


CO2-Responsive Vinyl Polymers: From Synthesis to Application



Abstract
1. Introduction
2. CO2-Responsive Polymer Mechanisms
2.1. Acidic Functional Groups
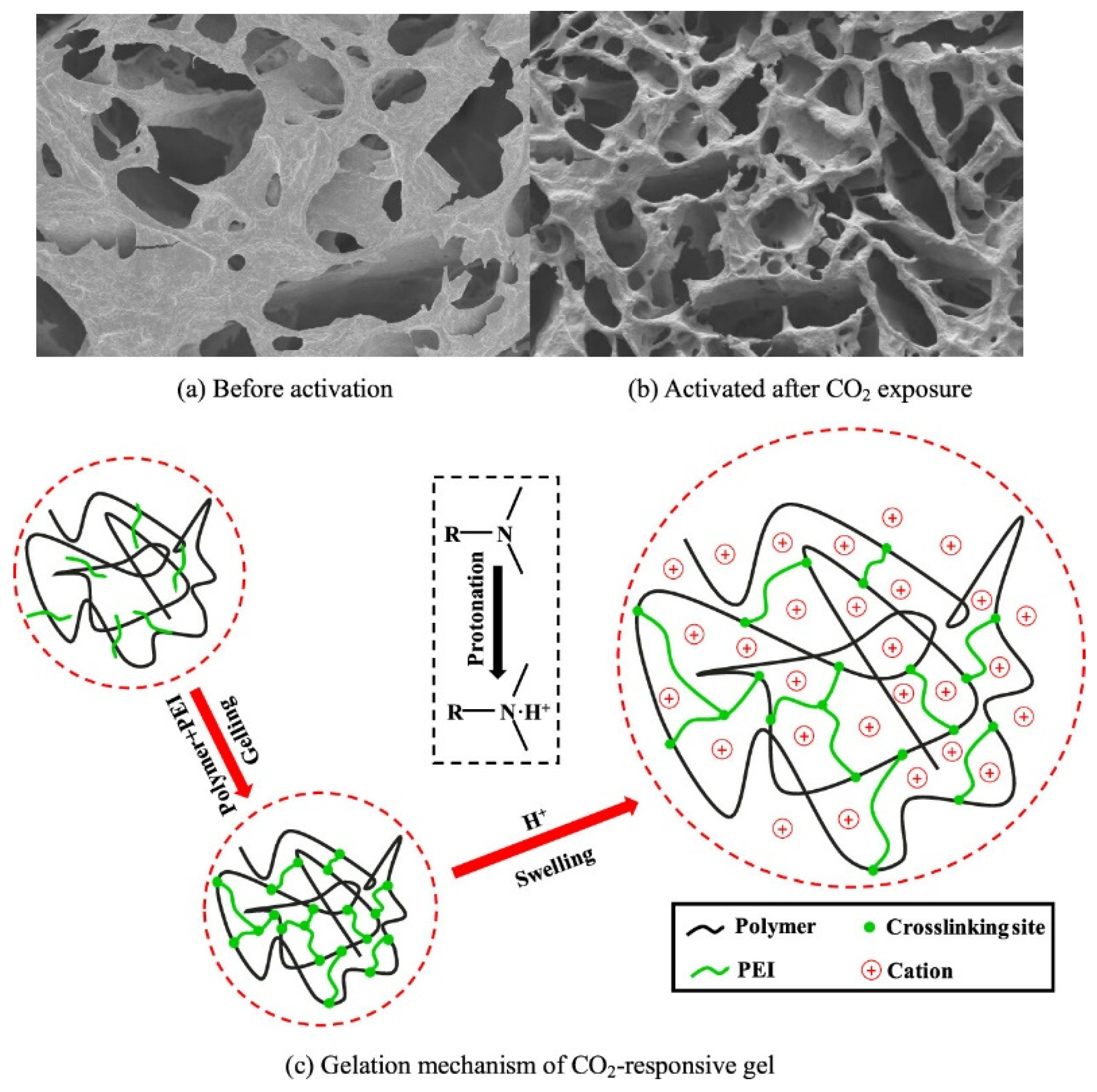
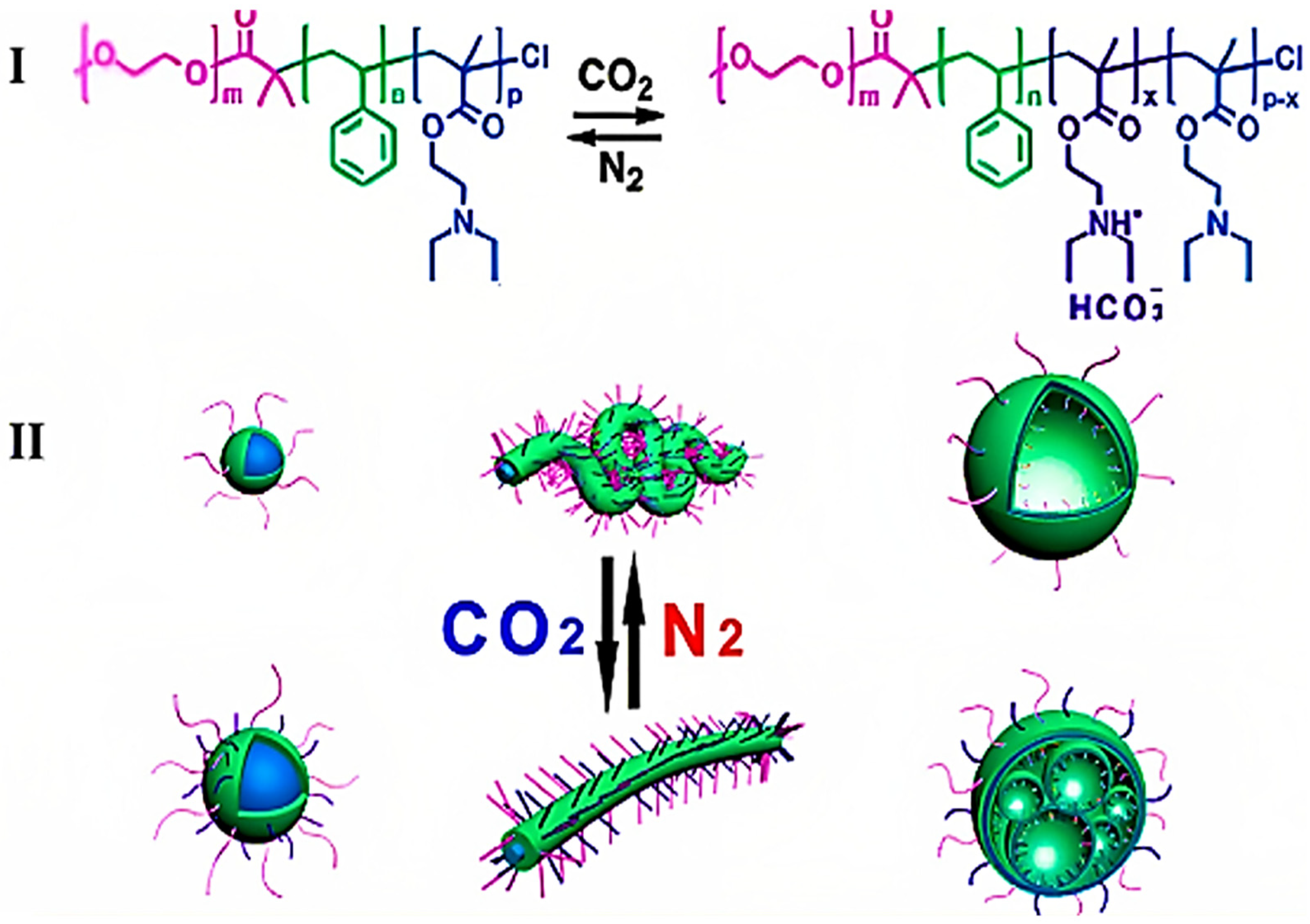
2.2. Basic Functional Groups
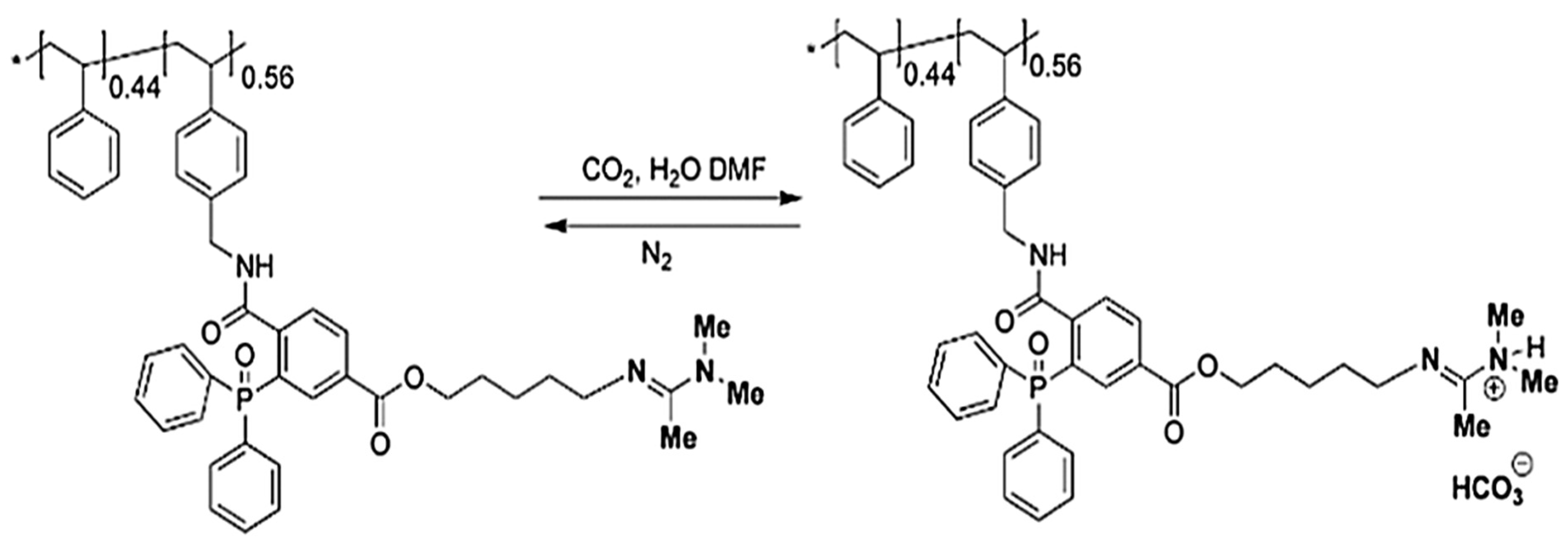
2.2.1. Amidines Groups
2.2.2. Imidazole Groups
2.2.3. Amino Groups
2.2.4. Guanidines Groups
3. Synthesis Methods of CO2-Responsive Polymers
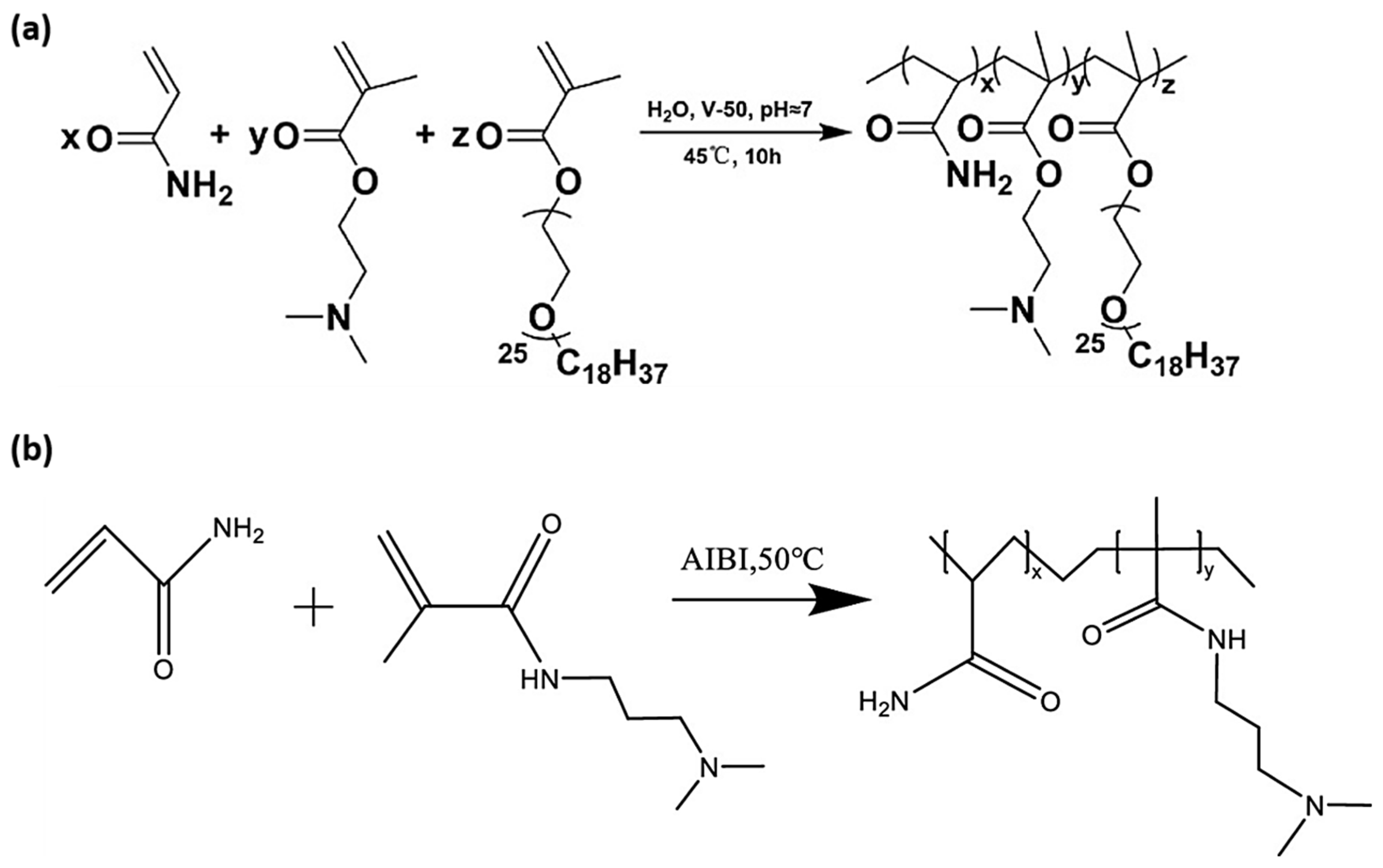
3.1. Free Radical Polymerization (FRP)
3.2. Controlled Radical Polymerization Techniques
3.2.1. Atomic Transfer Radical Polymerization (ATRP)
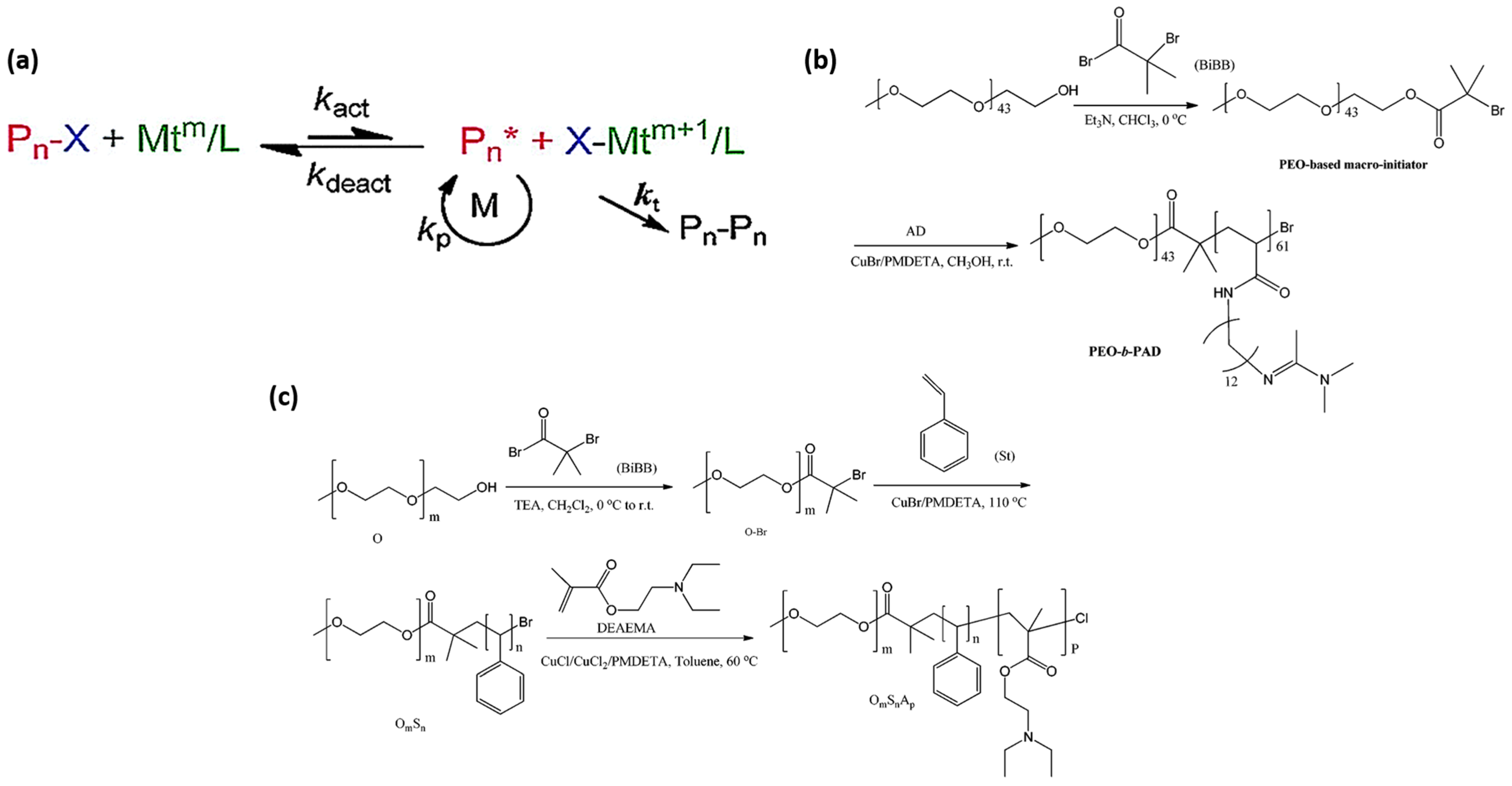
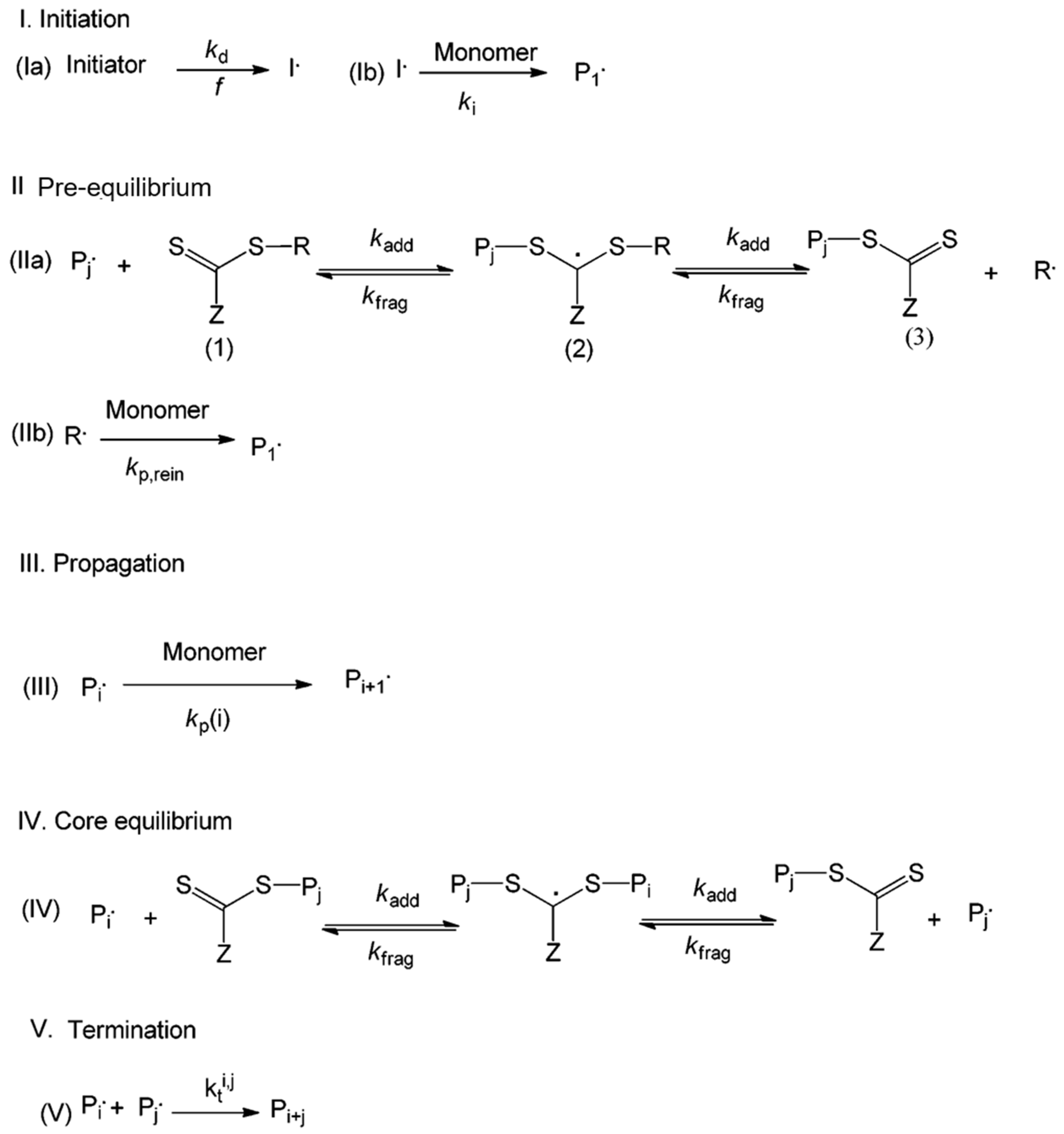
3.2.2. Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization
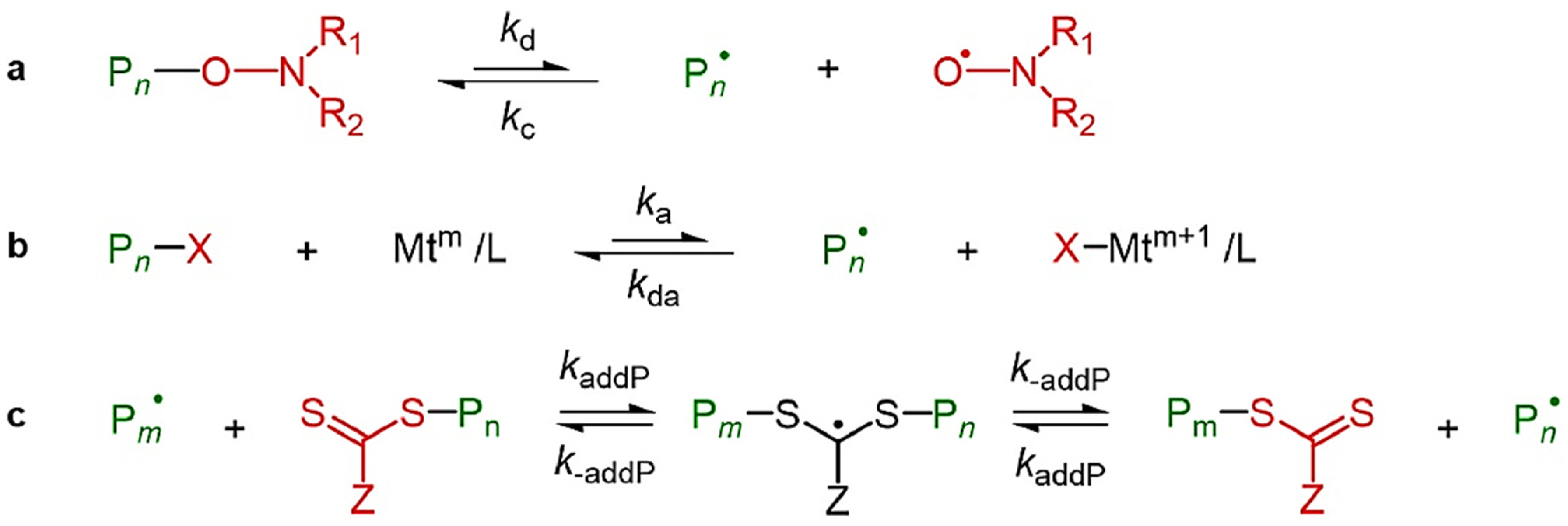
3.2.3. Nitroxide-Mediated Polymerization (NMP)
4. Applications of CO2-Responsive Polymers
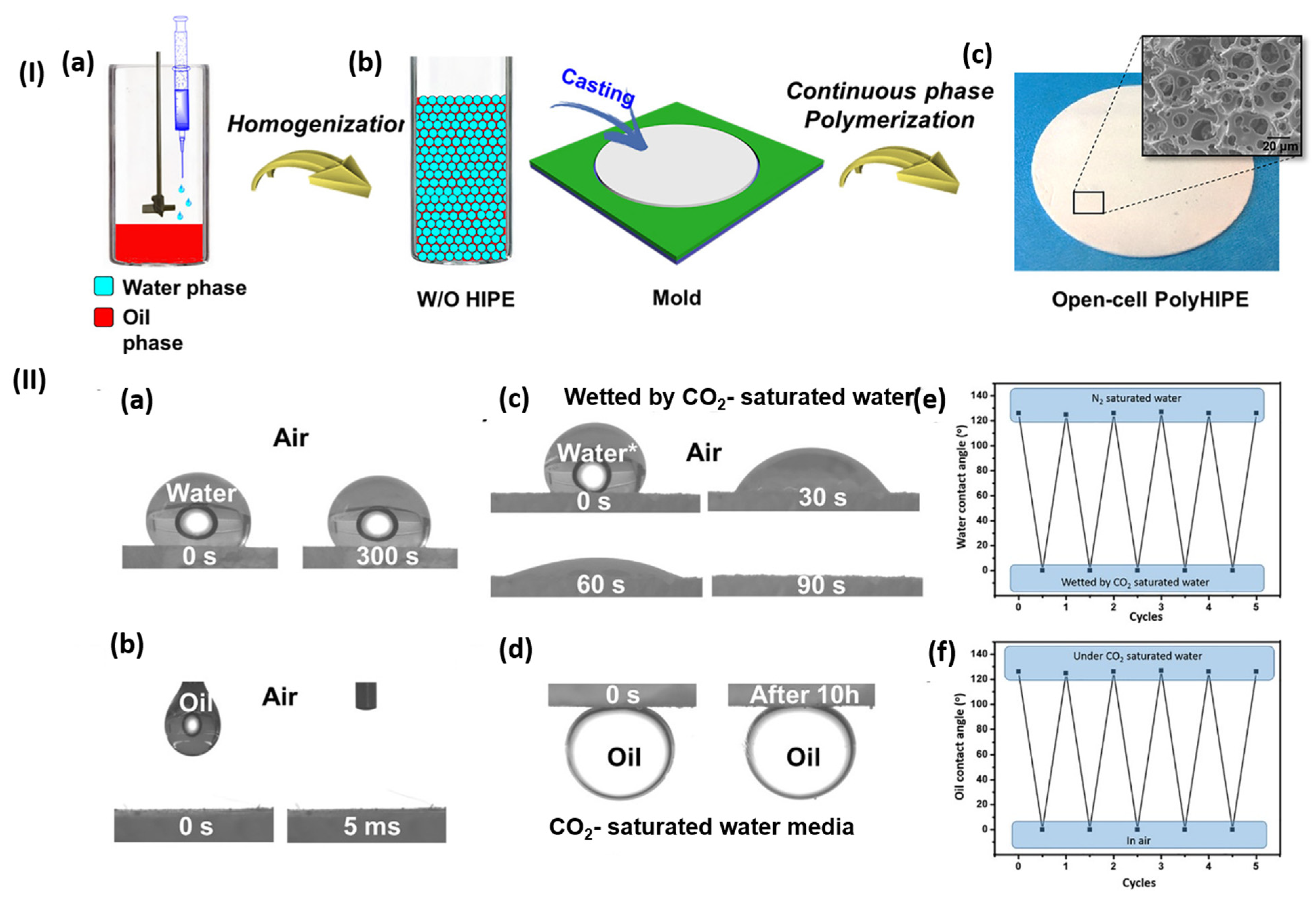
4.1. Oil-Water Separation
4.2. Carbon Capture and Storage
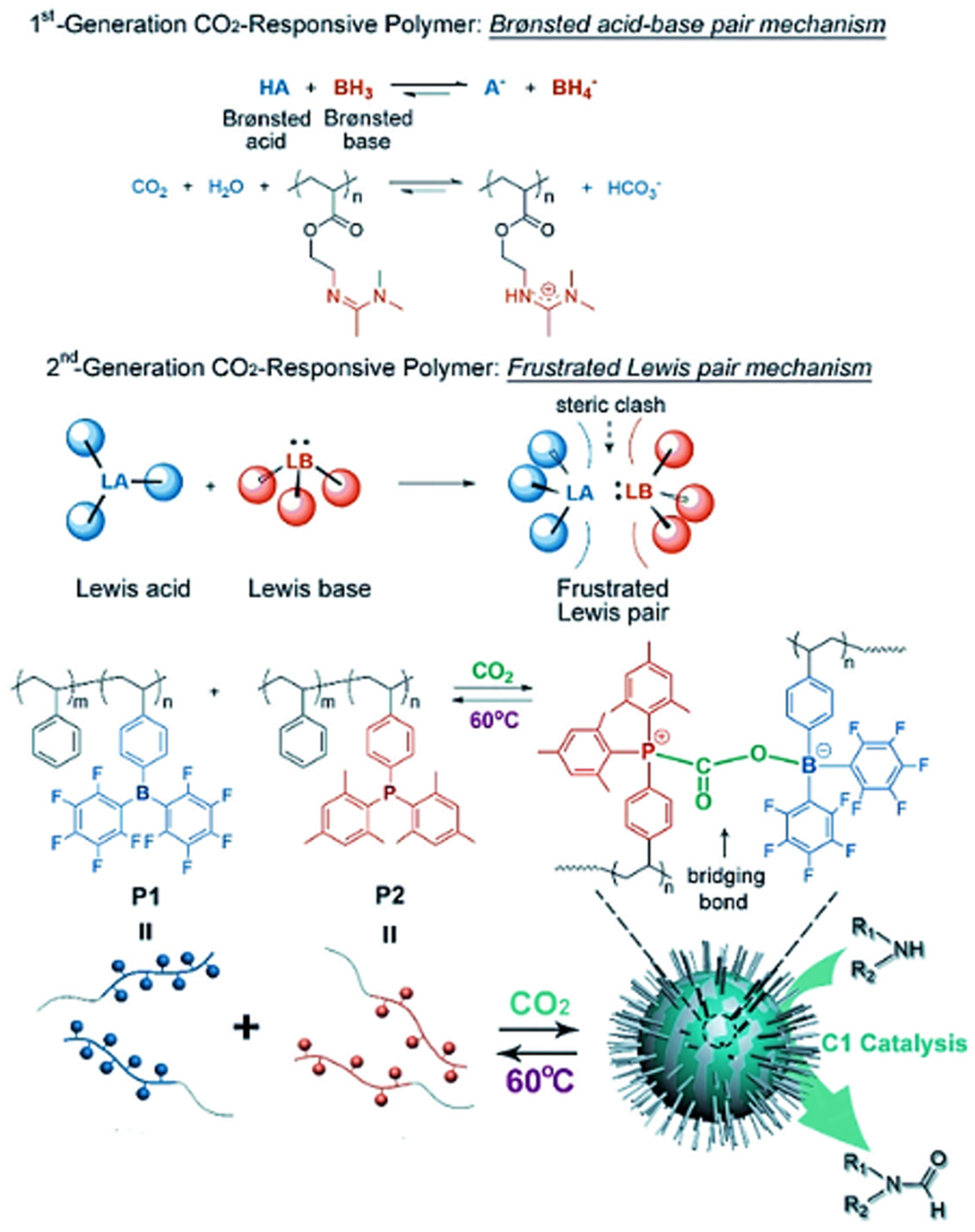
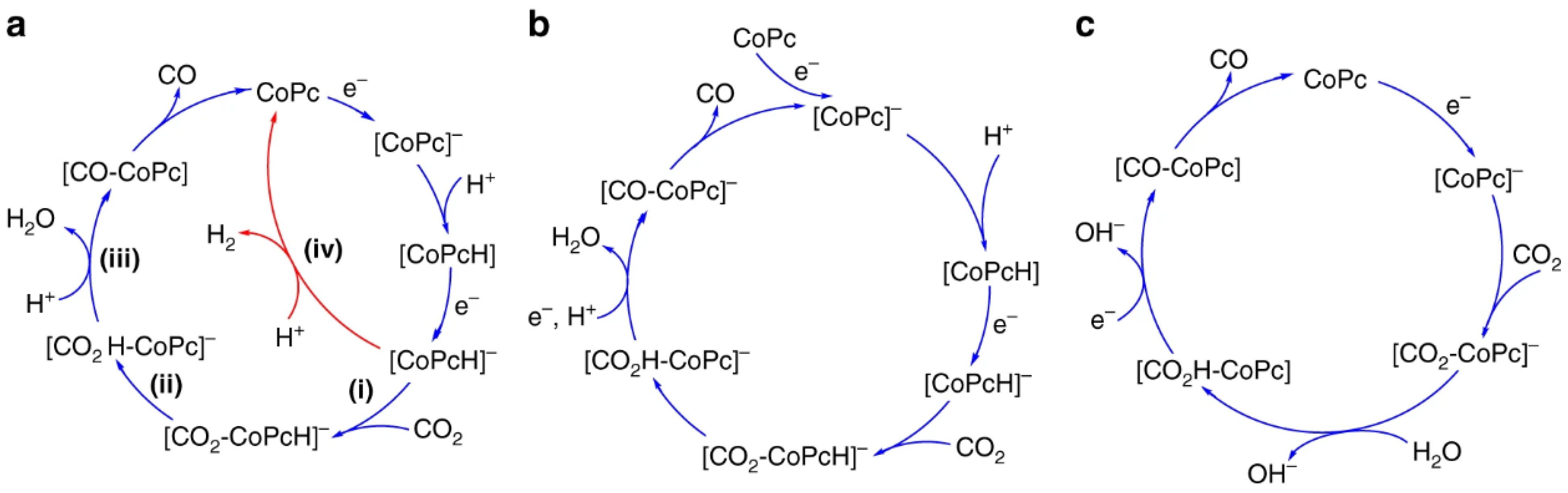
4.3. Polymer-Assisted CO2 Reduction
4.4. Drug Delivery System
4.5. Other Applications
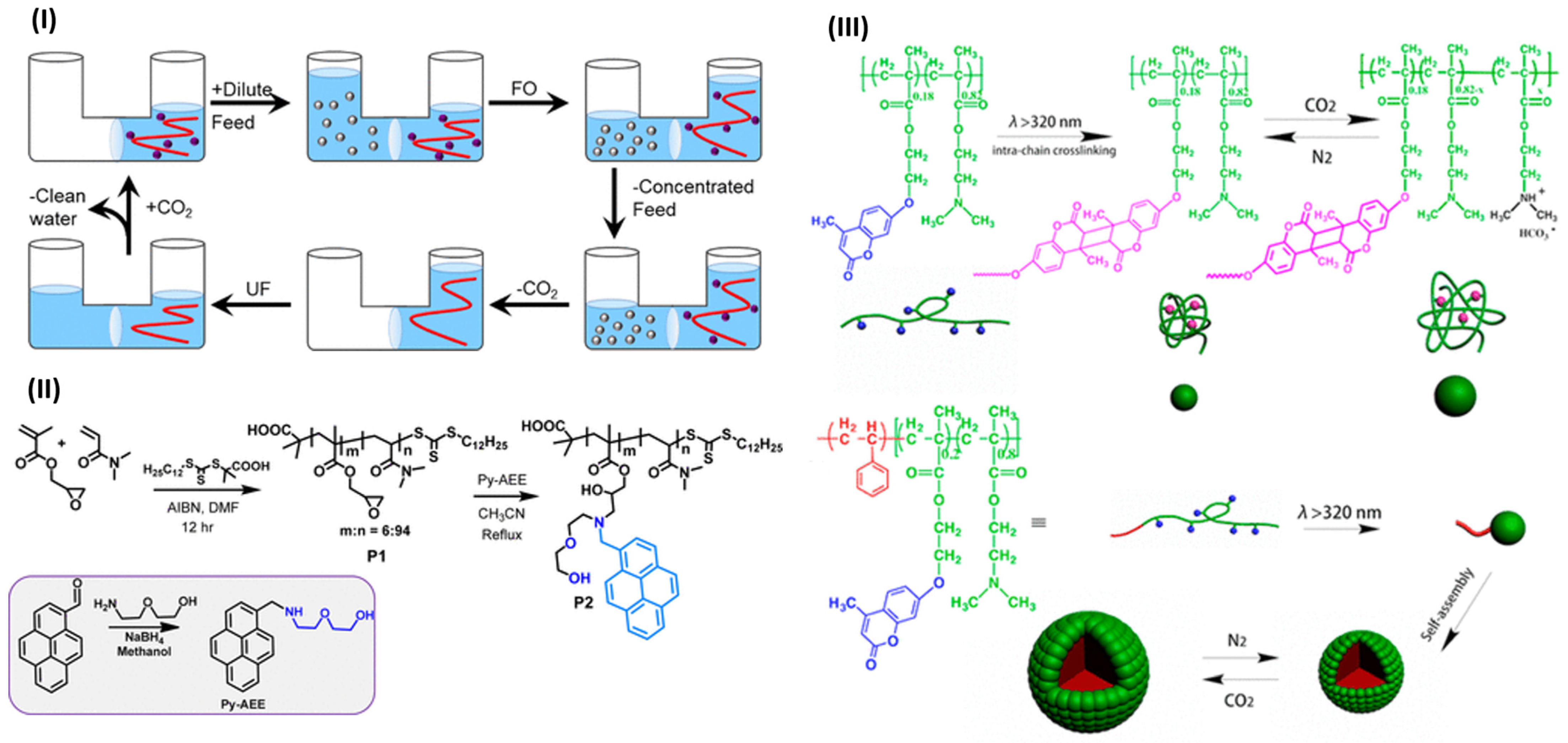
4.5.1. Forward Osmosis Desalination
4.5.2. Tissue Engineering
4.5.3. Smart Emulsions
4.5.4. CO2 Sensing
5. Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mrinalini, M.; Prasanthkumar, S. Recent Advances on Stimuli-Responsive Smart Materials and Their Applications. ChemPlusChem 2019, 84, 1103–1121. [Google Scholar] [CrossRef] [PubMed]
- Shafranek, R.T.; Millik, S.C.; Smith, P.T.; Lee, C.-U.; Boydston, A.J.; Nelson, A. Stimuli-Responsive Materials in Additive Manufacturing. Prog. Polym. Sci. 2019, 93, 36–67. [Google Scholar] [CrossRef]
- Theato, P.; Sumerlin, B.S.; O’Reilly, R.K.; Epps, T.H., III. Stimuli Responsive Materials. Chem. Soc. Rev. 2013, 42, 7055. [Google Scholar] [CrossRef] [PubMed]
- Moulin, E.; Faour, L.; Carmona-Vargas, C.C.; Giuseppone, N. From Molecular Machines to Stimuli-Responsive Materials. Adv. Mater. 2020, 32, 1906036. [Google Scholar] [CrossRef]
- Abdollahi, A.; Roghani-Mamaqani, H.; Razavi, B.; Salami-Kalajahi, M. The Light-Controlling of Temperature-Responsivity in Stimuli-Responsive Polymers. Polym. Chem. 2019, 10, 5686–5720. [Google Scholar] [CrossRef]
- Jochum, F.D.; Theato, P. Temperature- and Light-Responsive Smart Polymer Materials. Chem. Soc. Rev. 2013, 42, 7468–7483. [Google Scholar] [CrossRef]
- Ruan, C.; Zeng, K.; Grimes, C.A. A Mass-Sensitive PH Sensor Based on a Stimuli-Responsive Polymer. Anal. Chim. Acta 2003, 497, 123–131. [Google Scholar] [CrossRef]
- Guragain, S.; Bastakoti, B.P.; Malgras, V.; Nakashima, K.; Yamauchi, Y. Multi-Stimuli-Responsive Polymeric Materials. Chem.-A Eur. J. 2015, 21, 13164–13174. [Google Scholar] [CrossRef]
- Andrade, F.; Roca-Melendres, M.M.; Durán-Lara, E.F.; Rafael, D.; Schwartz, S. Stimuli-Responsive Hydrogels for Cancer Treatment: The Role of PH, Light, Ionic Strength and Magnetic Field. Cancers 2021, 13, 1164. [Google Scholar] [CrossRef]
- Zheng, R.; Wei, Y.; Zhang, Z.; Wang, Z.; Ma, L.; Wang, Y.; Huang, L.; Lu, Y. Stimuli-responsive Active Materials for Dynamic Control of Light Field. Responsive Mater. 2023, 1, 230017. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Future Perspectives and Recent Advances in Stimuli-Responsive Materials. Prog. Polym. Sci. 2010, 35, 278–301. [Google Scholar] [CrossRef]
- Hiratani, T.; Kose, O.; Hamad, W.Y.; MacLachlan, M.J. Stable and Sensitive Stimuli-Responsive Anisotropic Hydrogels for Sensing Ionic Strength and Pressure. Mater. Horiz. 2018, 5, 1076–1081. [Google Scholar] [CrossRef]
- Tan, B.H.; Tam, K.C.; Lam, Y.C.; Tan, C.B. Microstructure and Rheology of Stimuli-Responsive Nanocolloidal SystemsEffect of Ionic Strength. Langmuir 2004, 20, 11380–11386. [Google Scholar] [CrossRef]
- Qi, L.; Qiao, J. Design of Switchable Enzyme Carriers Based on Stimuli-Responsive Porous Polymer Membranes for Bioapplications. ACS Appl. Bio Mater. 2021, 4, 4706–4719. [Google Scholar] [CrossRef]
- Liu, Y.; Terrell, J.L.; Tsao, C.; Wu, H.; Javvaji, V.; Kim, E.; Cheng, Y.; Wang, Y.; Ulijn, R.V.; Raghavan, S.R.; et al. Biofabricating Multifunctional Soft Matter with Enzymes and Stimuli-Responsive Materials. Adv. Funct. Mater. 2012, 22, 3004–3012. [Google Scholar] [CrossRef]
- Wang, Y.; Shim, M.S.; Levinson, N.S.; Sung, H.; Xia, Y. Stimuli-Responsive Materials for Controlled Release of Theranostic Agents. Adv. Funct. Mater. 2014, 24, 4206–4220. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.-M.; Zhang, S.X.-A. Stimuli-Induced Reversible Proton Transfer for Stimuli-Responsive Materials and Devices. Acc. Chem. Res. 2021, 54, 2216–2226. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-Responsive Self-Healing Materials Formed from Host–Guest Polymers. Nat. Commun. 2011, 2, 511. [Google Scholar] [CrossRef]
