
CO2 Reforming of Methane over Ru Supported Catalysts Under Mild Conditions

 , ,
, ,  ,
,  and
and 
Abstract

1. Introduction
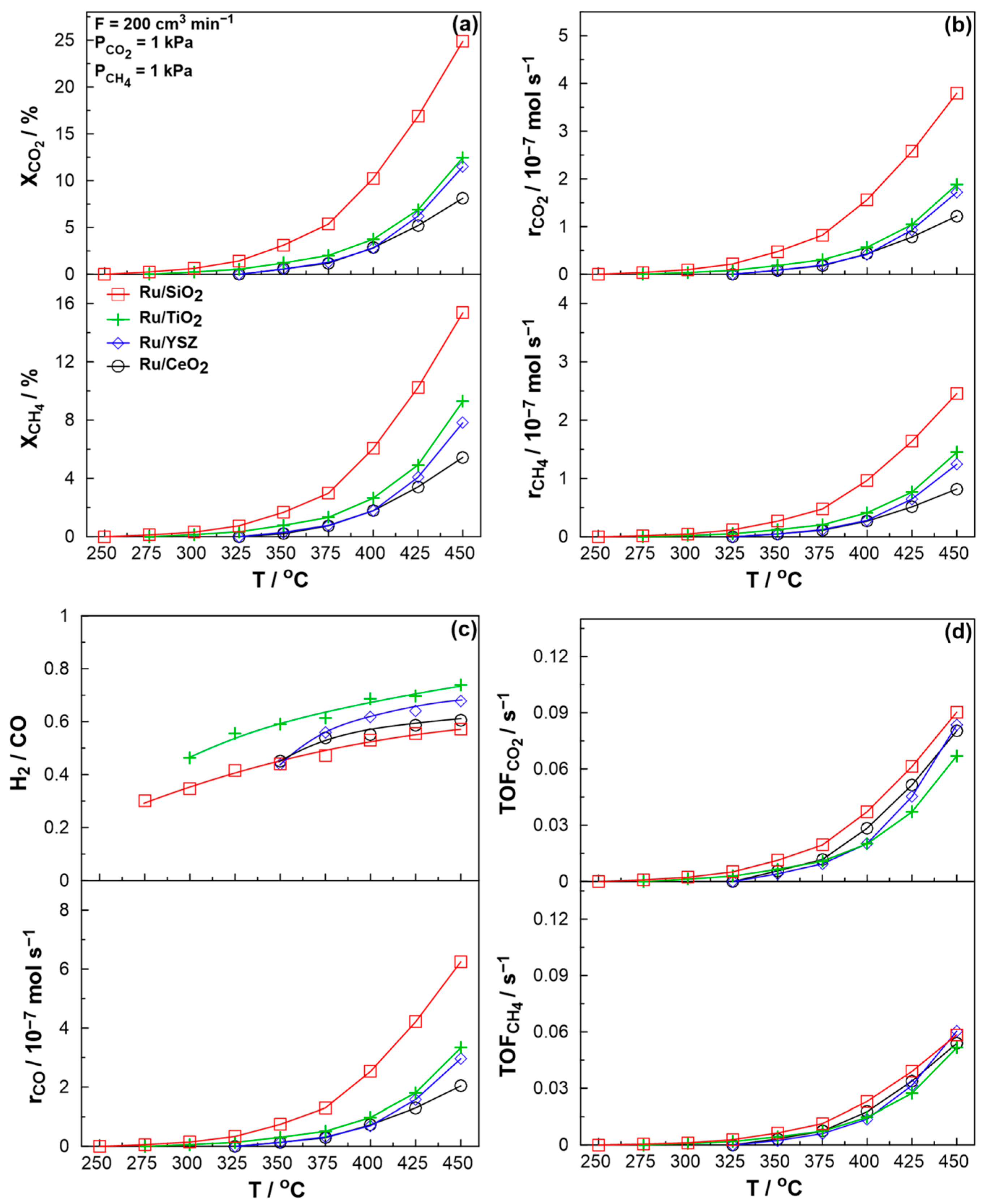
2. Results and Discussion
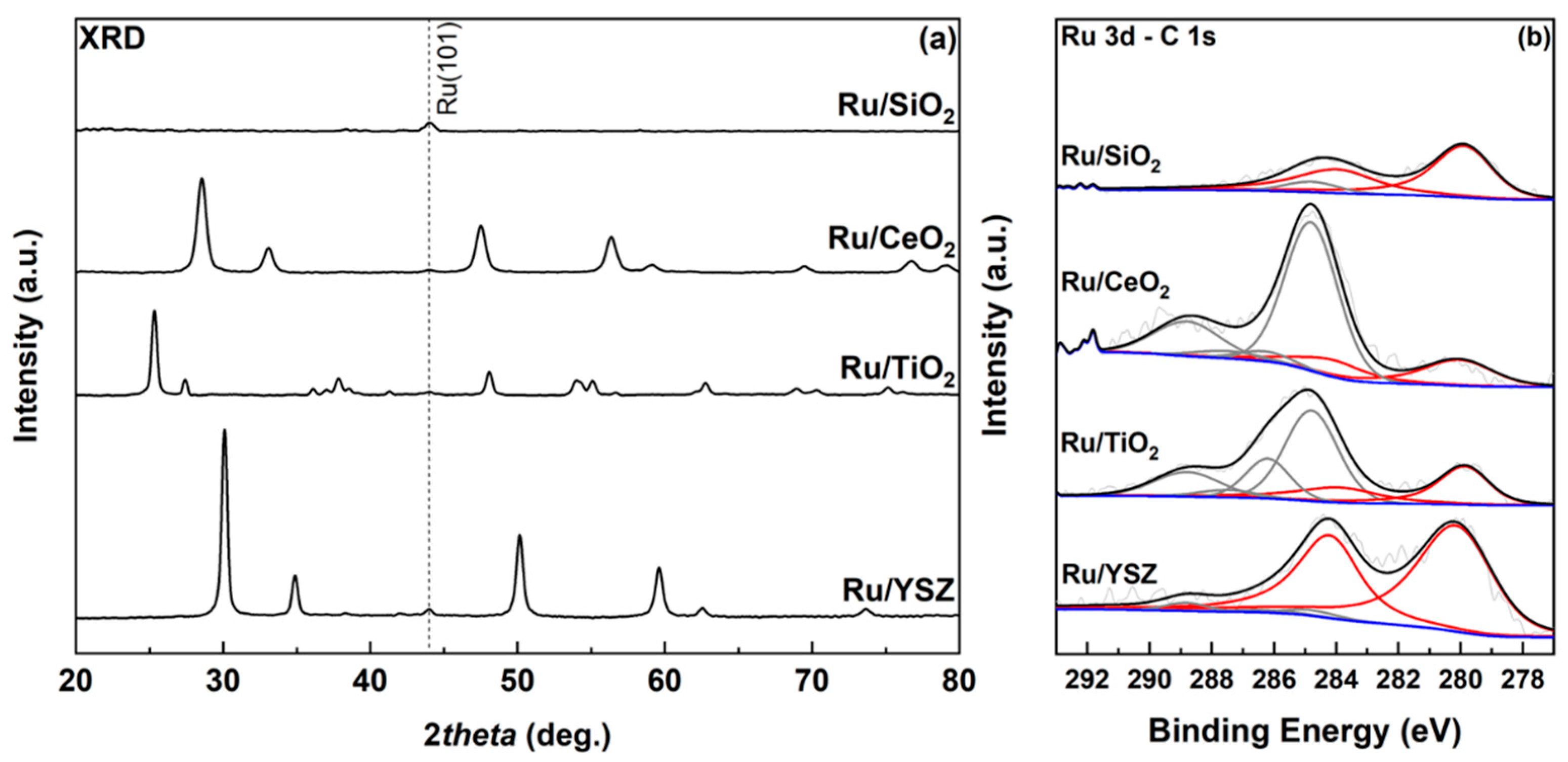
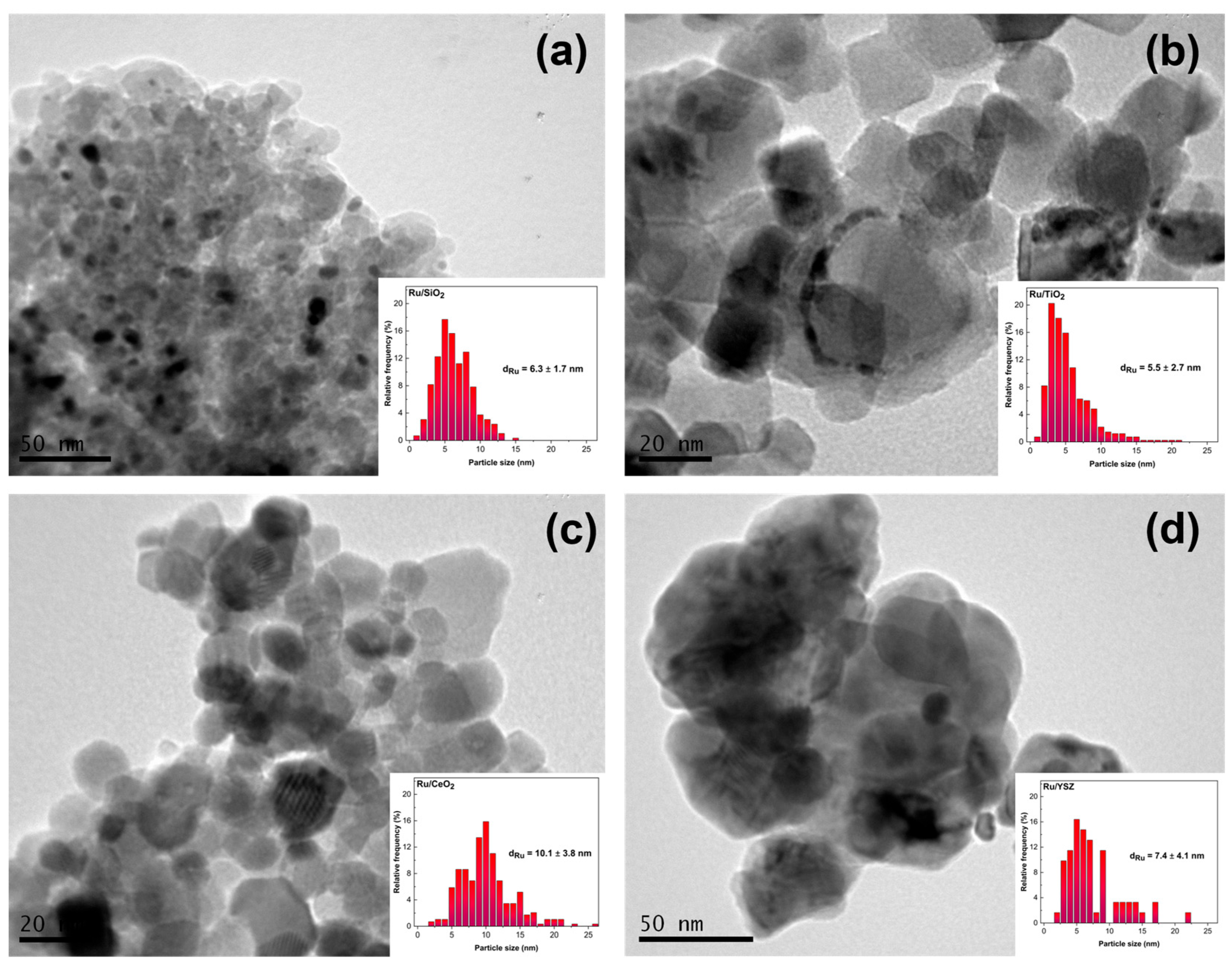
2.1. Physicochemical Characterization
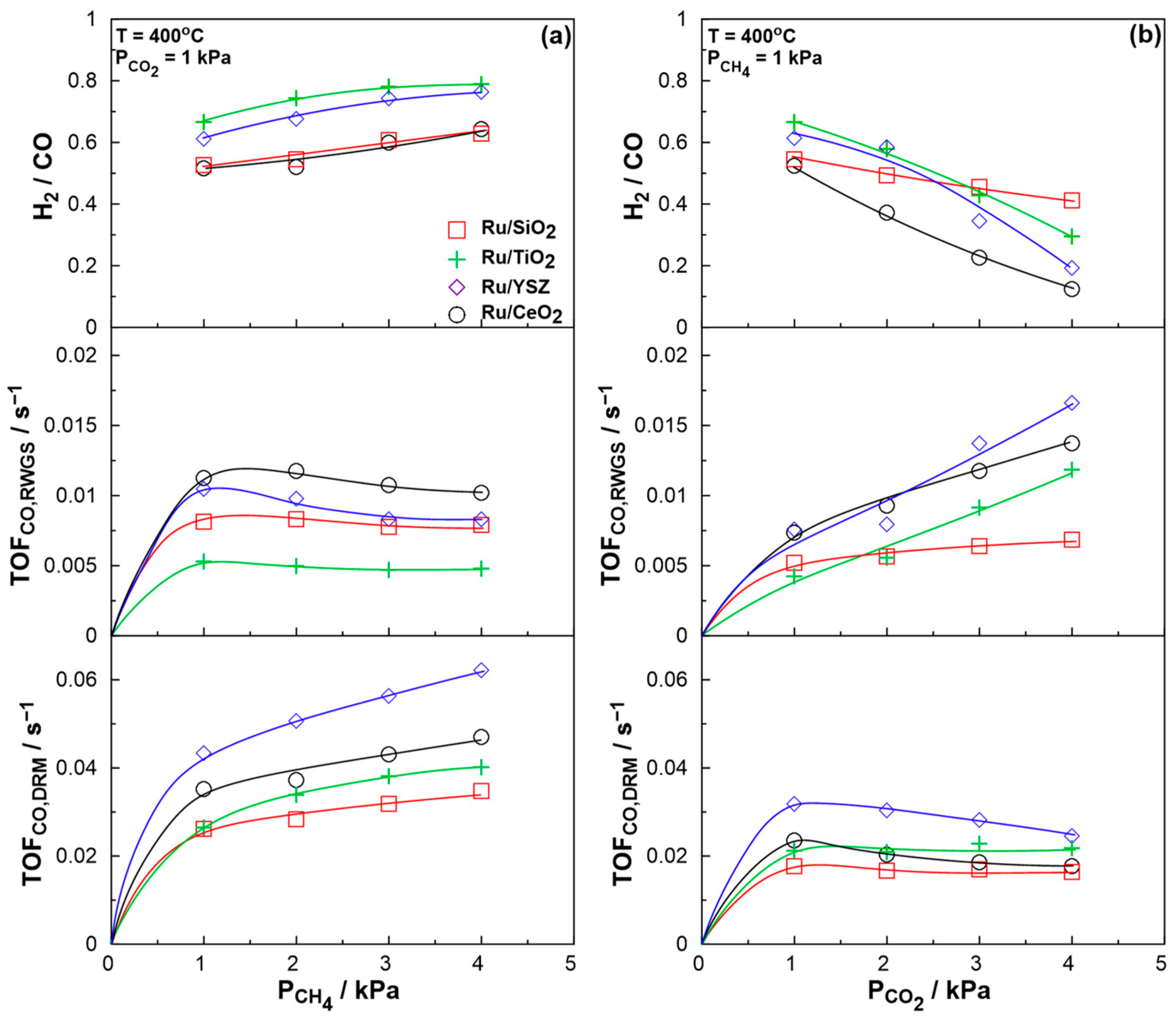
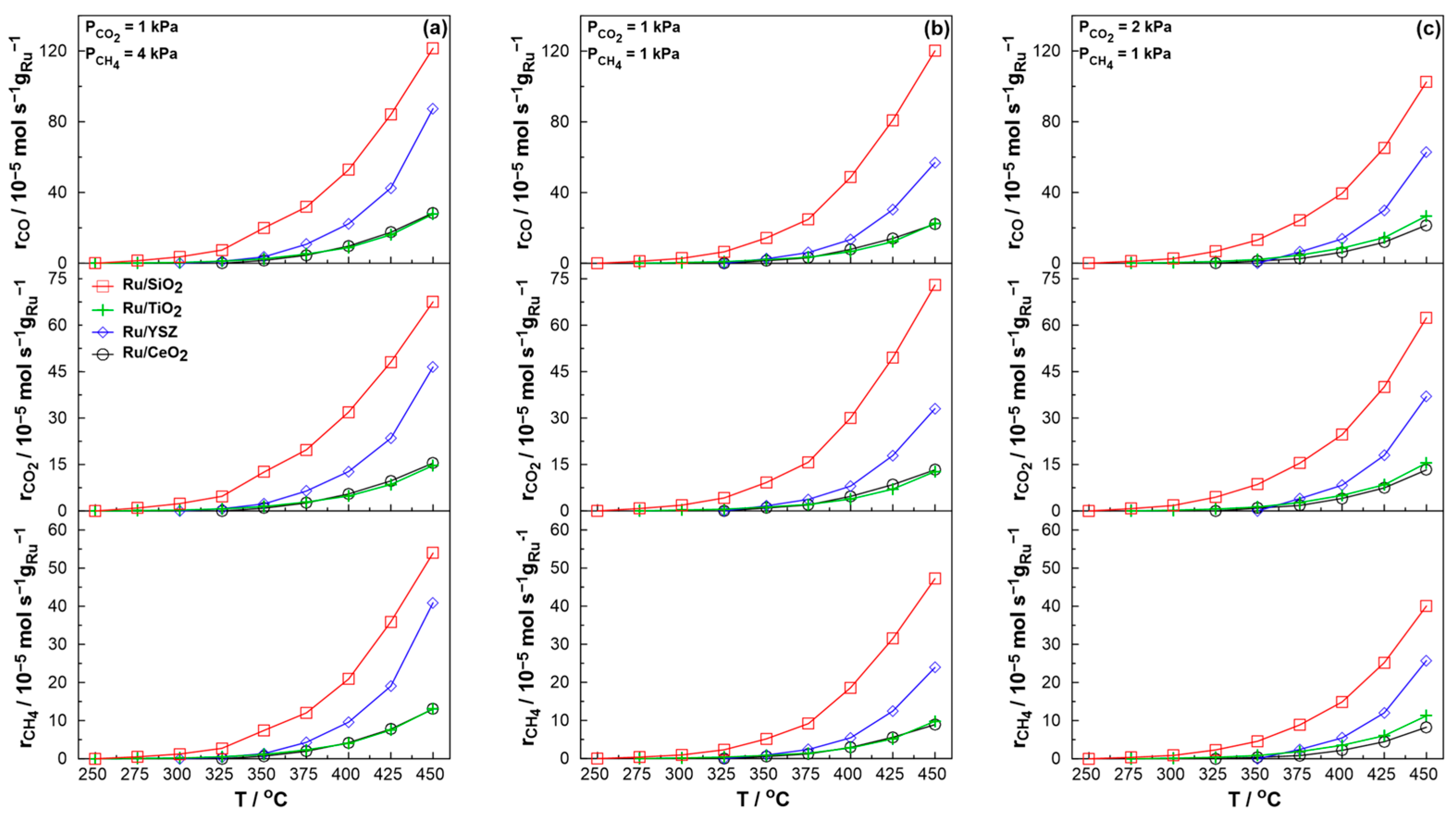
2.2. Catalytic Performance
3. Experimental
3.1. Catalyst Preparation
3.2. Physicochemical Characterization
3.3. Catalytic Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Paris Agreement, UNFCCC. Available online: https://unfccc.int/process-and-meetings/the-paris-agreement (accessed on 21 January 2025).
- WMO Greenhouse Gas Bulletin, No. 20, WMO. Available online: https://wmo.int/publication-series/wmo-greenhouse-gas-bulletin-no-20 (accessed on 21 January 2025).
- Chatzilias, C.; Martino, E.; Bikogiannakis, A.K.; Kyriakou, G.; Katsaounis, A. Unraveling the Role of EPOC during the Enhancement of RWGS Reaction in a Pt/YSZ/Au Single Chamber Reactor. J. CO2 Util. 2024, 90, 102980. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Zhang, Y.; He, Y. Chapter Seven—Biogas Upgrading. In Advances in Bioenergy; Li, Y., Khanal, S.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 5, pp. 309–344. [Google Scholar]
- Jung, S.; Lee, J.; Moon, D.H.; Kim, K.-H.; Kwon, E.E. Upgrading Biogas into Syngas through Dry Reforming. Renew. Sustain. Energy Rev. 2021, 143, 110949. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Papadam, T.; Goula, G. Electricity Production from Wastewater Treatment via a Novel Biogas-SOFC Aided Process. Solid State Ion. 2008, 179, 1521–1525. [Google Scholar] [CrossRef]
