 57.7, 53.6, 51.4, 49.9, 48.2, 41.4, 38.7, 37.2, 35.1, 34.9, 31.2, 29.8, 25.2, 24.8, 24.8, 24.6, 23.5, 23.4, 23.2, 22.1, 22.0, 21.9, 21.8, 19.5. HRMS (ESI) m/z calcd. for C36H59N5O5 [M + H]+: 664.4408; found: 664.4411.
- Cyclo(Me-L-Leu-L-Leu-4-Chl-D-Phe-Me-L-Leu-L-Leu) Gala17. The product 10q (149 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala17 (80 mg, 0.12 mmol, 55% yield) as a white solid. [α]25D = −75.6 (c = 1.0, CHCl3).−67.5 m.p. 235–236 °C. 1H NMR (600 MHz, DMSO-d6) δ7.60 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 8.6 Hz, 1H), 7.42 (dd, J = 20.0, 8.1 Hz, 1H), 7.30–7.16 (m, 4H), 5.04 (dt, J = 10.5, 5.2 Hz, 1H), 4.95–4.80 (m, 1H), 4.80–4.68 (m, 1H), 4.57 (dt, J = 11.6, 5.8 Hz, 1H), 4.18–3.81 (m, 1H), 3.03 (m, 1H), 2.97 (s, 3H), 2.76 (dd, J = 13.3, 6.4 Hz, 1H), 2.64 (d, J = 12.2 Hz, 3H), 1.76–1.20 (m, 12H), 1.03–0.53 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.8, 171.3, 170.9, 170.4, 138.2, 131.7, 129.7, 128.5, 128.4, 57.8, 57.7, 53.7, 51.5, 51.2, 48.2, 41.4, 38.7, 37.2, 35.2, 31.2, 29.9, 25.2, 24.8, 24.8, 24.7, 23.5, 23.4, 23.4, 23.2, 22.2, 22.0, 21.9, 21.9. HRMS (ESI) m/z calcd. for C35H56ClN5O5 [M + H]+: 684.3862; found: 684.3864.
- Cyclo(Me-L-Leu-L-Leu-4-Bro-D-Phe-Me-L-Leu-L-Leu) Gala18. The product 10r (159 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala18 (85 mg, 0.12 mmol, 55% yield) as a white solid. [α]25D = −65.6 (c = 1.0, CHCl3). m.p. 245–246 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.58 (dd, J = 17.6, 8.3 Hz, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.20 (m, 5H), 5.05 (t, J = 7.9 Hz, 1H), 4.87 (q, J = 8.0 Hz, 1H), 4.75 (q, J = 7.6 Hz, 1H), 4.57 (dd, J = 10.3, 5.6 Hz, 1H), 4.11 (q, J = 7.8 Hz, 1H), 3.04 (dd, J = 13.1, 8.5 Hz, 1H), 2.97 (s, 3H), 2.76 (dd, J = 13.3, 6.2 Hz, 1H), 2.63 (s, 3H), 1.92–1.08 (m, 13H), 1.04–0.53 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.8, 171.3, 170.9, 170.4, 138.2, 129.7, 128.5, 126.7, 57.7, 53.7, 51.2, 48.2, 41.4, 38.7, 37.8, 37.2, 35.2, 31.2, 29.9, 25.2, 24.8, 24.8, 24.7, 23.5, 23.4, 23.2, 22.2, 22.0, 21.9, 21.9. HRMS (ESI) m/z calcd. for C35H56BrN5O5 [M + H]+: 728.3357; found: 728.3360.