- Medeiros, S.F.; Santos, A.M.; Fessi, H.; Elaissari, A. Stimuli-Responsive Magnetic Particles for Biomedical Applications. Int. J. Pharm. 2011, 403, 139–161. [Google Scholar] [CrossRef]
- Zhang, W.; Choi, H. Stimuli-Responsive Polymers and Colloids under Electric and Magnetic Fields. Polymers 2014, 6, 2803–2818. [Google Scholar] [CrossRef]
- Manouras, T.; Vamvakaki, M. Field Responsive Materials: Photo-, Electro-, Magnetic- and Ultrasound-Sensitive Polymers. Polym. Chem. 2017, 8, 74–96. [Google Scholar] [CrossRef]
- Lu, S.; Sun, Y.-H.; Shi, R.; Clark, C.; Li, L.; Chiang, V.L. Novel and Mechanical Stress–Responsive MicroRNAs in Populus Trichocarpa That Are Absent from Arabidopsis. Plant Cell 2005, 17, 2186–2203. [Google Scholar] [CrossRef] [PubMed]
- Dolui, T.; Natarajan, T.S.; Aiswarya, S.; Chanda, J.; Ghosh, P.; Mukhopadhyay, R.; Wießner, S.; Heinrich, G.; Das, A.; Banerjee, S.S. Stimuli–Responsive Mechanoadaptive Elastomeric Composite Materials: Challenges, Opportunities, and New Approaches. Adv. Eng. Mater. 2023, 25, 2300584. [Google Scholar] [CrossRef]
- Mano, J.F. Stimuli-Responsive Polymeric Systems for Biomedical Applications. Adv. Eng. Mater. 2008, 10, 515–527. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.; Tang, K.; Verpoort, F.; Sun, T. Polymer-Based Stimuli-Responsive Recyclable Catalytic Systems for Organic Synthesis. Small 2014, 10, 32–46. [Google Scholar] [CrossRef]
- Blelloch, N.D.; Yarbrough, H.J.; Mirica, K.A. Stimuli-Responsive Temporary Adhesives: Enabling Debonding on Demand through Strategic Molecular Design. Chem. Sci. 2021, 12, 15183–15205. [Google Scholar] [CrossRef]
- Jessop, P.G.; Mercer, S.M.; Heldebrant, D.J. CO2-Triggered Switchable Solvents, Surfactants, and Other Materials. Energy Environ. Sci. 2012, 5, 7240. [Google Scholar] [CrossRef]
- Lin, S.; Theato, P. CO2-Responsive Polymers. Macromol. Rapid Commun. 2013, 34, 1118–1133. [Google Scholar] [CrossRef]
- Guo, Z.; Feng, Y.; He, S.; Qu, M.; Chen, H.; Liu, H.; Wu, Y.; Wang, Y. CO2-Responsive “Smart” Single-Walled Carbon Nanotubes. Adv. Mater. 2013, 25, 584–590. [Google Scholar] [CrossRef]
- Liu, H.; Lin, S.; Feng, Y.; Theato, P. CO2-Responsive Polymer Materials. Polym. Chem. 2017, 8, 12–23. [Google Scholar] [CrossRef]
- Darabi, A.; Jessop, P.G.; Cunningham, M.F. CO2-Responsive Polymeric Materials: Synthesis, Self-Assembly, and Functional Applications. Chem. Soc. Rev. 2016, 45, 4391–4436. [Google Scholar] [CrossRef] [PubMed]
- Schattling, P.; Pollmann, I.; Theato, P. Synthesis of CO2-Responsive Polymers by Post-Polymerization Modification. React. Funct. Polym. 2014, 75, 16–21. [Google Scholar] [CrossRef]
- Deng, J.-N.; Zhao, H.; Zheng, H.; Zhuang, Y.; Wei, K.; Yuan, H.; Deng, Z.; Gao, Y.; Zhou, X.; Yu, T.; et al. CO2-Responsive Preformed Particle Gels with High Strength for CO2 Conformance Control in Heterogeneous Reservoirs. Fuel 2025, 379, 133040. [Google Scholar] [CrossRef]
- Zhang, R.; Lu, H.; Ji, S.; Lin, X.; Yang, Z.; Zhang, Z. CO2-Triggered Synergistic Self-Assembly of Cationic Polymer and Anionic Surfactant Systems for Enhanced Profile Control and Carbon Storage. Colloids Surf. A Physicochem. Eng. Asp. 2025, 713, 136538. [Google Scholar] [CrossRef]
- Hicks, J.C.; Drese, J.H.; Fauth, D.J.; Gray, M.L.; Qi, G.; Jones, C.W. Designing Adsorbents for CO2 Capture from Flue Gas-Hyperbranched Aminosilicas Capable of Capturing CO2 Reversibly. J. Am. Chem. Soc. 2008, 130, 2902–2903. [Google Scholar] [CrossRef]
- Rochelle, G.T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, T.; Wang, B.; Qiu, D.; Ma, N. Highly Sensitive CO2-Responsive Polymeric Microgels That Respond Within Seconds. Langmuir 2015, 31, 8138–8145. [Google Scholar] [CrossRef]
- Darmayanti, M.G.; Tuck, K.L.; Thang, S.H. Carbon Dioxide Capture by Emerging Innovative Polymers: Status and Perspectives. Adv. Mater. 2024, 36, 2403324. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-N.; Ilhami, F.B.; Cheng, C.-C. CO2-Responsive Water-Soluble Conjugated Polymers as a Multifunctional Fluorescent Probe for Bioimaging Applications. Biomacromolecules 2024, 25, 997–1008. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Lai, Y.-C.; Shieh, Y.-T.; Chang, Y.-H.; Lee, A.-W.; Chen, J.-K.; Lee, D.-J.; Lai, J.-Y. CO2-Responsive Water-Soluble Conjugated Polymers for In Vitro and In Vivo Biological Imaging. Biomacromolecules 2020, 21, 5282–5291. [Google Scholar] [CrossRef]
