


Colchicine Binding Site Tubulin Inhibitors Impair Vincristine-Resistant Neuroblastoma Cell Function

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Impact of Colchicine Binding Site Inhibition on Neuroblastoma Cell Lines
2.1.1. Cell Viability Paneling Using Diverse Neuroblastoma Cells
2.1.2. CBSI Treatment Causes an Increase in Protein Markers of Mitosis
2.1.3. Inhibition of Colony Formation
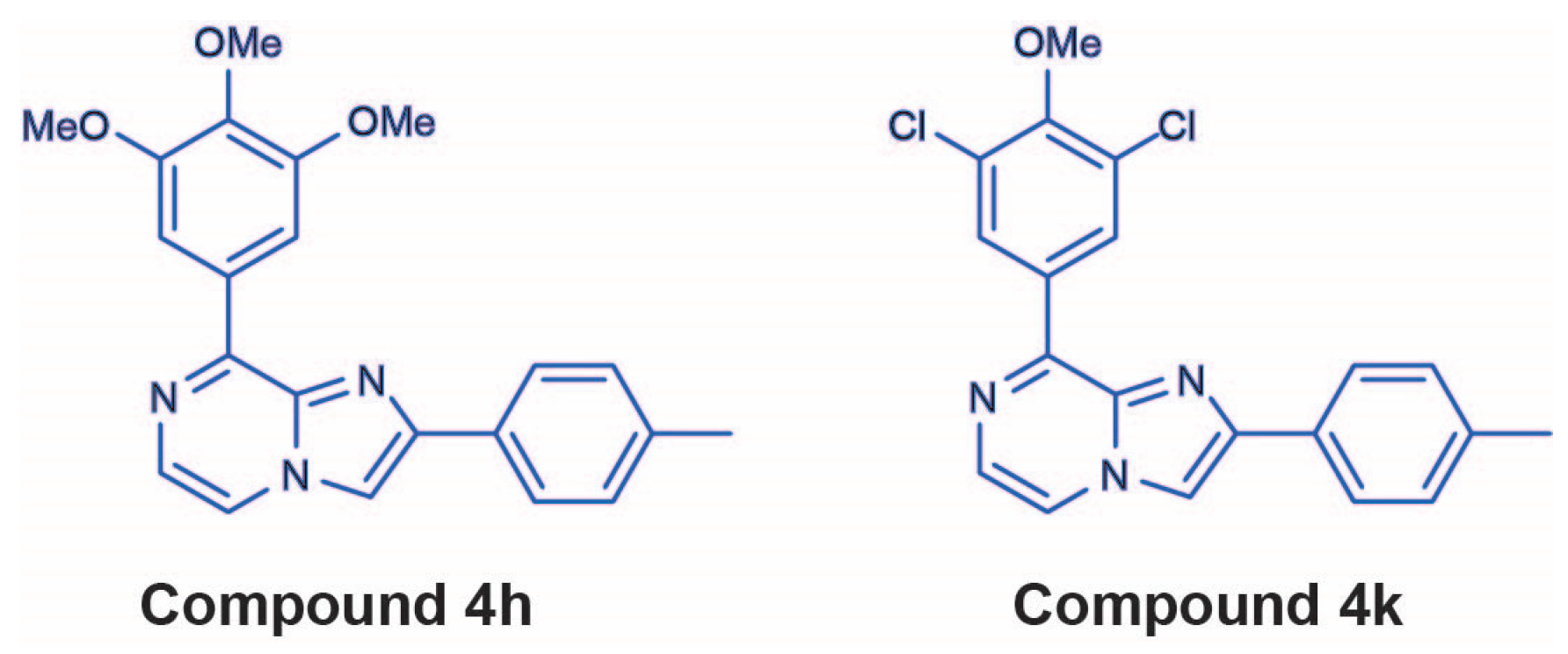
2.2. 4h and 4k Reduce Vincristine-Resistant Neuroblastoma Cell Viability in a Dose-Dependent Manner
2.2.1. Cell Viability in Vincristine-Resistant Neuroblastoma Cell Lines
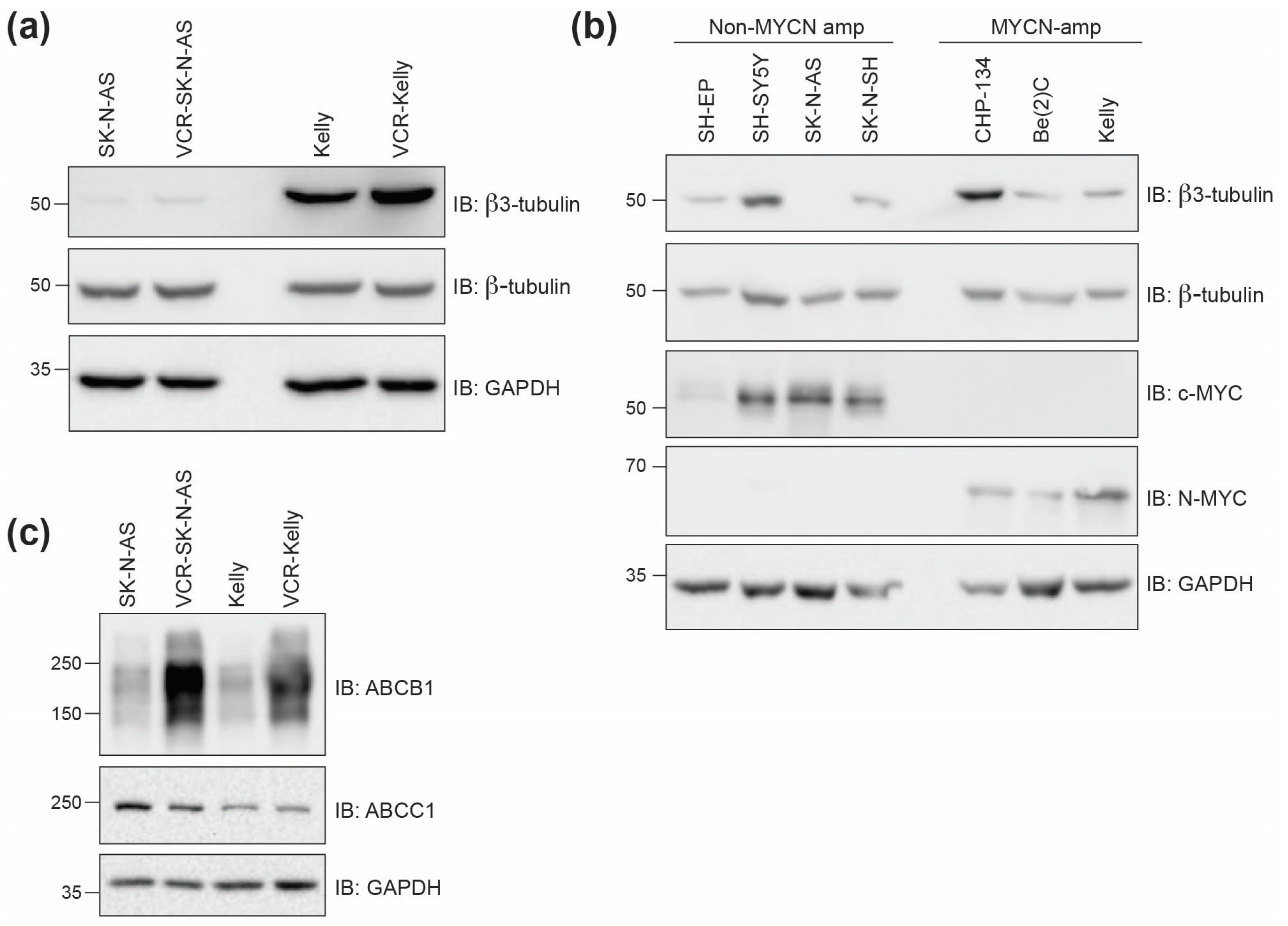
2.2.2. Beta3-Tubulin Expression Levels Do Not Correlate with Cell Line Response
2.3. 4h and 4k Treatment Impair Intracellular Microtubule Networks and Act as Antimitotics in Vincristine-Resistant Neuroblastoma Cell Lines
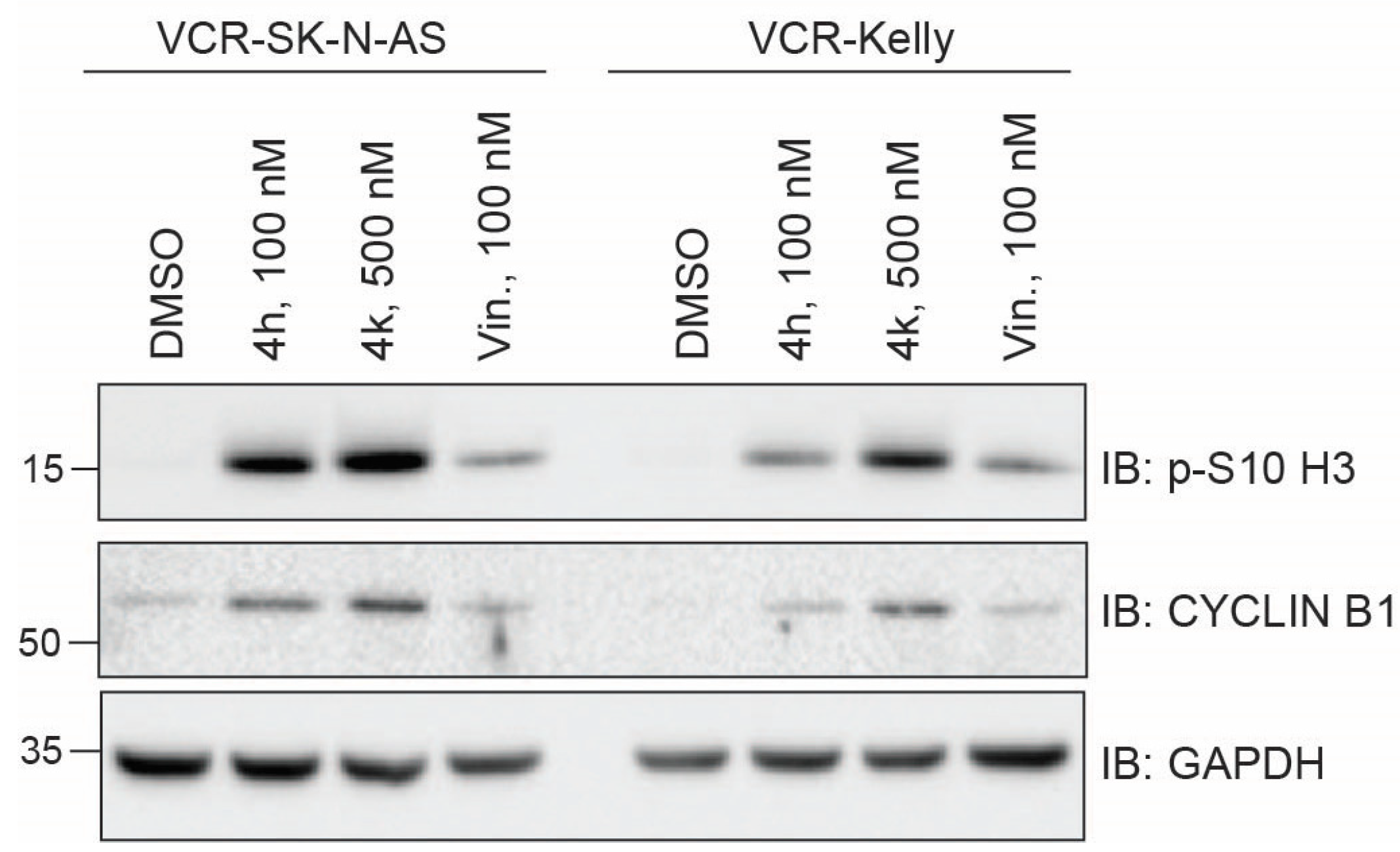
2.3.1. Impact of CBSIs on Intracellular Tubulin and Mitotic Marker Induction
2.3.2. Cell Cycle Phase Distribution Changes Induced by Treatment with CBSIs
2.4. Apoptotic Activity of 4h and 4k in Neuroblastoma Cells Compared to Vincristine-Resistant Neuroblastoma Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Protein Lysates and Western Blotting
4.3. Colony Formation Assay
4.4. Cell Viability Screening
4.5. Cell Cycle Analysis
4.6. Annexin V Staining
4.7. Immunofluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| VCR | Vincristine-resistant |
| CBSI | Colchicine binding site inhibitor |
| DMSO | Dimethyl sulfoxide |
| ABC | ATP-binding cassette |
References
- Zhou, X.; Wang, X.; Li, N.; Guo, Y.; Yang, X.; Lei, Y. Therapy resistance in neuroblastoma: Mechanisms and reversal strategies. Front. Pharmacol. 2023, 14, 1114295. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.; Foster, J.H. Treatment of high-risk neuroblastoma. Children 2023, 10, 1302. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-relevant ABC transporter for anti-cancer drug resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef] [PubMed]
- Thammathong, J.; Chisam, K.B.; Tessmer, G.E.; Womack, C.B.; Sidrak, M.M.; Weissmiller, A.M.; Banerjee, S. Fused imidazopyrazine-based tubulin polymerization inhibitors inhibit neuroblastoma cell function. ACS Med. Chem. Lett. 2023, 14, 1284–1294. [Google Scholar] [CrossRef]