- Cyclo(Me-L-Leu-L-Leu-D-cyclohexyl-Gly-Me-L-Leu-L-Leu) Gala19. The product 10s (140 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala19 (70 mg, 0.11 mmol, 51% yield) as a white solid. [α]25D = −35.6 (c = 1.0, CHCl3). m.p. 204–205 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.63 (d, J = 7.8 Hz, 1H), 7.45 (d, J = 9.1 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 5.10 (dd, J = 10.2, 6.0 Hz, 1H), 4.71 (t, J = 7.4 Hz, 1H), 4.69–4.54 (m, 1H), 4.43 (t, J = 9.5 Hz, 1H), 4.24 (q, J = 7.6 Hz, 1H), 2.95 (s, 3H), 2.81 (s, 3H), 1.77 (d, J = 11.1 Hz, 1H), 1.73–1.56 (m, 8H), 1.57–1.22 (m, 10H), 1.13 (p, J = 12.9, 12.4 Hz, 3H), 0.97–0.73 (m, 26H). 13C NMR (151 MHz, DMSO-d6) δ 174.0, 172.5, 171.1, 171.0, 170.7, 57.5, 54.1, 53.7, 51.3, 48.2, 41.2, 37.0, 35.6, 31.1, 30.3, 29.8, 28.6, 26.5, 25.9, 25.2, 25.0, 24.7, 23.5, 23.2, 22.2, 22.1, 21.9, 21.7. HRMS (ESI) m/z calcd. for C34H61N5O5 [M + Na]+: 642.4565; found: 642.4571.
- Cyclo(Me-L-Leu-L-Leu-D-phenyl-Gly-Me-L-Leu-L-Leu) Gala20. The product 10t (140 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala20 (70 mg, 0.11 mmol, 51% yield) as a white solid. [α]25D = −34.6 (c = 1.0, CHCl3). m.p. 204–205 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.89 (d, J = 8.2 Hz, 1H), 7.77 (d, J = 8.8 Hz, 1H), 7.31 (m, 5H), 5.88 (d, J = 8.8 Hz, 1H), 5.08 (t, J = 7.8 Hz, 1H), 4.86 (td, J = 9.4, 4.0 Hz, 1H), 4.47 (dd, J = 9.9, 5.4 Hz, 1H), 4.20 (td, J = 9.4, 8.8, 4.7 Hz, 1H), 3.03 (s, 3H), 2.80 (s, 3H), 1.84–1.13 (m, 13H), 0.89 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.8, 171.8, 171.3, 170.2, 138.8, 128.3, 128.1, 127.6, 58.5, 53.9, 53.4, 51.4, 48.5, 41.4, 38.4, 37.5, 35.0, 31.5, 30.1, 25.3, 24.9, 24.8, 24.8, 24.6, 23.6, 23.6, 23.3, 23.0, 22.3, 22.2, 21.9, 21.3. HRMS (ESI) m/z calcd. for C34H55N5O5 [M + Na]+: 636.4095; found: 636.4102.
- Cyclo(Me-L-Leu-L-Leu-D-Val-Me-L-Leu-L-Leu) Gala21. The product 10u (131 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala21 (65 mg, 0.11 mmol, 50% yield) as a white solid. [α]25D = −38.7 (c = 1.0, CHCl3). m.p. 192–193 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.62 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 9.1 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 5.12 (dd, J = 10.3, 5.7 Hz, 1H), 4.70 (d, J = 7.4 Hz, 1H), 4.63 (dd, J = 10.4, 5.7 Hz, 1H), 4.38 (t, J = 9.4 Hz, 1H), 4.24 (d, J = 7.4 Hz, 1H), 2.95 (s, 3H), 2.83 (s, 3H), 2.04 (dt, J = 9.7, 6.7 Hz, 1H), 1.76–1.18 (m, 12H), 0.94–0.77 (m, 26H). 13C NMR (151 MHz, DMSO-d6) δ 173.9, 172.7, 171.1, 171.0, 170.7, 57.5, 55.3, 53.7, 51.5, 48.2, 41.2, 39.1, 37.0, 35.7, 31.1, 30.3, 29.9, 25.2, 25.0, 25.0, 24.7, 23.5, 23.5, 23.3, 23.1, 22.3, 22.2, 21.9, 21.7, 19.6, 18.9. HRMS (ESI) m/z calcd. for C31H57N5O5 [M + H]+: 602.4252; found: 602.4261.