- Du, N.; Park, H.B.; Robertson, G.P.; Dal-Cin, M.M.; Visser, T.; Scoles, L.; Guiver, M.D. Polymer Nanosieve Membranes for CO2-Capture Applications. Nat. Mater. 2011, 10, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Zhang, J.; Chen, Z.; Zhu, B.; Li, J.; Liang, S.; Bai, Y.; Xu, J.; Rao, D.; et al. Scalable and Switchable CO2-Responsive Membranes with High Wettability for Separation of Various Oil/Water Systems. Nat. Commun. 2023, 14, 1108. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Liu, Y.; Gao, Q.; Tao, D.; Wang, Y.; Guo, J.; Yu, Y. CO2-Responsive Nanofibrous Membranes with Gas-Tunable Wettability for Switchable Oil/Water Separation. React. Funct. Polym. 2023, 182, 105481. [Google Scholar] [CrossRef]
- Lin, S.; Schattling, P.; Theato, P. Thermo- and CO2-Responsive Linear Polymers and Hydrogels as CO2 Capturing Materials. Sci. Adv. Mater. 2015, 7, 948–955. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, Y.; Wen, X.; Liu, Y.; Feng, M.; Rui, Z. Development and Applications of CO2-Responsive Gels in CO2 Flooding and Geological Storage. Gels 2023, 9, 936. [Google Scholar] [CrossRef]
- Frauholz, J.; Cunningham, M.F.; Jessop, P.G. Thermo- and CO2-Responsive Copolymers for Forward Osmosis Applications. Ind. Eng. Chem. Res. 2024, 63, 10713–10720. [Google Scholar] [CrossRef]
- Davies, O.R.; Lewis, A.L.; Whitaker, M.J.; Tai, H.; Shakesheff, K.M.; Howdle, S.M. Applications of Supercritical CO2 in the Fabrication of Polymer Systems for Drug Delivery and Tissue Engineering. Adv. Drug Deliv. Rev. 2008, 60, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ke, Y.; Hu, X.; Peng, F.; Yu, C.; Xing, L. Synthesis and Switchable Behavior of a CO2 Responsive Polymeric Surfactant Acting as Emulsifier. J. Dispers. Sci. Technol. 2021, 42, 407–415. [Google Scholar] [CrossRef]
- Ma, Y.; Promthaveepong, K.; Li, N. CO2-Responsive Polymer-Functionalized Au Nanoparticles for CO2 Sensor. Anal. Chem. 2016, 88, 8289–8293. [Google Scholar] [CrossRef]
- Zhang, Q.; Lei, L.; Zhu, S. Gas-Responsive Polymers. ACS Macro Lett. 2017, 6, 515–522. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, H.; Bai, Y.; Mei, K.; Zheng, Y.; Zhang, C.; Cheng, X. Study on the Effect of CO2-Responsive Hydrogel Prepared by Free Radical Polymerization on the Self-Healing of Cement Paste Fractures: Application in CCUS Cementing. Constr. Build. Mater. 2024, 435, 136651. [Google Scholar] [CrossRef]
- Abousalman-Rezvani, Z.; Eskandari, P.; Roghani-Mamaqani, H.; Mardani, H.; Salami-Kalajahi, M. Grafting Light-, Temperature, and CO2-Responsive Copolymers from Cellulose Nanocrystals by Atom Transfer Radical Polymerization for Adsorption of Nitrate Ions. Polymer 2019, 182, 121830. [Google Scholar] [CrossRef]
- Garcia-Valdez, O.; Brescacin, T.; Arredondo, J.; Bouchard, J.; Jessop, P.G.; Champagne, P.; Cunningham, M.F. Grafting CO2-Responsive Polymers from Cellulose Nanocrystals via Nitroxide-Mediated Polymerisation. Polym. Chem. 2017, 8, 4124–4131. [Google Scholar] [CrossRef]
- Quek, J.Y.; Roth, P.J.; Evans, R.A.; Davis, T.P.; Lowe, A.B. Reversible Addition–Fragmentation Chain Transfer Synthesis of Amidine-based, CO2-responsive Homo and AB Diblock (Co)Polymers Comprised of Histamine and Their Gas-triggered Self-assembly in Water. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 394–404. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, Y.; Huang, X.; Kang, T.; Severtson, S.J.; Wang, W.-J.; Liu, P. Tailoring CO2-Responsive Polymers and Nanohybrids for Green Chemistry and Processes. Ind. Eng. Chem. Res. 2019, 58, 15088–15108. [Google Scholar] [CrossRef]
- Lefevre, E.R.M.A. Method for Shear Coagulation of Latex Resins. U.S. Patent No. 4,623,678, 18 November 1986. [Google Scholar]
- Han, D.; Boissiere, O.; Kumar, S.; Tong, X.; Tremblay, L.; Zhao, Y. Two-Way CO2-Switchable Triblock Copolymer Hydrogels. Macromolecules 2012, 45, 7440–7445. [Google Scholar] [CrossRef]
- Fischer, V.; Landfester, K.; Muñoz-Espí, R. Molecularly Controlled Coagulation of Carboxyl-Functionalized Nanoparticles Prepared by Surfactant-Free Miniemulsion Polymerization. ACS Macro Lett. 2012, 1, 1371–1374. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, H.; Feng, Y. CO2-Responsive Anionic Wormlike Micelles Based on Natural Erucic Acid. Green Mater. 2014, 2, 95–103. [Google Scholar] [CrossRef]
- Shieh, Y.-T.; Yeh, Y.-C.; Cheng, C.-C. Two-Way CO2-Responsive Polymer Particles with Controllable Amphiphilic Properties. ACS Omega 2020, 5, 1862–1869. [Google Scholar] [CrossRef]
- Liu, P.; Lu, W.; Wang, W.-J.; Li, B.-G.; Zhu, S. Highly CO2 /N2-Switchable Zwitterionic Surfactant for Pickering Emulsions at Ambient Temperature. Langmuir 2014, 30, 10248–10255. [Google Scholar] [CrossRef]