- Hawash, M. Recent advances of tubulin inhibitors targeting the colchicine binding site for cancer therapy. Biomolecules 2022, 12, 1843. [Google Scholar] [CrossRef]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calo, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Banerjee, S.; Mahmud, F.; Deng, S.; Ma, L.; Yun, M.K.; Fakayode, S.O.; Arnst, K.E.; Yang, L.; Chen, H.; Wu, Z.; et al. X-ray crystallography-guided design, antitumor efficacy, and QSAR analysis of metabolically stable cyclopenta-pyrimidinyl dihydroquinoxalinone as a potent tubulin polymerization inhibitor. J. Med. Chem. 2021, 64, 13072–13095. [Google Scholar] [CrossRef]
- Lu, Y.; Li, C.M.; Wang, Z.; Chen, J.; Mohler, M.L.; Li, W.; Dalton, J.T.; Miller, D.D. Design, synthesis, and SAR studies of 4-substituted methoxylbenzoyl-aryl-thiazoles analogues as potent and orally bioavailable anticancer agents. J. Med. Chem. 2011, 54, 4678–4693. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Wang, J.; Ahn, S.; Li, C.M.; Lu, Y.; Loveless, V.S.; Dalton, J.T.; Miller, D.D.; Li, W. Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm. Res. 2012, 29, 3040–3052. [Google Scholar] [CrossRef]
- Meany, H.J.; Sackett, D.L.; Maris, J.M.; Ward, Y.; Krivoshik, A.; Cohn, S.L.; Steinberg, S.M.; Balis, F.M.; Fox, E. Clinical outcome in children with recurrent neuroblastoma treated with ABT-751 and effect of ABT-751 on proliferation of neuroblastoma cell lines and on tubulin polymerization in vitro. Pediatr. Blood Cancer 2010, 54, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Thirant, C.; Peltier, A.; Durand, S.; Kramdi, A.; Louis-Brennetot, C.; Pierre-Eugene, C.; Gautier, M.; Costa, A.; Grelier, A.; Zaidi, S.; et al. Reversible transitions between noradrenergic and mesenchymal tumor identities define cell plasticity in neuroblastoma. Nat. Commun. 2023, 14, 2575. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Chatten, J.; Newton, W.A., Jr.; Sachs, N.; Hamoudi, A.B.; Chiba, T.; Marsden, H.B.; Misugi, K. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J. Natl. Cancer Inst. 1984, 73, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.D.; Kattan, D.R.; Thomas, S.K.; Spengler, B.A.; Guo, H.F.; Biedler, J.L.; Cheung, N.K.; Ross, R.A. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004, 6, 838–845. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The expanding world of N-MYC-driven tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef]
- Trigg, R.M.; Turner, S.D. ALK in neuroblastoma: Biological and therapeutic implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voute, P.A.; Schwab, M.; et al. N-MYC enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef]
- Siaw, J.T.; Javanmardi, N.; Van den Eynden, J.; Lind, D.E.; Fransson, S.; Martinez-Monleon, A.; Djos, A.; Sjoberg, R.M.; Ostensson, M.; Caren, H.; et al. 11q deletion or ALK activity curbs DLG2 expression to maintain an undifferentiated state in neuroblastoma. Cell Rep. 2020, 32, 108171. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Jurkovic Mlakar, S.; Lopez, G.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. 11q deletion in neuroblastoma: A review of biological and clinical implications. Mol. Cancer 2017, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Vujic, I.; Posch, C.; Sanlorenzo, M.; Yen, A.J.; Tsumura, A.; Kwong, A.; Feichtenschlager, V.; Lai, K.; Arneson, D.V.; Rappersberger, K.; et al. Mutant NRASQ61 shares signaling similarities across various cancer types—Potential implications for future therapies. Oncotarget 2014, 5, 7936–7944. [Google Scholar] [CrossRef]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Ferrell, J.E., Jr. The roles of Cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 2010, 21, 3149–3161. [Google Scholar] [CrossRef]
- Gurley, L.R.; D’Anna, J.A.; Barham, S.S.; Deaven, L.L.; Tobey, R.A. Histone phosphorylation and chromatin structure during mitosis in chinese hamster cells. Eur. J. Biochem. 1978, 84, 1–15. [Google Scholar] [CrossRef]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447–456. [Google Scholar] [CrossRef]
- Kanakkanthara, A.; Miller, J.H. BetaIII-tubulin overexpression in cancer: Causes, consequences, and potential therapies. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188607. [Google Scholar] [CrossRef]
