
The Impact of Perfluoroalkyl Groups on Phosphane Basicity


Abstract

1. Introduction
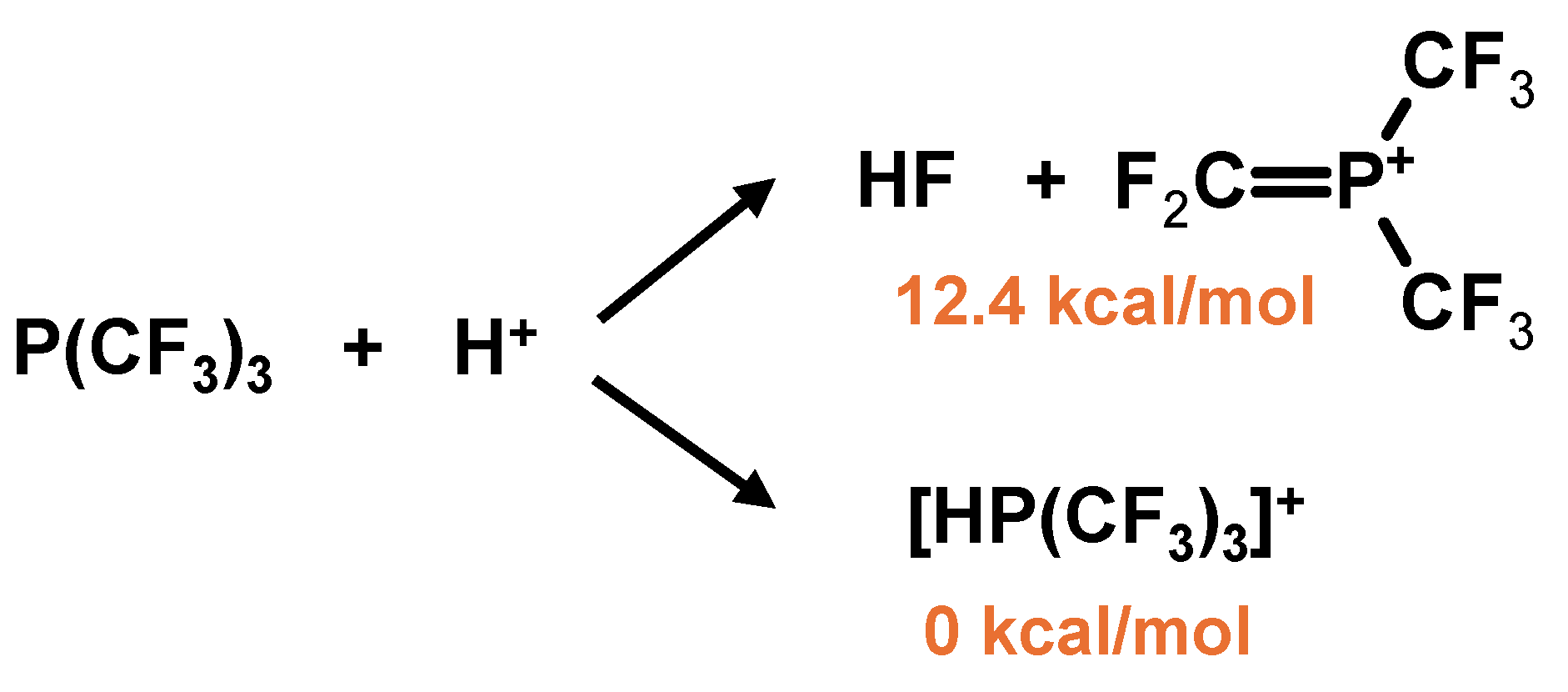
2. Results and Discussion
3. Computational Methods

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whitehurst, W.G.; Kim, J.; Koenig, S.G.; Chirik, P.J. Three-Component Coupling of Arenes, Ethylene, and Alkynes Catalyzed by a Cationic Bis(Phosphine) Cobalt Complex: Intercepting Metallacyclopentenes for C–H Functionalization. J. Am. Chem. Soc. 2022, 144, 4530–4540. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Yuzawa, K.; Bhanage, B.M.; Ikushima, Y.; Arai, M. Palladium-Catalyzed Heck Coupling Reactions Using Different Fluorinated Phosphine Ligands in Compressed Carbon Dioxide and Conventional Organic Solvents. J. Mol. Catal. A Chem. 2002, 180, 35–42. [Google Scholar] [CrossRef]
- Murai, M.; Takeshima, H.; Morita, H.; Kuninobu, Y.; Takai, K. Acceleration Effects of Phosphine Ligands on the Rhodium-Catalyzed Dehydrogenative Silylation and Germylation of Unactivated C(Sp3)–H Bonds. J. Org. Chem. 2015, 80, 5407–5414. [Google Scholar] [CrossRef]
- Pikma, M.-L.; Ilisson, M.; Zalite, R.; Lavogina, D.; Haljasorg, T.; Mäeorg, U. The Effect of Substituents on Carbon–Carbon Double Bond Isomerization in Heterocyclic Hydrazine Derivatives. Chem. Heterocycl. Compd. 2022, 58, 206–216. [Google Scholar] [CrossRef]
- Brisdon, A.K.; Herbert, C.J. Fluoroalkyl-Containing Phosphines. Coord. Chem. Rev. 2013, 257, 880–901. [Google Scholar] [CrossRef]
- Brisdon, A.K.; Herbert, C.J. A Generic Route to Fluoroalkyl-Containing Phosphanes. Chem. Commun. 2009, 6658–6660. [Google Scholar] [CrossRef]
- Choate, M.M.; Baughman, R.G.; Phelps, J.E.; Peters, R.G. Synthesis, Characterization, and Coordination Chemistry of Several Novel Electroneutral Phosphane Ligands. J. Organomet. Chem 2011, 696, 956–962. [Google Scholar] [CrossRef]
- Murphy-Jolly, M.B.; Lewis, L.C.; Caffyn, A.J.M. The Synthesis of Tris(Perfluoroalkyl)Phosphines. Chem. Commun. 2005, 4479–4480. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, W.; Li, S. Synthesis and Applications of Trifluoromethylphosphines. Eur. J. Org. Chem. 2024, 27, e202301169. [Google Scholar] [CrossRef]
- Hayes, S.A.; Berger, R.J.F.; Neumann, B.; Mitzel, N.W.; Bader, J.; Hoge, B. Molecular Structure of Tris(Pentafluoroethyl)Phosphane P(C2F5)3. Dalton Trans. 2010, 39, 5630–5636. [Google Scholar] [CrossRef]
