

From Aromatic Motifs to Cluster-Assembled Materials: Silicon–Lithium Nanoclusters for Hydrogen Storage Applications

 , , , , , , , and
, , , , , , , and
Abstract

1. Introduction
2. Results and Discussion
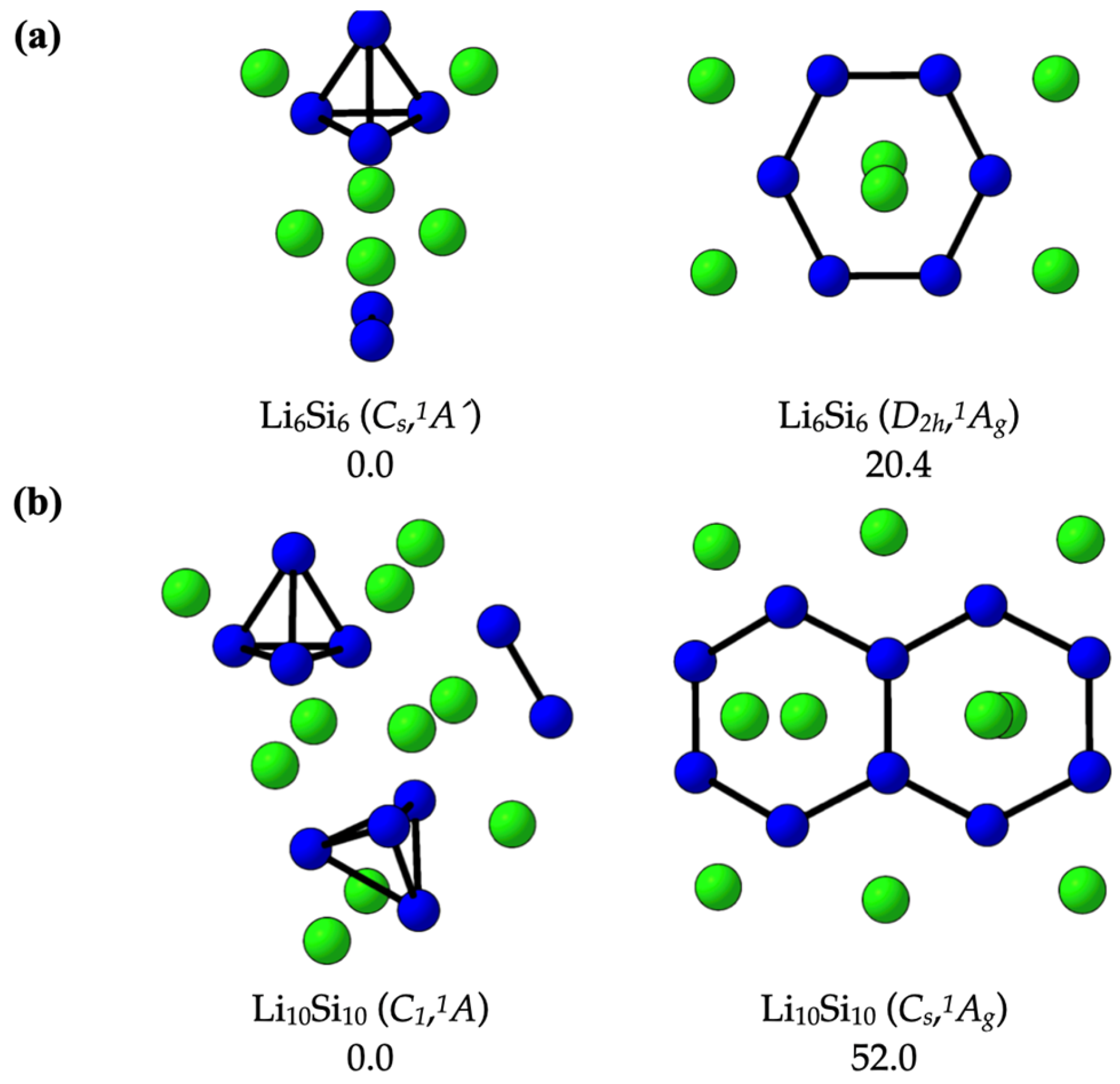
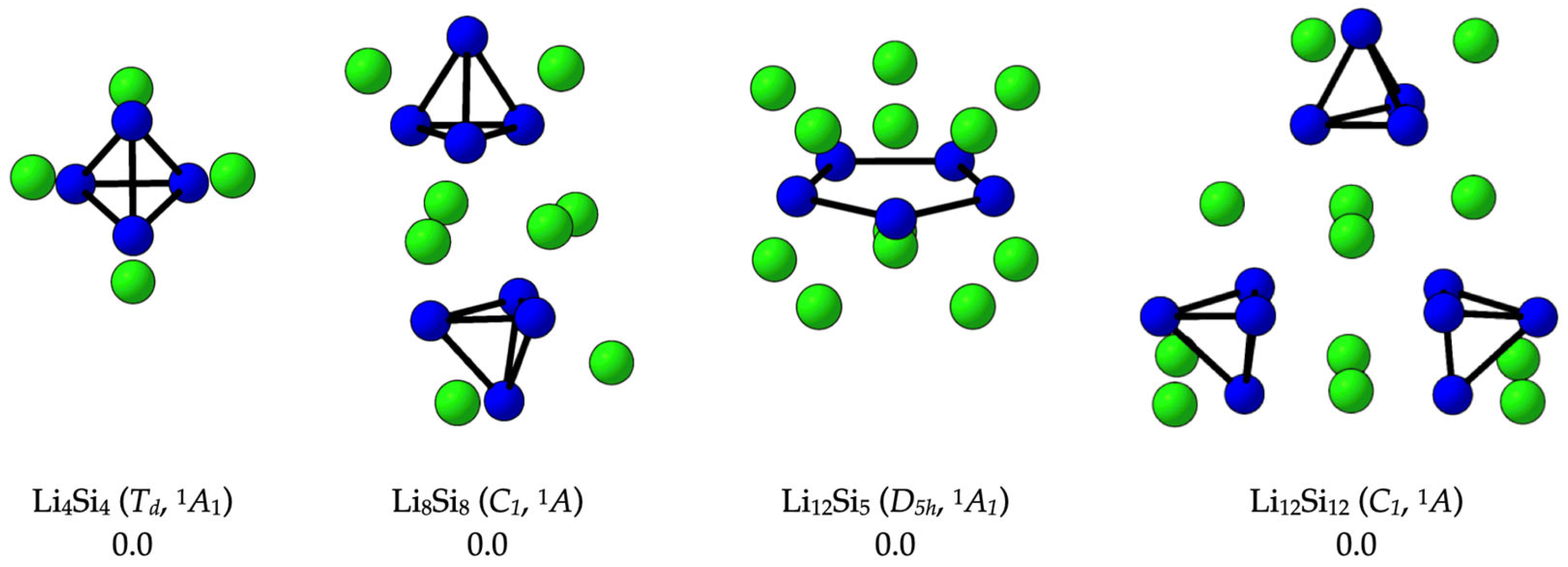
2.1. Confirming the Lowest-Energy Li-Si Structures
2.2. The Structural and Electronic Features of Bare and Hydrogen-Adsorbed Si–Li Clusters
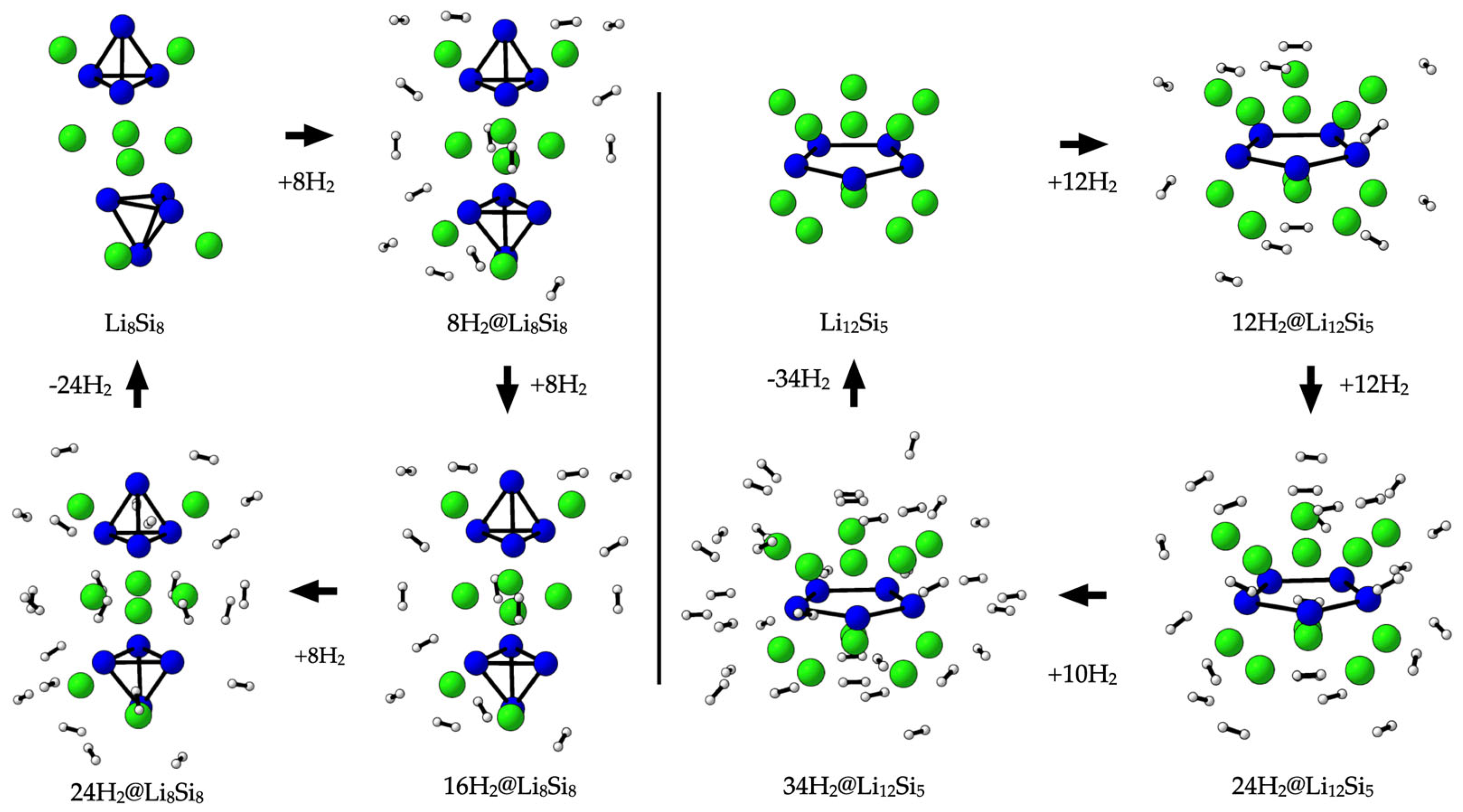
2.3. The Hydrogen Adsorption Energetics, Charge Redistribution, and Storage Capacity
2.4. Thermal Stability and Hydrogen Release Dynamics via BOMD Simulations
2.5. Visualization of Non-Covalent Interactions via IGMH Analysis
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Usman, M.R. Hydrogen storage methods: Review and current status. Renew. Sustain. Energy Rev. 2022, 167, 112743. [Google Scholar] [CrossRef]
- Kumar, N.; Lee, S.-Y.; Park, S.-J. Advancements in hydrogen storage technologies: A comprehensive review of materials, methods, and economic policy. Nano Today 2024, 56, 102302. [Google Scholar] [CrossRef]
- Bosu, S.; Rajamohan, N. Recent advancements in hydrogen storage—Comparative review on methods, operating conditions and challenges. Int. J. Hydrogen Energy 2024, 52, 352–370. [Google Scholar] [CrossRef]
- Hassan, I.A.; Ramadan, H.S.; Saleh, M.A.; Hissel, D. Hydrogen storage technologies for stationary and mobile applications: Review, analysis and perspectives. Renew. Sustain. Energy Rev. 2021, 149, 111311. [Google Scholar] [CrossRef]
- El-Adawy, M.; Dalha, I.B.; Ismael, M.A.; Al-Absi, Z.A.; Nemitallah, M.A. Review of Sustainable Hydrogen Energy Processes: Production, Storage, Transportation, and Color-Coded Classifications. Energy Fuels 2024, 38, 22686–22718. [Google Scholar] [CrossRef]
- Jesse, L.C.; Rowsell, O.M.Y.P.D. Strategies for Hydrogen Storage in Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2005, 44, 4670–4679. [Google Scholar]