- Cyclo(Me-L-Leu-L-Leu-D-Met-Me-L-Leu-L-Leu) Gala22. The product 10v (138 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala22 (70 mg, 0.11 mmol, 52% yield) as a white solid. [α]25D = −54.7 (c = 1.0, CHCl3). m.p. 174–175 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.71 (d, J = 8.1 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.26 (d, J = 8.9 Hz, 1H), 5.12 (dd, J = 9.5, 6.3 Hz, 1H), 4.81 (dq, J = 22.6, 7.3 Hz, 2H), 4.47 (dd, J = 10.1, 5.0 Hz, 1H), 4.16 (ddd, J = 10.7, 8.0, 5.0 Hz, 1H), 2.99 (s, 3H), 2.70 (s, 3H), 2.44–2.18 (m, 2H), 2.03 (s, 4H), 1.70 (m, 3H), 1.66–1.44 (m, 7H), 1.39 (m, 3H), 0.88 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.6, 171.6, 171.5, 171.1, 170.4, 58.0, 53.6, 51.4, 48.5, 48.4, 41.4, 38.5, 37.3, 35.1, 31.5, 31.4, 30.0, 29.8, 25.3, 24.9, 24.8, 24.8, 23.6, 23.5, 23.4, 23.1, 22.0, 22.0, 21.6, 14.9. HRMS (ESI) m/z calcd. for C31H57N5O5S [M + Na]+: 634.3973; found: 634.3981.
- Cyclo(Me-L-Leu-L-Leu-3-Me-D-Phe-Me-L-Leu-L-Leu) Gala23. The product 10w (145 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala23 (75 mg, 0.12 mmol, 53% yield) as a white solid. [α]25D = −45.6 (c = 1.0, CHCl3). m.p. 194–195 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.62 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.45 (d, J = 8.1 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 7.05–6.87 (m, 3H), 5.06 (dd, J = 9.8, 6.1 Hz, 1H), 4.84 (d, J = 7.7 Hz, 1H), 4.76 (t, J = 7.5 Hz, 1H), 4.57 (dd, J = 10.4, 5.5 Hz, 1H), 4.13 (d, J = 7.6 Hz, 1H), 3.01 (dd, J = 13.1, 8.5 Hz, 1H), 2.97 (s, 3H), 2.71 (dd, J = 13.3, 6.2 Hz, 1H), 2.62 (s, 3H), 2.25 (s, 3H), 1.64–1.31 (m, 10H), 1.31–1.10 (m, 2H), 1.00–0.50 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 171.9, 171.3, 170.9, 170.4, 138.1, 137.4, 130.2, 128.4, 127.3, 126.7, 57.7, 53.6, 51.5, 51.1, 48.2, 41.4, 38.7, 37.8, 37.2, 35.2, 31.2, 29.8, 26.8, 25.2, 24.8, 24.8, 24.7, 23.5, 23.4, 23.2, 22.2, 22.0, 21.9, 21.4. HRMS (ESI) m/z calcd. for C36H59N5O5 [M + Na]+: 664.4408; found: 664.4412.
- Cyclo(Me-L-Leu-L-Leu-3-Me-D-Phe-Me-L-Leu-L-Leu) Gala24. The product 10x (145 mg, 0.22 mmol, 1 equiv) was dissolved in MeCN (4.3 mM) and reacted according to route E for macrolactamization. PyBOP (229 mg, 0.44 mmol, 2 equiv), DMAP (80 mg, 0.66 mmol, 3 equiv). The crude residue was purified by column chromatography (hexane/EtOAc = 60:40, v/v), obtaining compound Gala24 (71 mg, 0.11 mmol, 50% yield) as a white solid. [α]25D = −65.6 (c = 1.0, CHCl3). m.p. 204–205 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.58 (t, J = 9.2 Hz, 2H), 7.45 (d, J = 8.1 Hz, 1H), 7.31–6.75 (m, 4H), 5.03 (dd, J = 9.8, 5.9 Hz, 1H), 4.83 (q, J = 7.9 Hz, 1H), 4.74 (q, J = 7.6 Hz, 1H), 4.57 (dd, J = 10.3, 5.6 Hz, 1H), 4.11 (q, J = 7.6 Hz, 1H), 3.02–2.97 (m, 1H), 2.96 (s, 3H), 2.71 (dd, J = 13.2, 6.2 Hz, 1H), 2.62 (s, 3H), 2.24 (s, 3H), 1.70–1.12 (m, 12H), 1.00–0.53 (m, 24H). 13C NMR (151 MHz, DMSO-d6) δ 174.1, 171.9, 171.3, 170.9, 170.4, 135.6, 135.0, 129.5, 129.1, 57.67, 53.6, 51.6, 51.2, 48.2, 41.4, 38.8, 37.4, 37.2, 35.3, 31.2, 29.9, 25.2, 24.9, 24.8, 24.6, 23.5, 23.4, 23.4, 23.2, 22.2, 22.0, 21.9, 21.1. HRMS (ESI) m/z calcd. for C36H59N5O5 [M + Na]+: 664.4408; found: 664.4412.