- Zhai, P.; Li, Y.; Wang, M.; Liu, J.; Cao, Z.; Zhang, J.; Xu, Y.; Liu, X.; Li, Y.-W.; Zhu, Q.; et al. Development of Direct Conversion of Syngas to Unsaturated Hydrocarbons Based on Fischer-Tropsch Route. Chem 2021, 7, 3027–3051. [Google Scholar] [CrossRef]
- Liu, G.; Yang, G.; Peng, X.; Wu, J.; Tsubaki, N. Recent Advances in the Routes and Catalysts for Ethanol Synthesis from Syngas. Chem. Soc. Rev. 2022, 51, 5606–5659. [Google Scholar] [CrossRef]
- Cheng, K.; Kang, J.; King, D.L.; Subramanian, V.; Zhou, C.; Zhang, Q.; Wang, Y. Chapter Three—Advances in Catalysis for Syngas Conversion to Hydrocarbons. In Advances in Catalysis; Song, C., Ed.; Academic Press: San Diego, CA, USA, 2017; Volume 60, pp. 125–208. [Google Scholar]
- An, Y.; Lin, T.; Yu, F.; Yang, Y.; Zhong, L.; Wu, M.; Sun, Y. Advances in Direct Production of Value-Added Chemicals via Syngas Conversion. Sci. China Chem. 2017, 60, 887–903. [Google Scholar] [CrossRef]
- Jang, W.J.; Shim, J.O.; Kim, H.M.; Yoo, S.Y.; Roh, H.S. A Review on Dry Reforming of Methane in Aspect of Catalytic Properties. Catal. Today 2019, 311, 15–26. [Google Scholar] [CrossRef]
- Androulakis, A.; Yentekakis, I.V.; Panagiotopoulou, P. Dry Reforming of Methane over Supported Rh and Ru Catalysts: Effect of the Support (Al2O3, TiO2, ZrO2, YSZ) on the Activity and Reaction Pathway. Int. J. Hydrogen Energy 2023, 48, 33886–33902. [Google Scholar] [CrossRef]