- Zhou, W.; Yildirim, T. Nature and Tunability of Enhanced Hydrogen Binding in Metal−Organic Frameworks with Exposed Transition Metal Sites. J. Phys. Chem. C 2008, 112, 8132–8135. [Google Scholar] [CrossRef]
- Jana, G.; Chattaraj, P.K. Exploring advanced nanostructures and functional materials for efficient hydrogen storage: A theoretical investigation on mechanisms, adsorption process, and future directions. Front. Chem. 2025, 13, 1525140. [Google Scholar] [CrossRef]
- Alabdulhadi, R.A.; Khan, S.; Khan, A.; Alfuhaid, L.T.; Khan, M.Y.; Usman, M.; Maity, N.; Helal, A. Potential Use of Reticular Materials (MOFs, ZIFs, and COFs) for Hydrogen Storage. ACS Appl. Energy Mater. 2025, 8, 1397–1413. [Google Scholar] [CrossRef]
- Fang, W.; Ding, C.; Chen, L.; Zhou, W.; Wang, J.; Huang, K.; Zhu, R.; Wu, J.; Liu, B.; Fang, Q.; et al. Review of Hydrogen Storage Technologies and the Crucial Role of Environmentally Friendly Carriers. Energy Fuels 2024, 38, 13539–13564. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, W.; Li, D.; Ding, S.; Xiao, C.; Zeng, L. Emerging Supported Metal Atomic Clusters for Electrocatalytic Renewable Conversions. ACS Catal. 2025, 15, 2434–2458. [Google Scholar] [CrossRef]
- Islam, S.; Weerasinghe, H.; Prado, D.M.; Gonzaga, A.N.; Burda, C. Diversifying the Materials and Technologies for the Future of Energy Storage. Energy Fuels 2025, 39, 8369–8390. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Matzger, A.J. Water Stability of Microporous Coordination Polymers and the Adsorption of Pharmaceuticals from Water. Langmuir 2010, 26, 17198–17202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, D.; Lai, C.; Zeng, G.; Qin, L.; Wang, H.; Yi, H.; Li, B.; Liu, S.; Zhang, M.; et al. Recent advances in covalent organic frameworks (COFs) as a smart sensing material. Chem. Soc. Rev. 2019, 48, 5266–5302. [Google Scholar] [CrossRef]
- Klontzas, E.; Mavrandonakis, A.; Tylianakis, E.; Froudakis, G.E. Improving Hydrogen Storage Capacity of MOF by Functionalization of the Organic Linker with Lithium Atoms. Nano Lett. 2008, 8, 1572–1576. [Google Scholar] [CrossRef]