- Cunningham, M.F.; Jessop, P.G. An Introduction to the Principles and Fundamentals of CO2-Switchable Polymers and Polymer Colloids. Eur. Polym. J. 2016, 76, 208–215. [Google Scholar] [CrossRef]
- Liu, Y.; Jessop, P.G.; Cunningham, M.; Eckert, C.A.; Liotta, C.L. Switchable Surfactants. Science 2006, 313, 958–960. [Google Scholar] [CrossRef]
- Scott, L.M.; Robert, T.; Harjani, J.R.; Jessop, P.G. Designing the Head Group of CO2-Triggered Switchable Surfactants. RSC Adv. 2012, 2, 4925. [Google Scholar] [CrossRef]
- Harjani, J.R.; Liang, C.; Jessop, P.G. A Synthesis of Acetamidines. J. Org. Chem. 2011, 76, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.Y.; Davis, T.P.; Lowe, A.B. Amidine Functionality as a Stimulus-Responsive Building Block. Chem. Soc. Rev. 2013, 42, 7326. [Google Scholar] [CrossRef]
- Chen, S.; Lin, X.; Zhai, Z.; Lan, R.; Li, J.; Wang, Y.; Zhou, S.; Farooqi, Z.H.; Wu, W. Synthesis and Characterization of CO2-Sensitive Temperature-Responsive Catalytic Poly(Ionic Liquid) Microgels. Polym. Chem. 2018, 9, 2887–2896. [Google Scholar] [CrossRef]
- Fowler, C.I.; Jessop, P.G.; Cunningham, M.F. Aryl Amidine and Tertiary Amine Switchable Surfactants and Their Application in the Emulsion Polymerization of Methyl Methacrylate. Macromolecules 2012, 45, 2955–2962. [Google Scholar] [CrossRef]
- Barzagli, F.; Mani, F.; Peruzzini, M. A 13C NMR Study of the Carbon Dioxide Absorption and Desorption Equilibria by Aqueous 2-Aminoethanol and N-Methyl-Substituted 2-Aminoethanol. Energy Environ. Sci. 2009, 2, 322. [Google Scholar] [CrossRef]
- Mercer, S.M.; Jessop, P.G. “Switchable Water”: Aqueous Solutions of Switchable Ionic Strength. ChemSusChem 2010, 3, 467–470. [Google Scholar] [CrossRef]
- Darabi, A.; Shirin-Abadi, A.R.; Pinaud, J.; Jessop, P.G.; Cunningham, M.F. Nitroxide-Mediated Surfactant-Free Emulsion Copolymerization of Methyl Methacrylate and Styrene Using Poly(2-(Diethyl)Aminoethyl Methacrylate-Co-Styrene) as a Stimuli-Responsive Macroalkoxyamine. Polym. Chem. 2014, 5, 6163–6170. [Google Scholar] [CrossRef]
- Adekomi, A.A. Enhanced CO2 Capture and Desorption Using Surface-Active Tertiary Amine. Doctoral Dissertation, The University of Texas at Austin, Austin, TX, USA, 2024. [Google Scholar]
- Phan, L.; Chiu, D.; Heldebrant, D.J.; Huttenhower, H.; John, E.; Li, X.; Pollet, P.; Wang, R.; Eckert, C.A.; Liotta, C.L.; et al. Switchable Solvents Consisting of Amidine/Alcohol or Guanidine/Alcohol Mixtures. Ind. Eng. Chem. Res. 2008, 47, 539–545. [Google Scholar] [CrossRef]
- Pashayev, E.; Georgopanos, P. CO2-Responsive Copolymers for Membrane Applications, Synthesis, and Performance Evaluation. Macromol. Mater. Eng. 2025, 310, 2400290. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, M.; Su, X.; Feng, Y. CO2-Responsive Polymer Promoted by Polyether to Efficient Viscosity Increase for CO2 Plugging. Polymer 2024, 306, 127227. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, W.-J.; Lu, Y.; Li, B.-G.; Zhu, S. Reversibly Coagulatable and Redispersible Polystyrene Latex Prepared by Emulsion Polymerization of Styrene Containing Switchable Amidine. Macromolecules 2011, 44, 6539–6545. [Google Scholar] [CrossRef]
- Asua, J.M. (Ed.) Polymer Reaction Engineering; Wiley: Hoboken, NJ, USA, 2007; ISBN 9781405144421. [Google Scholar]
- Fowler, C.I.; Muchemu, C.M.; Miller, R.E.; Phan, L.; O’Neill, C.; Jessop, P.G.; Cunningham, M.F. Emulsion Polymerization of Styrene and Methyl Methacrylate Using Cationic Switchable Surfactants. Macromolecules 2011, 44, 2501–2509. [Google Scholar] [CrossRef]
- Mihara, M.; Jessop, P.; Cunningham, M. Redispersible Polymer Colloids Using Carbon Dioxide as an External Trigger. Macromolecules 2011, 44, 3688–3693. [Google Scholar] [CrossRef]
- Che, N.; Yang, S.; Kang, H.; Liu, R.; Li, Z.; Liu, Z.; Li, P.; Qu, X.; Huang, Y. Synthesis and Properties of CO2-Switchable Dex-g-PAHMA Copolymers. Polym. Chem. 2014, 5, 7109–7120. [Google Scholar] [CrossRef]
- Morse, A.J.; Armes, S.P.; Thompson, K.L.; Dupin, D.; Fielding, L.A.; Mills, P.; Swart, R. Novel Pickering Emulsifiers Based on PH-Responsive Poly(2-(Diethylamino)Ethyl Methacrylate) Latexes. Langmuir 2013, 29, 5466–5475. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lou, Y.; Zhai, X.; Li, Z.; Liu, B. Development and Characterization of CO2 –Responsive Intelligent Polymer Sealant. ACS Omega 2023, 8, 35066–35076. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, Y.; Dai, X.; Xu, Q.; Zhang, L.; Tan, J. Open-Air Preparation of Cross-Linked CO2-Responsive Polymer Vesicles by Enzyme-Assisted Photoinitiated Polymerization-Induced Self-Assembly. Chem. Commun. 2019, 55, 11920–11923. [Google Scholar] [CrossRef]