- Schrenk, F.; Lindenthal, L.; Drexler, H.; Urban, G.; Rameshan, R.; Summerer, H.; Berger, T.; Ruh, T.; Opitz, A.K.; Rameshan, C. Impact of Nanoparticle Exsolution on Dry Reforming of Methane: Improving Catalytic Activity by Reductive Pre-Treatment of Perovskite-Type Catalysts. Appl. Catal. B Environ. 2022, 318, 121886. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, L.; Zheng, X.; Liu, W.; Cao, Z.; Peng, H. Coke-Resistance over Rh–Ni Bimetallic Catalyst for Low Temperature Dry Reforming of Methane. Int. J. Hydrogen Energy 2023, 48, 13890–13901. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An Overview on Dry Reforming of Methane: Strategies to Reduce Carbonaceous Deactivation of Catalysts. RSC Adv. 2016, 6, 108668–108688. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst Design for Dry Reforming of Methane: Analysis Review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Sharifianjazi, F.; Esmaeilkhanian, A.; Bazli, L.; Eskandarinezhad, S.; Khaksar, S.; Shafiee, P.; Yusuf, M.; Abdullah, B.; Salahshour, P.; Sadeghi, F. A Review on Recent Advances in Dry Reforming of Methane over Ni- and Co-Based Nanocatalysts. Int. J. Hydrogen Energy 2022, 47, 42213–42233. [Google Scholar] [CrossRef]
- Bian, Z.; Zhong, W.; Yu, Y.; Wang, Z.; Jiang, B.; Kawi, S. Dry Reforming of Methane on Ni/Mesoporous-Al2O3 Catalysts: Effect of Calcination Temperature. Int. J. Hydrogen Energy 2021, 46, 31041–31053. [Google Scholar] [CrossRef]