- Jena, N.K.; Srinivasu, K.; Ghosh, S.K. Computational investigation of hydrogen adsorption in silicon-lithium binary clusters. J. Chem. Sci. 2012, 124, 255–260. [Google Scholar] [CrossRef]
- Jaiswal, A.; Sahoo, R.K.; Ray, S.S.; Sahu, S. Alkali metals decorated silicon clusters (SinMn, n = 6, 10; M = Li, Na) as potential hydrogen storage materials: A DFT study. Int. J. Hydrogen Energy 2022, 47, 1775–1789. [Google Scholar] [CrossRef]
- Pan, S.; Merino, G.; Chattaraj, P.K. The hydrogen trapping potential of some Li-doped star-like clusters and super-alkali systems. Phys. Chem. Chem. Phys. 2012, 14, 10345–10350. [Google Scholar] [CrossRef]
- Guo, C.; Wang, C. Li center clusters MLi4+ (M = C, Si, Ge) for dihydrogen storage. Int. J. Hydrogen Energy 2020, 45, 24968–24979. [Google Scholar] [CrossRef]
- Lan, J.; Cao, D.; Wang, W. Li12Si60H60 Fullerene Composite: A Promising Hydrogen Storage Medium. ACS Nano 2009, 3, 3294–3300. [Google Scholar] [CrossRef]
- Manrique-de-la-Cuba, M.F.; Leyva-Parra, L.; Inostroza, D.; Gomez, B.; Vásquez-Espinal, A.; Garza, J.; Yañez, O.; Tiznado, W. Li8Si8, Li10Si9, and Li12Si10: Assemblies of Lithium-Silicon Aromatic Units. ChemPhysChem 2021, 22, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Inostroza, D.; Leyva-Parra, L.; Pino-Rios, R.; Solar-Encinas, J.; Vásquez-Espinal, A.; Pan, S.; Merino, G.; Yañez, O.; Tiznado, W. Li6E5Li6: Tetrel Sandwich Complexes with 10-π-Electrons. Angew. Chem. Int. Ed. 2024, 136, e202317848. [Google Scholar] [CrossRef]
- Yañez, O.; Garcia, V.; Garza, J.; Orellana, W.; Vásquez-Espinal, A.; Tiznado, W. (Li6Si5)2–5: The Smallest Cluster-Assembled Materials Based on Aromatic Si56− Rings. Chem. Eur. J. 2018, 25, 2467–2471. [Google Scholar] [CrossRef]
- Song, B.; Zhang, C.; He, P. Si20H20 cluster modified by small organic molecules and lithium atoms for high-capacity hydrogen storage. Int. J. Hydrogen Energy 2015, 40, 8093–8105. [Google Scholar] [CrossRef]
- Khanna, S.N.; Jena, P. Assembling crystals from clusters. Phys. Rev. Lett. 1992, 69, 1664–1667. [Google Scholar] [CrossRef]