- Ellis, S.N.; Riabtseva, A.; Dykeman, R.R.; Hargreaves, S.; Robert, T.; Champagne, P.; Cunningham, M.F.; Jessop, P.G. Nitrogen Rich CO2-Responsive Polymers as Forward Osmosis Draw Solutes. Ind. Eng. Chem. Res. 2019, 58, 22579–22586. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H. A Pyrene Derived CO2-Responsive Polymeric Probe for the Turn-On Fluorescent Detection of Nerve Agent Mimics with Tunable Sensitivity. Macromolecules 2017, 50, 6888–6895. [Google Scholar] [CrossRef]
- Fan, W.; Tong, X.; Farnia, F.; Yu, B.; Zhao, Y. CO2-Responsive Polymer Single-Chain Nanoparticles and Self-Assembly for Gas-Tunable Nanoreactors. Chem. Mater. 2017, 29, 5693–5701. [Google Scholar] [CrossRef]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications. Prog. Polym. Sci. 2020, 111, 101311. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- SZWARC, M. ‘Living’ Polymers. Nature 1956, 178, 1168–1169. [Google Scholar] [CrossRef]
- Cunningham, M.F. Controlled/Living Radical Polymerization in Aqueous Dispersed Systems. Prog. Polym. Sci. 2008, 33, 365–398. [Google Scholar] [CrossRef]
- Zetterlund, P.B.; Kagawa, Y.; Okubo, M. Controlled/Living Radical Polymerization in Dispersed Systems. Chem. Rev. 2008, 108, 3747–3794. [Google Scholar] [CrossRef] [PubMed]
- Asua, J.M. Miniemulsion Polymerization. Prog. Polym. Sci. 2002, 27, 1283–1346. [Google Scholar] [CrossRef]
- Qiu, J.; Charleux, B.; Matyjaszewski, K. Controlled/Living Radical Polymerization in Aqueous Media: Homogeneous and Heterogeneous Systems. Prog. Polym. Sci. 2001, 26, 2083–2134. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Yan, Q.; Zhou, R.; Fu, C.; Zhang, H.; Yin, Y.; Yuan, J. CO2-Responsive Polymeric Vesicles That Breathe. Angew. Chem. Int. Ed. 2011, 50, 4923–4927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qian, J.; Fan, Y.; Feng, W.; Tao, Z.; Yang, H. A Facile CO2 Switchable Nanocomposite with Reversible Transition from Sol to Self-Healable Hydrogel. RSC Adv. 2015, 5, 62229–62234. [Google Scholar] [CrossRef]
- Yan, Q.; Zhao, Y. CO2-Stimulated Diversiform Deformations of Polymer Assemblies. J. Am. Chem. Soc. 2013, 135, 16300–16303. [Google Scholar] [CrossRef]
- Xu, L.Q.; Zhang, B.; Sun, M.; Hong, L.; Neoh, K.-G.; Kang, E.-T.; Fu, G.D. CO2-Triggered Fluorescence “Turn-on” Response of Perylene Diimide-Containing Poly(N,N-Dimethylaminoethyl Methacrylate). J. Mater. Chem. A 2013, 1, 1207–1212. [Google Scholar] [CrossRef]
- Su, X.; Jessop, P.G.; Cunningham, M.F. Versatility of Organocatalyzed Atom Transfer Radical Polymerization and CO2-Switching for Preparing Both Hydrophobic and Hydrophilic Polymers with the Recycling of a Photocatalyst. Macromolecules 2019, 52, 6725–6733. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. (Ed.) Handbook of RAFT Polymerization; Wiley: Hoboken, NJ, USA, 2008; ISBN 9783527319244. [Google Scholar]
- Lin, S.; Das, A.; Theato, P. CO2-Responsive Graft Copolymers: Synthesis and Characterization. Polym. Chem. 2017, 8, 1206–1216. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, T.; Wu, Y.; Shi, W.; Choi, B.; Feng, A.; Thang, S.H. Synthesis of CO2-Responsive Gradient Copolymers by Switchable RAFT Polymerization and Their Controlled Self-Assembly. Polym. Chem. 2020, 11, 6794–6802. [Google Scholar] [CrossRef]
- Chen, L.; Liu, R.; Yan, Q. Polymer Meets Frustrated Lewis Pair: Second-Generation CO2-Responsive Nanosystem for Sustainable CO2 Conversion. Angew. Chem. 2018, 130, 9480–9484. [Google Scholar] [CrossRef]
- Abel, B.A.; Sims, M.B.; McCormick, C.L. Tunable PH- and CO2-Responsive Sulfonamide-Containing Polymers by RAFT Polymerization. Macromolecules 2015, 48, 5487–5495. [Google Scholar] [CrossRef]
- Feng, A.; Zhan, C.; Yan, Q.; Liu, B.; Yuan, J. A CO2—And Temperature-Switchable “Schizophrenic” Block Copolymer: From Vesicles to Micelles. Chem. Commun. 2014, 50, 8958. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Feng, Y.; Wang, Y.; Wang, J.; Wu, Y.; Zhang, Y. A Novel Smart Polymer Responsive to CO2. Chem. Commun. 2011, 47, 9348. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, Z.; He, S.; Yin, H.; Fei, C.; Feng, Y. CO2-Driven Vesicle to Micelle Regulation of Amphiphilic Copolymer: Random versus Block Strategy. Polym. Chem. 2014, 5, 4756–4763. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Y.; Dreiss, C.A.; Feng, Y. CO2-Switchable Multi-Compartment Micelles with Segregated Corona. Soft Matter 2014, 10, 6387–6391. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, G.; Wang, W.-J.; Yuan, H.; Li, B.-G.; Zhu, S. Switchable Block Copolymer Surfactants for Preparation of Reversibly Coagulatable and Redispersible Poly(Methyl Methacrylate) Latexes. Macromolecules 2013, 46, 1261–1267. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Zhou, S.; Zhang, H.; Feng, A.-C.; Jian, C.; Hu, J.; Gao, W.; Yuan, J. Synthesis and Self-Assembly of CO2 –Temperature Dual Stimuli-Responsive Triblock Copolymers. Macromolecules 2014, 47, 2938–2946. [Google Scholar] [CrossRef]