- Damaskinos, C.M.; Zavašnik, J.; Djinović, P.; Efstathiou, A.M. Dry Reforming of Methane over Ni/Ce0.8Ti0.2O2-δ: The Effect of Ni Particle Size on the Carbon Pathways Studied by Transient and Isotopic Techniques. Appl. Catal. B Environ. 2021, 296, 120321. [Google Scholar] [CrossRef]
- Pham, C.Q.; Cao, A.N.T.; Phuong, P.T.T.; Hoang Pham, L.K.; Vi Tran, T.T.; Trinh, T.H.; Vo, D.-V.N.; Bui, T.P.T.; Nguyen, T.M. Enhancement of Syngas Production from Dry Reforming of Methane over Co/Al2O3 Catalyst: Insight into the Promotional Effects of Europium and Neodymium. J. Energy Inst. 2022, 105, 314–322. [Google Scholar] [CrossRef]
- Chen, S.; Zaffran, J.; Yang, B. Dry Reforming of Methane over the Cobalt Catalyst: Theoretical Insights into the Reaction Kinetics and Mechanism for Catalyst Deactivation. Appl. Catal. B Environ. 2020, 270, 118859. [Google Scholar] [CrossRef]
- Rezaei, M.; Alavi, S.M.; Sahebdelfar, S.; Yan, Z.-F. Syngas Production by Methane Reforming with Carbon Dioxide on Noble Metal Catalysts. J. Nat. Gas Chem. 2006, 15, 327–334. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Rodríguez-Ramos, I.; Anderson, J.A.; Guerrero-Ruiz, A. Mechanistic Aspects of the Dry Reforming of Methane over Ruthenium Catalysts. Appl. Catal. Gen. 2000, 202, 183–196. [Google Scholar] [CrossRef]
- Qu, P.-F.; Wang, G.-C. Theoretical Insight into the Strong Size-Dependence of Dry Reforming of Methane over Ru/CeO2. J. CO2 Util. 2022, 65, 102221. [Google Scholar] [CrossRef]
- Derk, A.R.; Moore, G.M.; Sharma, S.; McFarland, E.W.; Metiu, H. Catalytic Dry Reforming of Methane on Ruthenium-Doped Ceria and Ruthenium Supported on Ceria. Top. Catal. 2014, 57, 118–124. [Google Scholar] [CrossRef]