- Jena, P.; Sun, Q. Super Atomic Clusters: Design Rules and Potential for Building Blocks of Materials. Chem. Rev. 2018, 118, 5755–5870. [Google Scholar] [CrossRef]
- Osorio, E.; Villalobos, V.; Santos, J.C.; Donald, K.J.; Merino, G.; Tiznado, W. Structure and stability of the Si4Lin (n=1–7) binary clusters. Chem. Phys. Lett. 2012, 522, 67–71. [Google Scholar] [CrossRef]
- Tiznado, W.; Perez-Peralta, N.; Islas, R.; Toro-Labbe, A.; Ugalde, J.M.; Merino, G. Designing 3-D Molecular Stars. J. Am. Chem. Soc. 2009, 131, 9426–9431. [Google Scholar] [CrossRef]
- Contreras, M.; Osorio, E.; Ferraro, F.; Puga, G.; Donald, K.J.; Harrison, J.G.; Merino, G.; Tiznado, W. Isomerization Energy Decomposition Analysis for Highly Ionic Systems: Case Study of Starlike E5Li7+ Clusters. Chem. Eur. J. 2013, 19, 2305–2310. [Google Scholar] [CrossRef]
- Nesper, R.; Curda, J.; Von Schnering, H.G. Li8MgSi6, a novel Zintl compound containing quasi-aromatic Si5 rings. J. Solid State Chem. 1986, 62, 199–206. [Google Scholar] [CrossRef]
- Kuhn, A.; Sreeraj, P.; Pöttgen, R.; Wiemhöfer, H.D.; Wilkening, M.; Heitjans, P. Li NMR Spectroscopy on Crystalline Li12Si7: Experimental Evidence for the Aromaticity of the Planar Cyclopentadienyl-Analogous Si56− Rings. Angew. Chem. Int. Ed. 2011, 50, 12099–12102. [Google Scholar] [CrossRef] [PubMed]
- Köster, T.K.J.; Salager, E.; Morris, A.J.; Key, B.; Seznec, V.; Morcrette, M.; Pickard, C.J.; Grey, C.P. Resolving the Different Silicon Clusters in Li12Si7 by 29Si and 6,7Li Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 2011, 50, 12591–12594. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.; Chen, Z.; Jiao, H. Spherical Aromaticity of Inorganic Cage Molecules. Angew. Chem. Int. Ed. 2001, 40, 2834–2838. [Google Scholar] [CrossRef]
- Doi, K.; Hino, S.; Miyaoka, H.; Ichikawa, T.; Kojima, Y. Hydrogen storage properties of lithium silicon alloy synthesized by mechanical alloying. J. Power Sources. 2011, 196, 504–507. [Google Scholar] [CrossRef]