- Taubenberger, A.V.; Baum, B.; Matthews, H.K. The mechanics of mitotic cell rounding. Front. Cell Dev. Biol. 2020, 8, 687. [Google Scholar] [CrossRef]
- Visconti, R.; Grieco, D. Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocr. Relat. Cancer 2017, 24, T107–T117. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Akerley, W.; Schabel, M.C.; Hong, D.S.; Uehara, C.; Chhabra, A.; Warren, T.; Mather, G.G.; Evans, B.A.; Woodland, D.P.; et al. Phase I clinical trial of MPC-6827 (Azixa), a microtubule destabilizing agent, in patients with advanced cancer. Mol. Cancer Ther. 2010, 9, 3410–3419. [Google Scholar] [CrossRef]
- Grossmann, K.F.; Colman, H.; Akerley, W.A.; Glantz, M.; Matsuoko, Y.; Beelen, A.P.; Yu, M.; De Groot, J.F.; Aiken, R.D.; Olson, J.J.; et al. Phase I trial of verubulin (MPC-6827) plus carboplatin in patients with relapsed glioblastoma multiforme. J. Neurooncol. 2012, 110, 257–264. [Google Scholar] [CrossRef]
- Fang, S.; Bi, S.; Li, Y.; Tian, S.; Xu, H.; Fu, L.; Wang, S.; Tang, Y.; Qiu, P. Design, synthesis and anti-tumor evaluation of plinabulin derivatives as potential agents targeting beta-tubulin. Bioorg Med. Chem. Lett. 2023, 91, 129370. [Google Scholar] [CrossRef]
- Han, B.; Feinstein, T.; Shi, Y.; Chen, G.; Yao, Y.; Hu, C.; Shi, J.; Feng, J.; Wu, H.; Cheng, Y.; et al. Plinabulin plus docetaxel versus docetaxel in patients with non-small-cell lung cancer after disease progression on platinum-based regimen (DUBLIN-3): A phase 3, international, multicentre, single-blind, parallel group, randomised controlled trial. Lancet Respir. Med. 2024, 12, 775–786. [Google Scholar] [CrossRef]
- Yakushiji, F.; Muguruma, K.; Hayashi, Y.; Shirasaka, T.; Kawamata, R.; Tanaka, H.; Yoshiwaka, Y.; Taguchi, A.; Takayama, K.; Hayashi, Y. Click strategy using disodium salts of amino acids improves the water solubility of plinabulin and KPU-300. Bioorg Med. Chem. 2017, 25, 3623–3630. [Google Scholar] [CrossRef]
- Chen, J.; Ahn, S.; Wang, J.; Lu, Y.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem. 2012, 55, 7285–7289. [Google Scholar] [CrossRef]