- Hu, J.; Whittaker, M.R.; Li, Y.; Quinn, J.F.; Davis, T.P. The Use of Endogenous Gaseous Molecules (NO and CO2) to Regulate the Self-Assembly of a Dual-Responsive Triblock Copolymer. Polym. Chem. 2015, 6, 2407–2415. [Google Scholar] [CrossRef]
- Yuan, W.; Shen, J.; Zou, H. Amphiphilic Block Copolymer Terminated with Pyrene Group: From Switchable CO2-Temperature Dual Responses to Tunable Fluorescence. RSC Adv. 2015, 5, 13145–13152. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, G.; Wei, Z.; Li, X.; Tang, B. Preparation and Drug Release Property of CO2 Stimulus-Sensitive Poly(N, N-Dimethylaminoethyl Methacrylate)-b-Polystyrene Nanoparticles. Eur. Polym. J. 2013, 49, 3165–3170. [Google Scholar] [CrossRef]
- Wang, T.-T.; Wu, Y.-Y.; Luo, Z.-H.; Zhou, Y.-N. “Living” Polymer Dispersity Quantification for Nitroxide-Mediated Polymerization Systems by Mimicking a Monodispersed Polymer Blending Strategy. Macromolecules 2020, 53, 10813–10822. [Google Scholar] [CrossRef]
- Solomon, D.H. Genesis of the CSIRO Polymer Group and the Discovery and Significance of Nitroxide-mediated Living Radical Polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5748–5764. [Google Scholar] [CrossRef]
- Georges, M.K.; Veregin, R.P.N.; Kazmaier, P.M.; Hamer, G.K. Narrow Molecular Weight Resins by a Free-Radical Polymerization Process. Macromolecules 1993, 26, 2987–2988. [Google Scholar] [CrossRef]
- Bertin, D.; Gigmes, D.; Marque, S.R.A.; Tordo, P. Kinetic Subtleties of Nitroxide Mediated Polymerization. Chem. Soc. Rev. 2011, 40, 2189. [Google Scholar] [CrossRef]
- Zhou, K.; Li, J.; Lu, Y.; Zhang, G.; Xie, Z.; Wu, C. Re-Examination of Dynamics of Polyeletrolytes in Salt-Free Dilute Solutions by Designing and Using a Novel Neutral−Charged−Neutral Reversible Polymer. Macromolecules 2009, 42, 7146–7154. [Google Scholar] [CrossRef]
- Glasing, J.; Bouchard, J.; Jessop, P.G.; Champagne, P.; Cunningham, M.F. Grafting Well-Defined CO2-Responsive Polymers to Cellulose Nanocrystals via Nitroxide-Mediated Polymerisation: Effect of Graft Density and Molecular Weight on Dispersion Behaviour. Polym. Chem. 2017, 8, 6000–6012. [Google Scholar] [CrossRef]
- Zehm, D.; Laschewsky, A.; Liang, H.; Rabe, J.P. Straightforward Access to Amphiphilic Dual Bottle Brushes by Combining RAFT, ATRP, and NMP Polymerization in One Sequence. Macromolecules 2011, 44, 9635–9641. [Google Scholar] [CrossRef]
- Muzammal, S.; Ahmad, A.; Sheraz, M.; Kim, J.; Ali, S.; Hanif, M.B.; Hussain, I.; Pandiaraj, S.; Alodhayb, A.; Javed, M.S.; et al. Polymer-Supported Nanomaterials for Photodegradation: Unraveling the Methylene Blue Menace. Energy Convers. Manag. X 2024, 22, 100547. [Google Scholar] [CrossRef]
- Stastny, V.; Rudkevich, D.M. Separations Using Carbon Dioxide. J. Am. Chem. Soc. 2007, 129, 1018–1019. [Google Scholar] [CrossRef]
- Lei, L.; Zhang, Q.; Shi, S.; Zhu, S. Highly Porous Poly(High Internal Phase Emulsion) Membranes with “Open-Cell” Structure and CO2-Switchable Wettability Used for Controlled Oil/Water Separation. Langmuir 2017, 33, 11936–11944. [Google Scholar] [CrossRef]
- Alex, J.P. Cormier CO2-Responsive Polymer Particles and Gels for Forward Osmosis; Queen’s University: Kingston, ON, Canada, 2023. [Google Scholar]
- Dou, B.; Lin, S.; Wang, Y.; Yang, L.; Yao, A.; Liao, H.; Tian, S.; Shang, J.; Lan, J. Versatile CO2-Responsive Sponges Decorated with ZIF-8 for Bidirectional Separation of Oil/Water and Controllable Removal of Dyes. ACS Appl. Mater. Interfaces 2023, 15, 37867–37883. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, L.; Grishkewich, N.; Tam, K.C.; Yuan, J.; Mao, Z.; Sui, X. CO2-Responsive Cellulose Nanofibers Aerogels for Switchable Oil–Water Separation. ACS Appl. Mater. Interfaces 2019, 11, 9367–9373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, Y.; Jiang, N.; Cheng, W.; Lu, D.; Zhang, T. Separate Reclamation of Oil and Surfactant from Oil-in-Water Emulsion with a CO2-Responsive Material. Environ. Sci. Technol. 2022, 56, 9651–9660. [Google Scholar] [CrossRef]
- Che, H.; Huo, M.; Peng, L.; Fang, T.; Liu, N.; Feng, L.; Wei, Y.; Yuan, J. CO2-Responsive Nanofibrous Membranes with Switchable Oil/Water Wettability. Angew. Chem. 2015, 127, 9062–9066. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, W.-Z.; Wang, Y.-M.; Qu, J.-P.; Lu, X.-B. N-Heterocyclic Carbene Functionalized Polymer for Reversible Fixation−Release of CO2. Macromolecules 2009, 42, 5419–5421. [Google Scholar] [CrossRef]