- Yang, J.; Wang, J.; Zhao, J.; Bai, Y.; Du, H.; Wang, Q.; Jiang, B.; Li, H. CO2 Conversion via Dry Reforming of Methane on a Core-Shell Ru@SiO2 Catalyst. J. CO2 Util. 2022, 57, 101893. [Google Scholar] [CrossRef]
- Nikolaraki, E.; Goula, G.; Panagiotopoulou, P.; Taylor, M.J.; Kousi, K.; Kyriakou, G.; Kondarides, D.I.; Lambert, R.M.; Yentekakis, I.V. Support Induced Effects on the Ir Nanoparticles Activity, Selectivity and Stability Performance under CO2 Reforming of Methane. Nanomaterials 2021, 11, 2880. [Google Scholar] [CrossRef] [PubMed]
- Gangarajula, Y.; Hong, F.; Li, Q.; Jiang, X.; Liu, W.; Akri, M.; Su, Y.; Zhang, Y.; Li, L.; Qiao, B. Operando Induced Strong Metal-Support Interaction of Rh/CeO2 Catalyst in Dry Reforming of Methane. Appl. Catal. B Environ. 2024, 343, 123503. [Google Scholar] [CrossRef]
- Shen, D.; Li, Z.; Shan, J.; Yu, G.; Wang, X.; Zhang, Y.; Liu, C.; Lyu, S.; Li, J.; Li, L. Synergistic Pt-CeO2 Interface Boosting Low Temperature Dry Reforming of Methane. Appl. Catal. B Environ. 2022, 318, 121809. [Google Scholar] [CrossRef]
- Colombo, R.; Moroni, G.; Negri, C.; Delen, G.; Monai, M.; Donazzi, A.; Weckhuysen, B.M.; Maestri, M. Surface Carbon Formation and Its Impact on Methane Dry Reforming Kinetics on Rhodium-Based Catalysts by Operando Raman Spectroscopy. Angew. Chem. Int. Ed. 2024, 63, e202408668. [Google Scholar] [CrossRef]
- Akpan, E.; Sun, Y.; Kumar, P.; Ibrahim, H.; Aboudheir, A.; Idem, R. Kinetics, Experimental and Reactor Modeling Studies of the Carbon Dioxide Reforming of Methane (CDRM) over a New Ni/CeO2-ZrO2 Catalyst in a Packed Bed Tubular Reactor. Chem. Eng. Sci. 2007, 62, 4012–4024. [Google Scholar] [CrossRef]