- Beg, M.A.A.; Clark, H.C. Chemistry of the Trifluoromethyl Group: Part i. Complex Formation by Phosphines Containing the Trifluoromethyl Group. Can. J. Chem. 1960, 38, 119–124. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Saga, Y.; Sato, Y.; Minamida, Y.; Nomoto, A.; Ogawa, A. P-Fluorous Phosphines as Electron-Poor/Fluorous Hybrid Functional Ligands for Precious Metal Catalysts: Synthesis of Rh(I), Ir(I), Pt(II), and Au(I) Complexes Bearing P-Fluorous Phosphine Ligands. Inorganics 2017, 5, 5. [Google Scholar] [CrossRef]
- Hope, E.G.; Simayi, R.; Stuart, A.M. Fluorous Organometallic Chemistry. In Organometallic Fluorine Chemistry; Braun, T., Hughes, R.P., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 217–240. ISBN 978-3-319-22096-3. [Google Scholar]
- Bader, J.; Maier, A.F.G.; Paradies, J.; Hoge, B. Perfluoroalkylated Main-Group Element Lewis Acids as Catalysts in Transfer Hydrogenation. Eur. J. Inorg. Chem. 2017, 2017, 3053–3056. [Google Scholar] [CrossRef]
- Altinel, H.; Avsar, G.; Guzel, B. Fluorinated Rhodium-Phosphine Complexes as Efficient Homogeneous Catalysts for the Hydrogenation of Styrene in Supercritical Carbon Dioxide. Transit. Met. Chem. 2009, 34, 331–335. [Google Scholar] [CrossRef]
- Horváth, I.T.; Kiss, G.; Cook, R.A.; Bond, J.E.; Stevens, P.A.; Rábai, J.; Mozeleski, E.J. Molecular Engineering in Homogeneous Catalysis: One-Phase Catalysis Coupled with Biphase Catalyst Separation. The Fluorous-Soluble HRh(CO){P[CH2CH2(CF2)5CF3]3}3 Hydroformylation System. J. Am. Chem. Soc. 1998, 120, 3133–3143. [Google Scholar] [CrossRef]
- Friesen, C.M.; Montgomery, C.D.; Temple, S.A.J.U. The First Fluorous Biphase Hydrogenation Catalyst Incorporating a Perfluoropolyalkylether: [RhCl(PPh2(C6H4C(O)OCH2CF(CF3)(OCF2CF(CF3))nF))3] with n = 4–9. J. Fluor. Chem. 2012, 144, 24–32. [Google Scholar] [CrossRef]
- Altinel, H.; Avsar, G.; Yilmaz, M.K.; Guzel, B. New Perfluorinated Rhodium–BINAP Catalysts and Hydrogenation of Styrene in Supercritical CO2. J. Supercrit. Fluids. 2009, 51, 202–208. [Google Scholar] [CrossRef]
- Begum, F.; Ikram, M.; Twamley, B.; Baker, R.J. Perfluorinated Phosphine and Hybrid P–O Ligands for Pd Catalysed C–C Bond Forming Reactions in Solution and on Teflon Supports. RSC Adv. 2019, 9, 28936–28945. [Google Scholar] [CrossRef]
- Matsubara, K.; Fujii, T.; Hosokawa, R.; Inatomi, T.; Yamada, Y.; Koga, Y. Fluorine-Substituted Arylphosphine for an NHC-Ni(I) System, Air-Stable in a Solid State but Catalytically Active in Solution. Molecules 2019, 24, 3222. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Bour, J.R.; Canty, A.J.; Kampf, J.W.; Sanford, M.S. Stoichiometric and Catalytic Aryl–Perfluoroalkyl Coupling at Tri-Tert-Butylphosphine Palladium(II) Complexes. J. Am. Chem. Soc. 2017, 139, 11662–11665. [Google Scholar] [CrossRef]