- Yañez, O.; Báez-Grez, R.; Inostroza, D.; Rabanal-León, W.A.; Pino-Rios, R.; Garza, J.; Tiznado, W. AUTOMATON: A Program That Combines a Probabilistic Cellular Automata and a Genetic Algorithm for Global Minimum Search of Clusters and Molecules. J. Chem. Theory Comput. 2019, 15, 1463–1475. [Google Scholar] [CrossRef]
- García-Argote, W.; Ruiz, L.; Inostroza, D.; Cardenas, C.; Yañez, O.; Tiznado, W. Introducing KICK-MEP: Exploring potential energy surfaces in systems with significant non-covalent interactions. J. Mol. Model. 2024, 30, 369–382. [Google Scholar] [CrossRef]
- Yañez, O.; Báez-Grez, R.; Inostroza, D.; Pino-Rios, R.; Rabanal-León, W.A.; Contreras-García, J.; Cardenas, C.; Tiznado, W. Kick–Fukui: A Fukui Function-Guided Method for Molecular Structure Prediction. J. Chem. Inf. Model. 2021, 61, 3955–3963. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Fuentealba, P.; Von-Szentpaly, L.; Preuss, H.; Stoll, H. Pseudopotential calculations for alkaline-earth atoms. J. Phys. B Atom. Mol. Phys. 1985, 18, 1287–1296. [Google Scholar] [CrossRef]
- Grimme, S.; Jens, A.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Truhlar, D.G. Basis-set extrapolation. Chem. Phys. Lett. 1998, 294, 45–48. [Google Scholar] [CrossRef]
- Frisch, M.E.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.G.A.; Petersson, G.A.; Nakatsuji, H.J.R.A.; et al. Gaussian 16 Rev. C.01, B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mayer, I.; Surján, P.R. Improved intermolecular SCF theory and the BSSE problem. Int. J. Quantum Chem. 1989, 36, 225–240. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Hehre, W.J. Ab initio molecular orbital theory. Acc. Chem. Res. 1976, 9, 399–406. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born–Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Voth, G.A. Atom-Centered Density Matrix Propagation (ADMP): Generalizations Using Bohmian Mechanics. J. Phys. Chem. A 2003, 107, 7269–7277. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | (Å) | (Å) | (Å) | (Å) |
|---|---|---|---|---|
| H2 | - | - | - | 0.74 |
| Li4Si4 | 2.44 | 2.51 | - | - |
| 4H2@Li4Si4 | 2.44 | 2.51–2.53 | 2.10 | 0.75 |