- Wittich, K.; Krämer, M.; Bottke, N.; Schunk, S.A. Catalytic Dry Reforming of Methane: Insights from Model Systems. ChemCatChem 2020, 12, 2130–2147. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Verykios, X.E. Carbon Dioxide Reforming of Methane to Synthesis Gas over Supported Ni Catalysts. Catal. Today 1994, 21, 589–595. [Google Scholar] [CrossRef]
- Zhu, Y.-A.; Chen, D.; Zhou, X.-G.; Yuan, W.-K. DFT Studies of Dry Reforming of Methane on Ni Catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Isotopic and Kinetic Assessment of the Mechanism of Reactions of CH4 with CO2 or H2O to Form Synthesis Gas and Carbon on Nickel Catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Mechanism and Site Requirements for Activation and Chemical Conversion of Methane on Supported Pt Clusters and Turnover Rate Comparisons among Noble Metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Becerra, A.M.; Iriarte, M.E.; Castro Luna, A.E. Catalytic Activity of a Nickel on Alumina Catalyst in the CO2 Reforming of Methane. React. Kinet. Catal. Lett. 2003, 79, 119–125. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Matralis, H.; Verykios, X. Utilization of Biogas as a Renewable Carbon Source: Dry Reforming of Methane. In Catalysis for Alternative Energy Generation; Guczi, L., Erdôhelyi, A., Eds.; Springer: New York, NY, USA, 2012; pp. 57–127. [Google Scholar]