3.2. Biological Evaluations
3.2.1. Cell Culture
3.2.2. CCK-8 Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bliss, M. Rewriting Medical History: Charles Best and the Banting and Best Myth. J. Hist. Med. Allied Sci. 1993, 48, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. [Google Scholar] [CrossRef]
- Craik, D.J. Seamless Proteins Tie up Their Loose Ends. Science 2006, 311, 1563–1564. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Cendron, L.; Chen, S.; Touati, J.; Winter, G.; Zanotti, G.; Heinis, C. Bicyclic Peptide Inhibitor Reveals Large Contact Interface with a Protease Target. ACS Chem. Biol. 2012, 7, 817–821. [Google Scholar] [CrossRef]
- Appavoo, S.D.; Huh, S.; Diaz, D.B.; Yudin, A.K. Conformational Control of Macrocycles by Remote Structural Modification. Chem. Rev. 2019, 119, 9724–9752. [Google Scholar] [CrossRef]
- Haubner, R.; Finsinger, D.; Kessler, H. Stereoisomeric Peptide Libraries and Peptidomimetics for Designing Selective Inhibitors of the αvβ3 Integrin for a New Cancer Therapy. Angew. Chem. Int. Ed. 1997, 36, 1374–1389. [Google Scholar] [CrossRef]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Aβ1–40 fibrillogenesis. J. Peptide Res. 2002, 60, 37–55. [Google Scholar] [CrossRef]
- Kessler, H. Detection of Hindered Rotation and Inversion by NMR Spectroscopy. Angew. Chem. Int. Ed. 1997, 9, 219–235. [Google Scholar] [CrossRef]
- Chatterjee, J.; Mierke, D.F.; Kessler, H. Conformational Preference and Potential Templates of N-Methylated Cyclic Pentaalanine Peptides. Chem. Eur. J. 2008, 14, 1508–1517. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef]