| 8H2@Li4Si4 | 2.44 | 2.52–2.53 | 2.12 | 0.75 |
| 12H2@Li4Si4 | 2.44 | 2.54–2.55 | 2.15–2.18 | 0.75 |
| Li6Si6 (*) | 2.31–2.36 | 2.40–2.76 | - | - |
| 6H2@Li6Si6 | 2.31–2.35 | 2.41–2.76 | 2.09–2.16 | 0.75 |
| 12H2@Li6Si6 | 2.31–2.34 | 2.42–2.73 | 2.08–3.58 | 0.75 |
| 18H2@Li6Si6 | 2.31–2.34 | 2.42–2.75 | 2.09–3.47 | 0.75 |
| Li6Si6 | 2.12–2.49 | 2.52–2.68 | - | - |
| 6H2@Li6Si6 | 2.12–2.48 | 2.50–2.69 | 2.10–2.20 | 0.75 |
| 12H2@Li6Si6 | 2.12–2.47 | 2.50–2.71 | 2.11–2.42 | 0.75 |
| 18H2@Li6Si6 | 2.12–2.48 | 2.52–2.73 | 2.14–3.41 | 0.75 |
| Li8Si8 | 2.35–2.54 | 2.44–2.89 | - | - |
| 8H2@Li8Si8 | 2.36–2.52 | 2.47–2.73 | 2.09–2.26 | 0.75 |
| 16H2@Li8Si8 | 2.36–2.51 | 2.50–2.72 | 2.12–2.29 | 0.75 |
| 24H2@Li8Si8 | 2.36–2.51 | 2.49–2.71 | 2.14–3.46 | 0.75 |
| Li10Si10 (*) | 2.26–2.47 | 2.42–3.35 | - | - |
| 10H2@Li10Si10 | 2.26–2.47 | 2.42–3.23 | 2.09–2.22 | 0.75 |
| 20H2@Li10Si10 | 2.27–2.43 | 2.46–3.14 | 2.09–3.72 | 0.75 |
| 30H2@Li10Si10 | 2.27–2.43 | 2.44–3.21 | 2.08–3.78 | 0.75 |
| Li10Si10 | 2.13–2.53 | 2.47–2.85 | - | - |
| 10H2@Li10Si10 | 2.13–2.52 | 2.46–2.94 | 2.09–2.25 | 0.75 |
| 20H2@Li10Si10 | 2.13–2.51 | 2.49–2.85 | 2.12–3.55 | 0.75 |
| 30H2@Li10Si10 | 2.13–2.50 | 2.51–2.85 | 2.13–3.90 | 0.75 |
| Li12Si5 | 2.57 | 2.51–2.56 | - | - |
| 12H2@Li12Si5 | 2.46–2.57 | 2.49–2.59 | 1.93–2.17 | 0.75 |
| 22H2@Li12Si5 | 2.46–2.56 | 2.50–2.58 | 1.91–3.65 | 0.75 |
| 24H2@Li12Si5 | 2.44–2.56 | 2.50–2.60 | 2.13–3.81 | 0.75 |
| 32H2@Li12Si5 | 2.45–2.56 | 2.50–2.59 | 1.94–3.76 | 0.75 |
| 34H2@Li12Si5 | 2.46–2.55 | 2.50–2.60 | 2.00–3.50 | 0.75 |
| Li12Si12 | 2.39–2.52 | 2.47–2.71 | - | - |
| 12H2@Li12Si12 | 2.38–2.48 | 2.47–2.72 | 2.10–2.30 | 0.75 |
| 24H2@Li12Si12 | 2.37–2.52 | 2.47–2.67 | 2.11–3.20 | 0.75 |
| 36H2@Li12Si12 | 2.36–2.52 | 2.49–2.67 | 2.13–3.47 | 0.75 |
| System | EHOMO | ELUMO | ΔEH-L |
|---|---|---|---|
| Li4Si4 | −4.4 | −1.3 | 3.1 |
| 4H2@Li4Si4 | −4.3 | −1.0 | 3.3 |
| 8H2@Li4Si4 | −4.3 | −0.9 | 3.4 |
| 12H2@Li4Si4 | −4.2 | −1.0 | 3.2 |
| Li6Si6 (*) | −3.6 | −1.4 | 2.2 |
| 6H2@Li6Si6 | −3.6 | −1.4 | 2.2 |