- Topham, C.; Tighe, A.; Ly, P.; Bennett, A.; Sloss, O.; Nelson, L.; Ridgway, R.A.; Huels, D.; Littler, S.; Schandl, C.; et al. MYC is a major determinant of mitotic cell fate. Cancer Cell 2015, 28, 129–140. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. Cell Syst. 2018, 6, 282–300.E2. [Google Scholar] [CrossRef]
- Jemaa, M.; Sime, W.; Abassi, Y.; Lasorsa, V.A.; Bonne Kohler, J.; Michaelis, M.; Cinatl, J., Jr.; Capasso, M.; Massoumi, R. Gene expression signature of acquired chemoresistance in neuroblastoma cells. Int. J. Mol. Sci. 2020, 21, 6811. [Google Scholar] [CrossRef]
- Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 82–106. [Google Scholar] [CrossRef]
- Van Maerken, T.; Rihani, A.; Dreidax, D.; De Clercq, S.; Yigit, N.; Marine, J.C.; Westermann, F.; De Paepe, A.; Vandesompele, J.; Speleman, F. Functional analysis of the p53 pathway in neuroblastoma cells using the small-molecule MDM2 antagonist nutlin-3. Mol. Cancer Ther. 2011, 10, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wunschel, J.; Toedling, J.; Deubzer, H.E.; Kunkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Cherney, B.; Reinhold, W.; Rucker, K.; O’Connor, P.M. Disruption of p53 function in immortalized human cells does not affect survival or apoptosis after taxol or vincristine treatment. Clin. Cancer Res. 1998, 4, 1047–1054. [Google Scholar]
- Zdioruk, M.; Want, A.; Mietelska-Porowska, A.; Laskowska-Kaszub, K.; Wojsiat, J.; Klejman, A.; Uzarowska, E.; Koza, P.; Olejniczak, S.; Pikul, S.; et al. A new inhibitor of tubulin polymerization kills multiple cancer cell types and reveals p21-mediated mechanism determining cell death after mitotic catastrophe. Cancers 2020, 12, 2161. [Google Scholar] [CrossRef]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.K.; Boos, J.; et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Frommann, K.; Appl, B.; Hundsdoerfer, P.; Reinshagen, K.; Eschenburg, G. Vincristine resistance in relapsed neuroblastoma can be efficiently overcome by Smac mimetic LCL161 treatment. J. Pediatr. Surg. 2018, 53, 2059–2064. [Google Scholar] [CrossRef]