- Burton, D.J.; Lu, L. Fluorinated Organometallic Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R.D., Ed.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 45–89. ISBN 978-3-540-69197-6. [Google Scholar]
- Duan, L.; Nesterov, V.; Runyon, J.W.; Schnakenburg, G.; Iii, A.J.A.; Streubel, R. Synthesis of Stabilized Phosphinidenoid Complexes Using Weakly Coordinating Cations1. Aust. J. Chem. 2011, 64, 1583–1586. [Google Scholar] [CrossRef]
- Bennett, J.A.; Hope, E.G.; Singh, K.; Stuart, A.M. Synthesis and Coordination Chemistry of Fluorinated Phosphonic Acids. J. Fluor. Chem. 2009, 130, 615–620. [Google Scholar] [CrossRef]
- Wehbi, M.; Mehdi, A.; Negrell, C.; David, G.; Alaaeddine, A.; Améduri, B. Phosphorus-Containing Fluoropolymers: State of the Art and Applications. ACS Appl. Mater. Interfaces 2020, 12, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Pikma, M.-L.; Tshepelevitsh, S.; Selberg, S.; Kaljurand, I.; Leito, I.; Kütt, A. PKaH Values and θ H Angles of Phosphanes to Predict Their Electronic and Steric Parameters. Dalton Trans. 2024, 53, 14226–14236. [Google Scholar] [CrossRef]
- Klamt, A. The COSMO and COSMO-RS Solvation Models. Comput. Mol. Sci. 2018, 8, e1338. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on Ab Initio Reaction Field Calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM Ab Initio Level. J. Mol. Struct. THEOCHEM. 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-Consistent Reaction Field Model for Aqueous and Nonaqueous Solutions Based on Accurate Polarized Partial Charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef]
- Chipman, D.M. Charge Penetration in Dielectric Models of Solvation. J. Chem. Phys. 1997, 106, 10194–10206. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Bentley, J.; Chipman, D.M. Volume Polarization in Reaction Field Theory. J. Chem. Phys. 1998, 108, 177–192. [Google Scholar] [CrossRef]
- Chipman, D.M. New Formulation and Implementation for Volume Polarization in Dielectric Continuum Theory. J. Chem. Phys. 2006, 124, 224111. [Google Scholar] [CrossRef]
- Kaupmees, K.; Trummal, A.; Leito, I. Basicities of Strong Bases in Water: A Computational Study. Croat. Chem. Acta 2014, 87, 385–395. [Google Scholar] [CrossRef]