- Tee, S.Y.; Lee, C.J.J.; Dinachali, S.S.; Lai, S.C.; Williams, E.L.; Luo, H.-K.; Chi, D.; Hor, T.S.A.; Han, M.-Y. Amorphous Ruthenium Nanoparticles for Enhanced Electrochemical Water Splitting. Nanotechnology 2015, 26, 415401. [Google Scholar] [CrossRef]
- Sayyed, S.A.A.R.; Beedri, N.I.; Kadam, V.S.; Pathan, H.M. Rose Bengal-Sensitized Nanocrystalline Ceria Photoanode for Dye-Sensitized Solar Cell Application. Bull. Mater. Sci. 2016, 39, 1381–1387. [Google Scholar] [CrossRef]
- Tang, M.; Xia, Y.; Yang, D.; Liu, J.; Zhu, X.; Tang, R. Effects of Hydrothermal Time on Structure and Photocatalytic Property of Titanium Dioxide for Degradation of Rhodamine B and Tetracycline Hydrochloride. Materials 2021, 14, 5674. [Google Scholar] [CrossRef]
- Sarjono; Riyanto, S.; Mutiara, E.; Yusnitha, E.; Yulianto, T.; Langenati, R.; Sumaryanto, A. R&D on Surrogate Kernel Fabrication in Support of Reaktor Daya Eksperimental (RDE) Project. J. Phys. Conf. Ser. 2021, 2048, 012011. [Google Scholar] [CrossRef]
- Maridurai, T.; Balaji, D.; Sagadevan, S. Synthesis and Characterization of Yttrium Stabilized Zirconia Nanoparticles. Mater. Res. 2016, 19, 812–816. [Google Scholar] [CrossRef]
- Pinder, J.W.; Major, G.H.; Baer, D.R.; Terry, J.; Whitten, J.E.; Čechal, J.; Crossman, J.D.; Lizarbe, A.J.; Jafari, S.; Easton, C.D.; et al. Avoiding Common Errors in X-Ray Photoelectron Spectroscopy Data Collection and Analysis, and Properly Reporting Instrument Parameters. Appl. Surf. Sci. Adv. 2024, 19, 100534. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving Ruthenium: XPS Studies of Common Ruthenium Materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Sexton, B.A.; Hughes, A.E.; Foger, K. XPS Investigation of Strong Metal-Support Interactions on Group IIIa–Va Oxides. J. Catal. 1982, 77, 85–93. [Google Scholar] [CrossRef]