- Kainz, J.; Werz, P.D.L.; Troll, C.; Rieger, B. Temperature and CO2 Responsive Polyethylenimine for Highly Efficient Carbon Dioxide Release. RSC Adv. 2015, 5, 9556–9560. [Google Scholar] [CrossRef]
- Endo, T.; Nagai, D.; Monma, T.; Yamaguchi, H.; Ochiai, B. A Novel Construction of a Reversible Fixation−Release System of Carbon Dioxide by Amidines and Their Polymers. Macromolecules 2004, 37, 2007–2009. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Z.; Thanneeru, S.; Meng, M.; Kruzyk, M.; Ung, G.; Liu, B.; He, J. A Polymer Solution To Prevent Nanoclustering and Improve the Selectivity of Metal Nanoparticles for Electrocatalytic CO2 Reduction. Angew. Chem. 2019, 131, 15981–15987. [Google Scholar] [CrossRef]
- Pham, T.H.M.; Zhang, J.; Li, M.; Shen, T.; Ko, Y.; Tileli, V.; Luo, W.; Züttel, A. Enhanced Electrocatalytic CO2 Reduction to C2+ Products by Adjusting the Local Reaction Environment with Polymer Binders. Adv. Energy Mater. 2022, 12, 2103663. [Google Scholar] [CrossRef]
- Soucy, T.L.; Dean, W.S.; Zhou, J.; Rivera Cruz, K.E.; McCrory, C.C.L. Considering the Influence of Polymer–Catalyst Interactions on the Chemical Microenvironment of Electrocatalysts for the CO2 Reduction Reaction. Acc. Chem. Res. 2022, 55, 252–261. [Google Scholar] [CrossRef]
- Liu, Y.; McCrory, C.C.L. Modulating the Mechanism of Electrocatalytic CO2 Reduction by Cobalt Phthalocyanine through Polymer Coordination and Encapsulation. Nat. Commun. 2019, 10, 1683. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Huo, M.; Peng, L.; Ye, Q.; Guo, J.; Wang, K.; Wei, Y.; Yuan, J. CO2-Switchable Drug Release from Magneto-Polymeric Nanohybrids. Polym. Chem. 2015, 6, 2319–2326. [Google Scholar] [CrossRef]
- Shigemura, M.; Lecuona, E.; Sznajder, J.I. Effects of Hypercapnia on the Lung. J. Physiol. 2017, 595, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Huang, B.; Xiao, C.; Vana, P. Intelligent CO2—And Photo-Dual-Responsive Polymer Vesicles with Tunable Wall Thickness. Polym. Chem. 2019, 10, 1610–1618. [Google Scholar] [CrossRef]
- Rabiee, H.; Jin, B.; Yun, S.; Dai, S. Gas-Responsive Cationic Microgels for Forward Osmosis Desalination. Chem. Eng. J. 2018, 347, 424–431. [Google Scholar] [CrossRef]
- Riabtseva, A.; Ellis, S.N.; Champagne, P.; Jessop, P.G.; Cunningham, M.F. CO2-Responsive Branched Polymers for Forward Osmosis Applications: The Effect of Branching on Draw Solute Properties. Ind. Eng. Chem. Res. 2021, 60, 9807–9816. [Google Scholar] [CrossRef]
- Jessop, P.G.; Mercer, S.M.; Robert, T.; Brown, R.S.; Clark, T.J.; Mariampillai, B.E.; Resendes, R.; Wechsler, D. Systems and Methods for Use of Water with Switchable Ionic Strength. U.S. Patent No. 10,377,647, 13 August 2019. [Google Scholar]
- Cai, Y.; Shen, W.; Wang, R.; Krantz, W.B.; Fane, A.G.; Hu, X. CO2 Switchable Dual Responsive Polymers as Draw Solutes for Forward Osmosis Desalination. Chem. Commun. 2013, 49, 8377. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, E.; Wang, J. Gas-Breathing Polymer Film for Constructing Switchable Ionic Diodes. RSC Adv. 2015, 5, 35622–35630. [Google Scholar] [CrossRef]
- Bhamidipati, M.; Scurto, A.M.; Detamore, M.S. The Future of Carbon Dioxide for Polymer Processing in Tissue Engineering. Tissue Eng. Part B Rev. 2013, 19, 221–232. [Google Scholar] [CrossRef]
- Sheridan, M.; Shea, L.; Peters, M.; Mooney, D. Bioabsorbable Polymer Scaffolds for Tissue Engineering Capable of Sustained Growth Factor Delivery. J. Control. Release 2000, 64, 91–102. [Google Scholar] [CrossRef]
- Shieh, Y.-T.; Lin, Y.-T.; Cheng, C.-C. CO2-Switchable Behavior of Chitosan-g-Poly[(2-Dimethylamino)Ethyl Methacrylate] as an Emulsifier. Carbohydr. Polym. 2017, 170, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, S.; Mokhtari, R.; Hohl, L.; Miller, R.; Kraume, M. Advances in CO2-Switchable Surfactants towards the Fabrication and Application of Responsive Colloids. Adv. Colloid Interface Sci. 2023, 315, 102907. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yung, L.-Y.L. Detection of Dissolved CO2 Based on the Aggregation of Gold Nanoparticles. Anal. Chem. 2014, 86, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, J.; Yin, Y.; Yuan, J. Breathing Polymersomes: CO2-Tuning Membrane Permeability for Size-Selective Release, Separation, and Reaction. Angew. Chem. Int. Ed. 2013, 52, 5070–5073. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method Name | CO2-Responsive Polymer Type | Key Findings | References |
|---|---|---|---|
| RAFT Polymerization | PDEAEMA-b-PNIPAM (block copolymer) | Dual CO2- and temperature-responsive block copolymer synthesized via RAFT. Shows switchable solubility and hydrophilicity under CO2. | [106] |