- Li, J.-N.; Fu, Y.; Liu, L.; Guo, Q.-X. First-Principle Predictions of Basicity of Organic Amines and Phosphines in Acetonitrile. Tetrahedron 2006, 62, 11801–11813. [Google Scholar] [CrossRef]
- Ding, F.; Smith, J.M.; Wang, H. First-Principles Calculation of pKa Values for Organic Acids in Nonaqueous Solution. J. Org. Chem. 2009, 74, 2679–2691. [Google Scholar] [CrossRef] [PubMed]
- Pliego, J.R.; Riveros, J.M. Theoretical Calculation of pKa Using the Cluster−Continuum Model. J. Phys. Chem. A 2002, 106, 7434–7439. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Gusev, D.G. Donor Properties of a Series of Two-Electron Ligands. Organometallics 2009, 28, 763–770. [Google Scholar] [CrossRef]
- Haav, K.; Saame, J.; Kütt, A.; Leito, I. Basicity of Phosphanes and Diphosphanes in Acetonitrile. Eur. J. Org. Chem. 2012, 2012, 2167–2172. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Greb, L.; Tussing, S.; Schirmer, B.; Oña-Burgos, P.; Kaupmees, K.; Lõkov, M.; Leito, I.; Grimme, S.; Paradies, J. Electronic Effects of Triarylphosphines in Metal-Free Hydrogen Activation: A Kinetic and Computational Study. Chem. Sci. 2013, 4, 2788–2796. [Google Scholar] [CrossRef]
- Mehlmann, P.; Mück-Lichtenfeld, C.; Tan, T.T.Y.; Dielmann, F. Tris(Imidazolin-2-Ylidenamino)Phosphine: A Crystalline Phosphorus(III) Superbase That Splits Carbon Dioxide. Chem. Eur. J. 2017, 23, 5929–5933. [Google Scholar] [CrossRef]
- Himmel, D.; Goll, S.K.; Leito, I.; Krossing, I. Anchor Points for the Unified Brønsted Acidity Scale: The rCCC Model for the Calculation of Standard Gibbs Energies of Proton Solvation in Eleven Representative Liquid Media. Chem. Eur. J. 2011, 17, 5808–5826. [Google Scholar] [CrossRef]
- Pikma, M.-L.; Lõkov, M.; Tshepelevitsh, S.; Saame, J.; Haljasorg, T.; Toom, L.; Selberg, S.; Leito, I.; Kütt, A. Tris(Benzophenoneimino)Phosphane and Related Compounds. Eur. J. Org. Chem. 2023, 26, e202300453. [Google Scholar] [CrossRef]
- Malloum, A.; Conradie, J. Solvation Free Energy of the Proton in Acetonitrile. J. Mol. Liq. 2021, 335, 116032. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- TURBOMOLE V7.8 2023, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: https://www.turbomole.org/turbomole/release-notes-turbomole-7-8/ (accessed on 16 May 2025).
- BIOVIA. Dassault Systèmes, TmoleX 2024; BIOVIA: San Diego, CA, USA, 2024. [Google Scholar]
- BIOVIA. Dassault Systèmes, COSMOconfX 2024; BIOVIA: San Diego, CA, USA, 2024. [Google Scholar]