- Xu, W.; Liao, X.; Xu, S.; Diao, J.; Du, B.; Zhou, X.; Pan, S. Isolation, Structure Determination, and Synthesis of Galaxamide, A Rare Cytotoxic Cyclic Pentapeptide from a Marine Algae Galaxaura filamentosa. Org. Lett. 2008, 10, 4569–4572. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yu, S.; Zhong, S.; Zhao, B.; Qiu, S.; Chen, J.; Lunagariya, J.; Liao, X.; Xu, S. d-Amino Acid Position Influences the Anticancer Activity of Galaxamide Analogs: An Apoptotic Mechanism Study. Int. J. Mol. Sci. 2017, 18, 544. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liao, X.; Qiu, S.; Liu, Z.; Du, B.; Xu, S. Paper Synthesis, Cytotoxicity and Apoptosis Induction in Human Tumor Cells by Galaxamide and Its Analogues. Mar. Drugs 2014, 12, 4521–4538. [Google Scholar] [CrossRef]
- Li, D.; Liao, X.; Zhong, S.; Zhao, B.; Xu, S. Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents. Mar. Drugs 2022, 20, 158. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, T.; Lv, R.; Cao, Y.; Zheng, L.; Cheng, Y.; Yang, J.; Zhou, S. Ni-catalyzed enantio- stereoselective synthesis of chiral chromans with quaternary allylic siloxanes and their antitumor activity. ChemRxiv 2023. [Google Scholar] [CrossRef]
- He, W.; Qiu, H.; Chen, Y.; Xi, J.; Yao, Z. Total synthesis of proposed structure of coibamide A, a highly N- and O-methylated cytotoxic marine cyclodepsipeptide. Tetrahedron Lett. 2014, 55, 6109–6112. [Google Scholar] [CrossRef]
- Jia, X.; Wang, Y.; Wang, N. Nickel-Catalyzed sp3 C–H Bond Activation from Decarboxylative Cross Coupling of α,β-Unsaturated Carboxylic Acids with Amides. Synlett 2014, 25, 1621–1625. [Google Scholar]
- Xiong, X.; Zhang, H.; Underwood, C.R.; Harpsøe, K.; Gardella, T.J.; Wöldike, M.F.; Mannstadt, M.; Gloriam, D.E.; Bräuner-Osborne, H.; Strømgaard, K. Total synthesis and structure–activity relationship studies of a series of selective G protein inhibitors. Nat. Chem. 2016, 8, 1035–1041. [Google Scholar] [CrossRef]