- Rovsing, A.B.; Thomsen, E.A.; Nielsen, I.; Skov, T.W.; Luo, Y.; Dybkaer, K.; Mikkelsen, J.G. Resistance to vincristine in DLBCL by disruption of p53-induced cell cycle arrest and apoptosis mediated by KIF18b and USP28. Br. J. Haematol. 2023, 202, 825–839. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | MYCN Status | Genetic Risk Factor | 4h (GI50, nM) | 4k (GI50, nM) | Vincristine |
|---|---|---|---|---|---|
| SK-N-SH | Single copy | ALK F1174L | 1.05 ± 0.15 | 55.23 ± 10.34 | NT |
| SK-N-AS | Single copy | 11q deletion, NRASQ61K | 1.02 ± 0.28 | 98.63 ± 12.75 | 1.83 ± 0.28 |
| SH-SY5Y | Single copy | ALKF1174L | 0.95 ± 0.17 | 59.15 ± 12.23 | NT |
| SH-EP | Single copy | N/A | 12.92 ± 4.38 | 389.80 ± 75.21 | NT |
| IMR-32 | Amplified | MYCN amp | 1.57 ± 0.36 | 69.13 ± 12.10 | NT |
| Kelly | Amplified | MYCN amp, ALKF1174L | 13.90 ± 2.32 | 300.00 ± 93.40 | 3.44 ± 0.21 |
| CHP-134 | Amplified | MYCN amp | 1.72 ± 0.28 | 45.19 ± 8.71 | NT |
| VCR-SK-N-AS | Single copy | 11q deletion, NRASQ61K | 15.90 ± 2.16 | 203.00 ± 20.40 | 206.00 ± 26.80 |
| VCR-Kelly | Amplified | MYCN amp, ALKF1174L | 8.30 ± 1.12 | 233.00 ± 55.90 | 45.40 ± 5.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reed, C.N.; Garrison, K.B.; Thammathong, J.; Cinatl, J., Jr.; Michaelis, M.; Banerjee, S.; Weissmiller, A.M. Colchicine Binding Site Tubulin Inhibitors Impair Vincristine-Resistant Neuroblastoma Cell Function. Molecules 2025, 30, 2186. https://doi.org/10.3390/molecules30102186
Reed CN, Garrison KB, Thammathong J, Cinatl J Jr., Michaelis M, Banerjee S, Weissmiller AM. Colchicine Binding Site Tubulin Inhibitors Impair Vincristine-Resistant Neuroblastoma Cell Function. Molecules. 2025; 30(10):2186. https://doi.org/10.3390/molecules30102186
Chicago/Turabian StyleReed, Cinthia N., Kaylee B. Garrison, Joshua Thammathong, Jindrich Cinatl, Jr., Martin Michaelis, Souvik Banerjee, and April M. Weissmiller. 2025. "Colchicine Binding Site Tubulin Inhibitors Impair Vincristine-Resistant Neuroblastoma Cell Function" Molecules 30, no. 10: 2186. https://doi.org/10.3390/molecules30102186
APA StyleReed, C. N., Garrison, K. B., Thammathong, J., Cinatl, J., Jr., Michaelis, M., Banerjee, S., & Weissmiller, A. M. (2025). Colchicine Binding Site Tubulin Inhibitors Impair Vincristine-Resistant Neuroblastoma Cell Function. Molecules, 30(10), 2186. https://doi.org/10.3390/molecules30102186