| 12H2@Li6Si6 | −3.5 | −1.2 | 2.3 |
| 18H2@Li6Si6 | −3.6 | −1.3 | 2.3 |
| Li6Si6 | −4.6 | −1.8 | 2.8 |
| 6H2@Li6Si6 | −4.5 | −1.6 | 2.9 |
| 12H2@Li6Si6 | −4.4 | −1.5 | 2.9 |
| 18H2@Li6Si6 | −4.6 | −1.8 | 2.8 |
| Li8Si8 | −4.4 | −1.7 | 2.7 |
| 8H2@Li8Si8 | −4.4 | −1.3 | 3.1 |
| 16H2@Li8Si8 | −4.3 | −1.2 | 3.1 |
| 24H2@Li8Si8 | −3.4 | −1.3 | 2.1 |
| Li10Si10 (*) | −3.3 | −1.5 | 1.8 |
| 10H2@Li10Si10 | −3.2 | −1.4 | 1.8 |
| 20H2@Li10Si10 | −3.2 | −1.4 | 1.8 |
| 30H2@Li10Si10 | −3.2 | −1.4 | 1.8 |
| Li10Si10 | −4.3 | −1.7 | 2.6 |
| 10H2@Li10Si10 | −4.2 | −1.9 | 2.2 |
| 20H2@Li10Si10 | −4.1 | −1.2 | 2.9 |
| 30H2@Li10Si10 | −4.1 | −1.2 | 2.9 |
| Li12Si5 | −3.0 | −1.7 | 1.3 |
| 12H2@Li12Si5 | −2.8 | −1.2 | 1.6 |
| 22H2@Li12Si5 | −2.7 | −1.1 | 1.6 |
| 24H2@Li12Si5 | −2.7 | −1.1 | 1.6 |
| 32H2@Li12Si5 | −2.8 | −1.1 | 1.7 |
| 34H2@Li12Si5 | −2.9 | −1.2 | 1.7 |
| Li12Si12 | −4.0 | −1.6 | 2.4 |
| 12H2@Li12Si12 | −3.9 | −1.3 | 2.6 |
| 24H2@Li12Si12 | −3.9 | −1.3 | 2.6 |
| 36H2@Li12Si12 | −3.9 | −1.3 | 2.6 |
| System | Q (Li) | (eV) | (eV) | |
|---|---|---|---|---|
| Li4Si4 | 0.86 | - | - | - |
| 4H2@Li4Si4 | 0.84 | −0.12 | −0.12 | 5.44 |
| 8H2@Li4Si4 | 0.82 | −0.12 | −0.12 | 10.32 |
| 12H2@Li4Si4 | 0.81 | −0.11 | −0.12 | 14.72 |
| Li6Si6 (*) | 0.83–0.84 | - | - | - |
| 6H2@Li6Si6 | 0.81–0.85 | −0.13 | −0.14 | 5.44 |
| 12H2@Li6Si6 | 0.81–0.82 | −0.13 | −0.13 | 10.30 |
| 18H2@Li6Si6 | 0.82–0.83 | −0.11 | −0.11 | 14.72 |
| Li6Si6 | 0.70–0.87 | - | - | - |
| 6H2@Li6Si6 | 0.70–0.84 | −0.12 | −0.13 | 5.44 |
| 12H2@Li6Si6 | 0.72–0.82 | −0.11 | −0.12 | 10.30 |
| 18H2@Li6Si6 | 0.72–0.81 | −0.10 | −0.11 | 14.72 |
| Li8Si8 | 0.71–0.88 | - | - | - |
| 8H2@Li8Si8 | 0.72–0.85 | −0.12 | −0.13 | 5.44 |
| 16H2@Li8Si8 | 0.73–0.81 | −0.11 | −0.12 | 10.32 |
| 24H2@Li8Si8 | 0.77–0.81 | −0.10 | −0.11 | 14.72 |
| Li10Si10 (*) | 0.74–0.87 | - | - | - |
| 10H2@Li10Si10 | 0.75–0.85 | −0.14 | −0.14 | 5.44 |
| 20H2@Li10Si10 | 0.75–0.83 | −0.13 | −0.13 | 10.32 |
| 30H2@Li10Si10 | 0.76–0.83 | −0.11 | −0.12 | 14.72 |
| Li10Si10 | 0.71–0.89 | - | - | - |