- Nemoto, H.; Ma, R.; Suzuki, I.; Shibuya, M. A New One-Pot Method for the Synthesis of α-Siloxyamides from Aldehydes or Ketones and Its Application to the Synthesis of (−)-Bestatin. Org. Lett. 2000, 2, 4245–4247. [Google Scholar] [CrossRef]
- Han, F.; Liu, G.; Jin, C.; Wang, J.; Liu, J.; Wang, L.; Chen, Y. Total Synthesis and Determination of the Absolute Configuration of Rakicidin C. Org. Lett. 2021, 23, 7069–7073. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Barrows, R.D.; Emge, T.J.; Knapp, S. Stereochemical Aspects of T3P Amidations. Org. Process Res. Dev. 2017, 21, 399–407. [Google Scholar] [CrossRef]
- Reddy, D.N.; Ballante, F.; Chuang, T.; Pirolli, A.; Marrocco, B.; Marshall, G.R. Design and Synthesis of Simplified Largazole Analogues as Isoform-Selective Human Lysine Deacetylase Inhibitors. J. Med. Chem. 2016, 59, 1613–1633. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Ma, J.; Mao, Z.; Liu, Y.; Wei, B. Asymmetric synthesis of emericellamide B. Chin. Chem. Lett. 2015, 26, 1209–1215. [Google Scholar] [CrossRef]
- Ben, L.; Gaster, E.; Libman, A.; Pappo, D. Synthesis of Biaryl-Bridged Cyclic Peptides via Catalytic Oxidative Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2020, 59, 4835–4839. [Google Scholar]



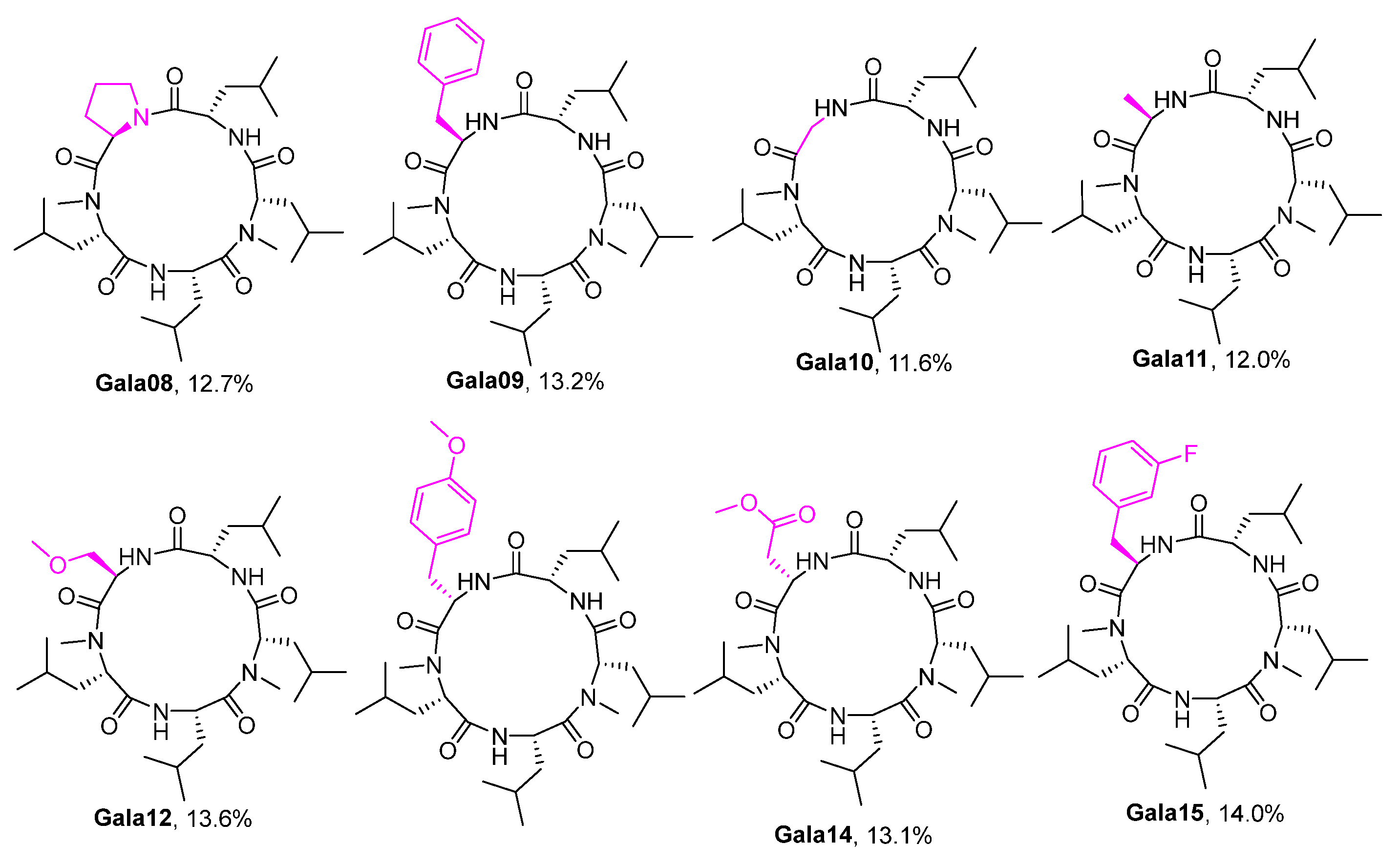
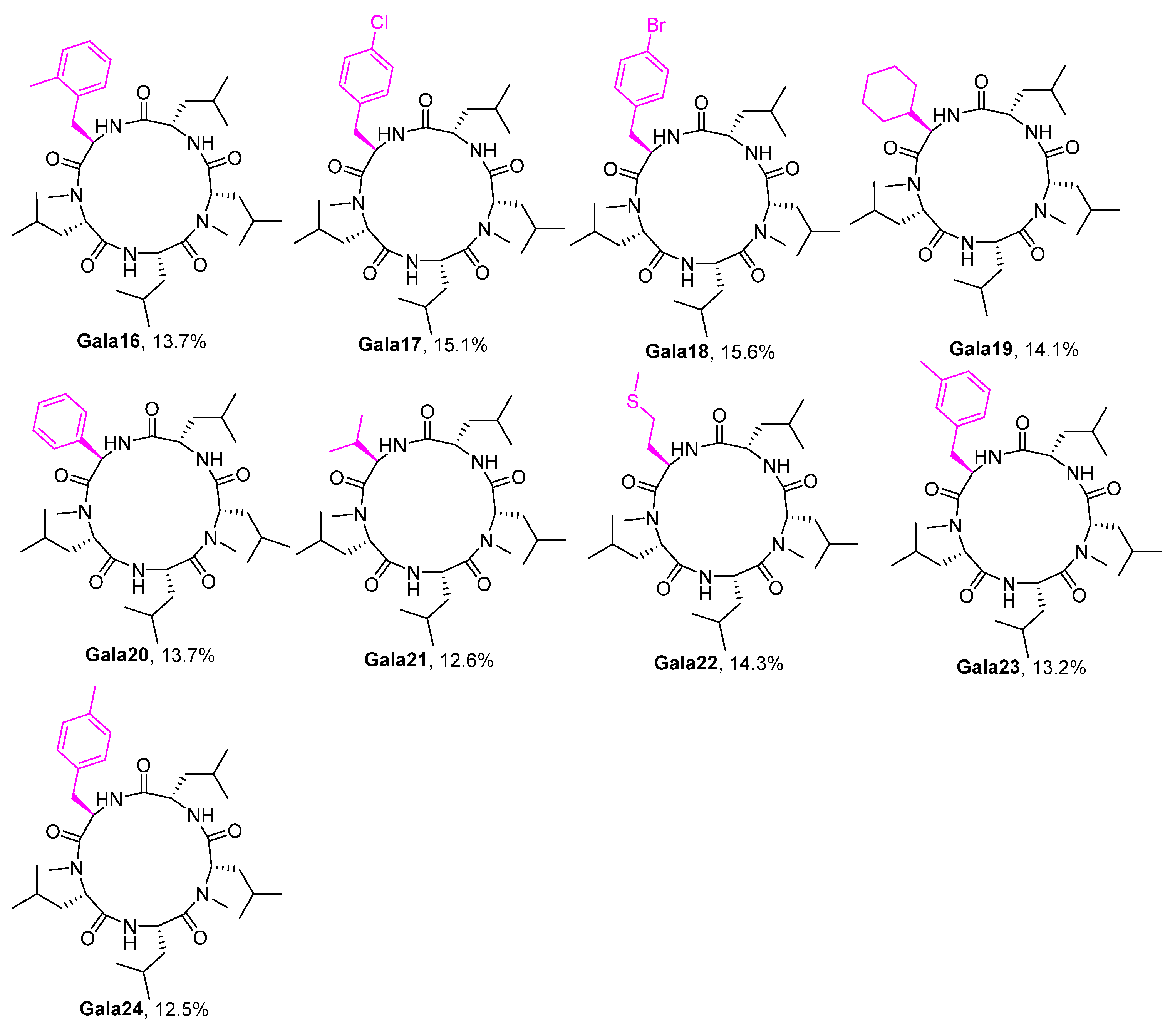

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Coupling Reagent | Solvent | Base | Yield |
| 1 | HATU | DMF | DIPEA | 28% |
| 2 | T3P | DMF | DIPEA | 33% |
| 3 | DPPA | DMF | DIPEA | 37% |
| 4 | FDPP | DMF | DIPEA | 41% |
| 5 | PyBOP | CH3CN | DMAP | 52% |
| Compounds | IC50 | ||