- BIOVIA. Dassault Systèmes, COSMOtherm 2025; BIOVIA: San Diego, CA, USA, 2025. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2025. [Google Scholar]
- University of Tartu “UT Rocket”. Share.Neic.No. Available online: https://doi.org/10.23673/PH6N-0144 (accessed on 16 May 2025).
- Pikma, M.-L.; Trummal, A.; Leito, I.; Kütt, A. The Impact of Perfluoroalkyl Groups on Phosphane Basicity. 2025. Available online: https://doi.org/10.23673/re-508 (accessed on 16 May 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
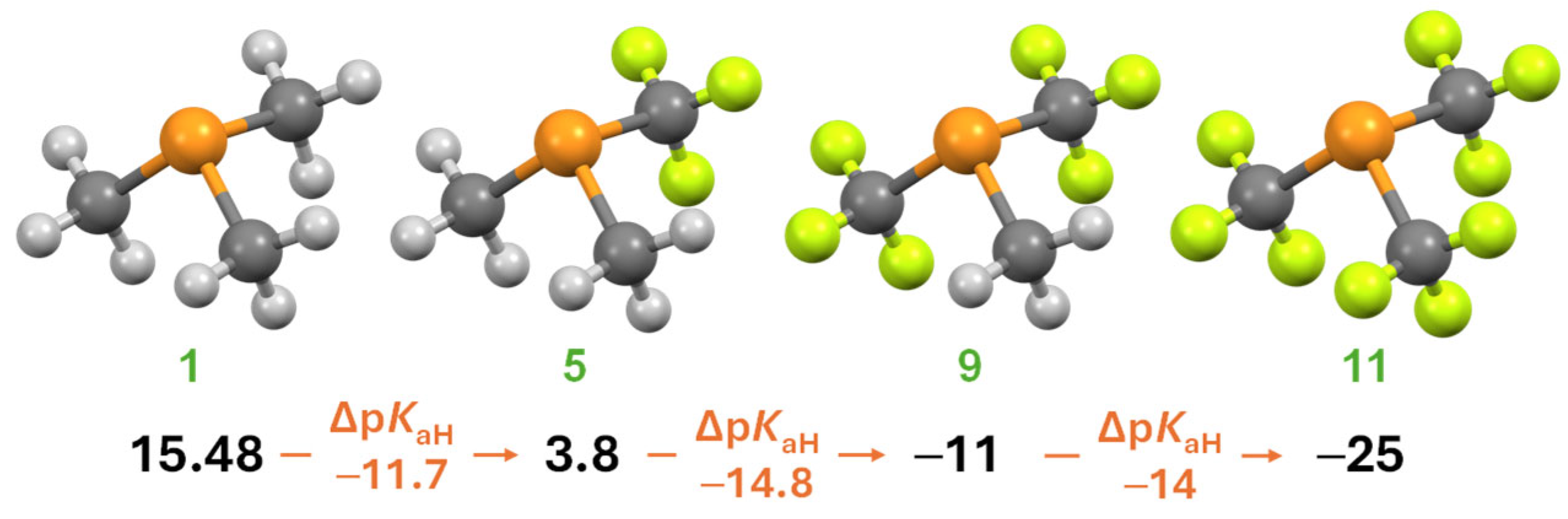
| No | Compound | pKaH(MeCN) | GB [kcal·mol−1] | TEP [cm−1] | θCPC [°] b | θH [°] b | θTolman [°] | ||
|---|---|---|---|---|---|---|---|---|---|
| Experimental | SMD | COSMO-RS a | |||||||
| 1 | PMe3 | 15.48 c | 18.5 | 15.6 f | 220.3 | 2064.1 | 110.4 | 163 | 118 |
| 2 | P(2-F-C6H4)Ph2 | 6.11 d | 6.3 | 5.9 f | 221.1 | 110.8 | 220 | ||
| 3 | P(2,6-F2-C6H3)Ph2 | 5.17 d | 5.17 e | 5.5 f | 220.7 | 111.4 | 221 | ||
| 4 | P(2-F-C6H4)2Ph | 4.56 d | 4.6 | 4.5 f | 219.4 | 110.5 | 226 | ||
| 5 | P(CF3)Me2 | 3.8 | 1.7 | 196.5 | 109.8 | 169 | 124 | ||
| 6 | P(C6F5)Ph2 | 2.54 d | 2.7 | 2.7 f | 212.7 | 2074.8 | 111.6 | 222 | 158 |
| 7 | P(2,6-F2-C6H3)2Ph | 2.5 d | 2.4 | 2.5 f | 217.0 | 111.7 | 232 | ||
| 8 | P(C6F5)3 | −8 | −7 f | 190.4 | 2090.9 | 112.6 g | 256 g | 184 | |
| 9 | P(CF3)2Me | −11 | −13 | 174.4 | 109.0 | 174 | |||
| 10 | PF3 | −24 | −23 | 146.2 | 2110.8 | 108.0 | 149 | 104 | |
| 11 | P(CF3)3 | −25 | −27 | 156.4 | 2104.4 | 109.0 g | 185 g | 137 | |
| 12 | P(C2F5)3 | −29 | −27 | 158.8 | 2102.9 | 109.0 | 217 | 171 | |
| 13 | P(C3F7)3 | −28 | −27 | 162.1 | 110.2 | 227 | |||
| 14 | P(C4F9)3 | −32 | −28 | 159.8 | 110.3 | 215 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pikma, M.-L.; Trummal, A.; Leito, I.; Kütt, A. The Impact of Perfluoroalkyl Groups on Phosphane Basicity. Molecules 2025, 30, 2220. https://doi.org/10.3390/molecules30102220
Pikma M-L, Trummal A, Leito I, Kütt A. The Impact of Perfluoroalkyl Groups on Phosphane Basicity. Molecules. 2025; 30(10):2220. https://doi.org/10.3390/molecules30102220
Chicago/Turabian StylePikma, Marta-Lisette, Aleksander Trummal, Ivo Leito, and Agnes Kütt. 2025. "The Impact of Perfluoroalkyl Groups on Phosphane Basicity" Molecules 30, no. 10: 2220. https://doi.org/10.3390/molecules30102220
APA StylePikma, M.-L., Trummal, A., Leito, I., & Kütt, A. (2025). The Impact of Perfluoroalkyl Groups on Phosphane Basicity. Molecules, 30(10), 2220. https://doi.org/10.3390/molecules30102220