| 10H2@Li10Si10 | 0.72–0.85 | −0.12 | −0.13 | 5.44 |
| 20H2@Li10Si10 | 0.73–0.83 | −0.11 | −0.12 | 10.32 |
| 30H2@Li10Si10 | 0.74–0.82 | −0.10 | −0.10 | 14.72 |
| Li12Si5 | 0.30–0.78 | - | - | - |
| 12H2@Li12Si5 | 0.63–0.84 | −0.16 | −0.17 | 9.76 |
| 22H2@Li12Si5 | 0.60–0.82 | −0.13 | −0.14 | 16.54 |
| 24H2@Li12Si5 | 0.59–0.81 | −0.14 | −0.14 | 17.78 |
| 32H2@Li12Si5 | 0.60–0.82 | −0.12 | −0.13 | 22.38 |
| 34H2@Li12Si5 | 0.63–0.82 | −0.11 | −0.12 | 23.45 |
| Li12Si12 | 0.75–0.89 | |||
| 12H2@Li12Si12 | 0.76–0.85 | −0.11 | −0.12 | 5.44% |
| 24H2@Li12Si12 | 0.77–0.84 | −0.11 | −0.12 | 10.32% |
| 36H2@Li12Si12 | 0.78–0.83 | −0.11 | −0.11 | 14.72% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Argote, W.; Medel, E.; Inostroza, D.; Vásquez-Espinal, A.; Solar-Encinas, J.; Leyva-Parra, L.; Ruiz, L.M.; Yañez, O.; Tiznado, W. From Aromatic Motifs to Cluster-Assembled Materials: Silicon–Lithium Nanoclusters for Hydrogen Storage Applications. Molecules 2025, 30, 2163. https://doi.org/10.3390/molecules30102163
García-Argote W, Medel E, Inostroza D, Vásquez-Espinal A, Solar-Encinas J, Leyva-Parra L, Ruiz LM, Yañez O, Tiznado W. From Aromatic Motifs to Cluster-Assembled Materials: Silicon–Lithium Nanoclusters for Hydrogen Storage Applications. Molecules. 2025; 30(10):2163. https://doi.org/10.3390/molecules30102163
Chicago/Turabian StyleGarcía-Argote, Williams, Erika Medel, Diego Inostroza, Alejandro Vásquez-Espinal, José Solar-Encinas, Luis Leyva-Parra, Lina María Ruiz, Osvaldo Yañez, and William Tiznado. 2025. "From Aromatic Motifs to Cluster-Assembled Materials: Silicon–Lithium Nanoclusters for Hydrogen Storage Applications" Molecules 30, no. 10: 2163. https://doi.org/10.3390/molecules30102163
APA StyleGarcía-Argote, W., Medel, E., Inostroza, D., Vásquez-Espinal, A., Solar-Encinas, J., Leyva-Parra, L., Ruiz, L. M., Yañez, O., & Tiznado, W. (2025). From Aromatic Motifs to Cluster-Assembled Materials: Silicon–Lithium Nanoclusters for Hydrogen Storage Applications. Molecules, 30(10), 2163. https://doi.org/10.3390/molecules30102163