|---|---|---|---|
| A549 | K562 | 293T | |
| (−)-β-elemene | >100.0 | >100.0 | >100.0 |
| Gala01 | 14.0 | >80.0 | 19.9 |
| Gala02 | >20.0 | 8.6 | NA |
| Gala03 | >20.0 | >80.0 | NA |
| Gala04 | 19.0 | 4.2 | 16.3 |
| Gala05 | >20.0 | 10.2 | NA |
| Gala06 | >20.0 | 28.8 | NA |
| Gala07 | >20.0 | >80 | NA |
| Gala08 | 18.5 | 16.8 | 16.9 |
| Gala09 | >20.0 | 14.9 | NA |
| Gala10 | >20.0 | 27.2 | NA |
| Gala11 | 19.5 | 33.8 | 13.31 |
| Gala12 | 17.0 | 61.8 | 14.1 |
| Gala13 | >20.0 | >80.0 | NA |
| Gala14 | 20.0 | 29.6 | 22.2 |
| Gala15 | >20.0 | 13.0 | NA |
| Gala16 | >20.0 | >80.0 | NA |
| Gala17 | >20.0 | 77.7 | NA |
| Gala18 | 15.5 | 24.2 | 14.1 |
| Gala19 | >20.0 | 75.2 | NA |
| Gala20 | >20.0 | >80.0 | NA |
| Gala21 | >20.0 | 49.3 | NA |
| Gala22 | 19.8 | 8.5 | 12.9 |
| Gala23 | >20.0 | 6.9 | NA |
| Gala24 | >20.0 | 14.4 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Fan, H.; Luo, Z.; Yang, J.; Liu, G. Design, Synthesis, and Antitumor Biological Evaluation of Galaxamide and Its Analogs. Molecules 2025, 30, 2362. https://doi.org/10.3390/molecules30112362
Guo Y, Fan H, Luo Z, Yang J, Liu G. Design, Synthesis, and Antitumor Biological Evaluation of Galaxamide and Its Analogs. Molecules. 2025; 30(11):2362. https://doi.org/10.3390/molecules30112362
Chicago/Turabian StyleGuo, Yanyan, Huixia Fan, Zhiqiang Luo, Jian Yang, and Guodu Liu. 2025. "Design, Synthesis, and Antitumor Biological Evaluation of Galaxamide and Its Analogs" Molecules 30, no. 11: 2362. https://doi.org/10.3390/molecules30112362
APA StyleGuo, Y., Fan, H., Luo, Z., Yang, J., & Liu, G. (2025). Design, Synthesis, and Antitumor Biological Evaluation of Galaxamide and Its Analogs. Molecules, 30(11), 2362. https://doi.org/10.3390/molecules30112362