| RAFT Polymerization | Poly(4-chloromethylstyrene)—PCO2-switchable amidine-based polymer | PCMS prepared by RAFT, then converted to PAMS via click chemistry, followed by amidine functionalization, resulting in CO2-responsive behavior. | [107] |
| RAFT Polymerization | CO2-switchable single-walled carbon nanotubes (SWCNTs) | SWCNTs were functionalized with CO2-responsive polymeric brushes using RAFT polymerization for improved environmental adaptability and selectivity under CO2. | [29] |
| RAFT Polymerization | PEO-b-DEAEMA-r-S and PEO-b-DEAEMA-b-S (diblock copolymers) | PEO-based block copolymers with CO2-switchable units (DEAEMA) designed for morphological transitions in aqueous solution. | [108] |
| RAFT Polymerization | PEO-b-(PDEAEMA-r-S) (triblock copolymer) | CO2-responsive triblock copolymer with segregated corona synthesized by sequential RAFT, showcasing morphological transitions on CO2 bubbling. | [109] |
| RAFT Polymerization | Poly(pentafluorophenyl acrylate)—PDMAEMA and PEGA block copolymers | Dual-responsive copolymers synthesized by RAFT, functionalized with histamine and arginine to achieve CO2-switchability. | [54] |
| RAFT Polymerization | PMMA-b-PDMAEMA (diblock copolymer) | CO2-responsive PMMA-b-PDMAEMA block copolymer synthesized via RAFT. Demonstrated self-assembly behavior in aqueous media under CO2–N2 cycling. | [110] |
| RAFT Polymerization | PNIPAM-b-PCL-b-PDMAEMA (triblock copolymer) | Dual-responsive PNIPAM-b-PCL-b-PDMAEMA copolymer synthesized by RAFT, responsive to temperature and CO2. | [111] |
| RAFT Polymerization | Poly(oligo(ethylene glycol)methyl ether methacrylate)-b-PDEAEMA-b-PAPUEMA | CO2- and temperature-responsive triblock copolymer synthesized by sequential RAFT polymerization for responsive polymeric brushes. | [112] |
| RAFT Polymerization | CO2-switchable pyrene-containing copolymer (Py-PCL-b-P(NIPAM-co-DMAEMA)) | Amphiphilic copolymer synthesized by RAFT for micelle formation in response to CO2. | [113] |
| RAFT Polymerization | CO2-responsive polystyrene (PS)-PDEAEMA nanoparticles | Nanoparticles prepared via surfactant-free mini-emulsion RAFT polymerization. Exhibited reversible self-assembly in response to CO2. | [114] |
| Polymerization Method | Key Advantages | Key Disadvantages |
|---|---|---|
| Free radical polymerization (FRP) |
|
|
| Reversible deactivation radical polymerization (RDRP) |
|
|
| Atom transfer radical polymerization (ATRP) |
|
|
| Reversible addition-fragmentation chain transfer (RAFT) |
|
|
| Nitroxide-mediated polymerization (NMP) |
|
|
| Polymer/Material | CO2 Stimulus | Method of CO2-Responsive Polymer Preparation | Oil–Water Separation Key Findings | Reference |
|---|---|---|---|---|
| Poly(styrene-co-N,N-(diethylamino)ethyl methacrylate) | CO2 switches wettability from hydrophobic–superoleophilic to hydrophilic–superoleophobic | Free radical polymerization (FRP) | Efficient and recyclable switchable oil–water separation driven by gravity | [124] |
| PDEAEMA polymer on PU sponge | CO2 induces protonation, switching from hydrophobic to hydrophilic | Free radical polymerization (FRP) | Demonstrates a high oil adsorption capacity of 14 g/g for emulsified oil, achieving a separation efficiency of 97.5% | [126] |
| Polymerized PDEAEMA on CNFs aerogels | CO2 induces protonation in PDEAEMA, altering wettability between hydrophobic and hydrophilic | Surface-Initiated Atom-Transfer Radical Polymerization (SI-ATRP) | Achieves oil–water separation efficiency of up to 99.96% | [127] |
| CO2-responsive cellulose nanofibers aerogels | CO2-induced wettability change from hydrophobic to hydrophilic for oil–water separation | Surface-Initiated Atom-Transfer Radical Polymerization (SI-ATRP) | Cellulose aerogels that are recyclable and have switchable wettability for the separation of oil–water mixtures | [128] |
| CO2-responsive nanofibrous membranes | CO2 causes a switchable hydrophilic to hydrophobic transition in nanofibers | Electrospinning (free radical polymerization) | Efficient selective separation of oil and water deploying switchable wettability | [129] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheraz, M.; Wang, R. CO2-Responsive Vinyl Polymers: From Synthesis to Application. Molecules 2025, 30, 2350. https://doi.org/10.3390/molecules30112350
Sheraz M, Wang R. CO2-Responsive Vinyl Polymers: From Synthesis to Application. Molecules. 2025; 30(11):2350. https://doi.org/10.3390/molecules30112350
Chicago/Turabian StyleSheraz, Mahshab, and Rui Wang. 2025. "CO2-Responsive Vinyl Polymers: From Synthesis to Application" Molecules 30, no. 11: 2350. https://doi.org/10.3390/molecules30112350
APA StyleSheraz, M., & Wang, R. (2025). CO2-Responsive Vinyl Polymers: From Synthesis to Application. Molecules, 30(11), 2350. https://doi.org/10.3390/molecules30112350