- Chen, J.Z.; Gao, J.; Probus, P.R.; Liu, W.; Wu, X.; Wegener, E.C.; Kropf, A.J.; Zemlyanov, D.; Zhang, G.; Yang, X.; et al. The Effect of Strong Metal–Support Interaction (SMSI) on Pt–Ti/SiO2 and Pt–Nb/SiO2 Catalysts for Propane Dehydrogenation. Catal. Sci. Technol. 2020, 10, 5973–5982. [Google Scholar] [CrossRef]
- Kyriakou, G.; Davis, D.J.; Grant, R.B.; Watson, D.J.; Keen, A.; Tikhov, M.S.; Lambert, R.M. Electron Impact-Assisted Carbon Film Growth on Ru(0001): Implications for Next-Generation EUV Lithography. J. Phys. Chem. C 2007, 111, 4491–4494. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.; Fang, D. A Review on C1s XPS-Spectra for Some Kinds of Carbon Materials. Fuller. Nanotub. Carbon. Nanostructures 2020, 28, 1048–1058. [Google Scholar] [CrossRef]
- Tsoukala, N.; Papadopoulos, A.-A.; Premeti, V.; Bikogiannakis, A.K.; Martino, E.; Amoiridis, A.; Kordouli, E.; Govatsi, K.; Manariotis, I.D.; Kyriakou, G.; et al. Biochar Made from Luffa Cylindrica and Applied as a Bifunctional Electrocatalyst in Zn–Air Batteries. RSC Adv. 2024, 14, 38924–38933. [Google Scholar] [CrossRef]
- Tzevelekidis, P.; Theodosiou, M.; Papadopoulou, A.; Sakellis, E.; Boukos, N.; Bikogiannakis, A.K.; Kyriakou, G.; Efthimiadou, E.K.; Mitsopoulou, C.A. Visible-Light-Activated Antibacterial and Antipollutant Properties of Biocompatible Cu-Doped and Ag-Decorated TiO2 Nanoparticles. Heliyon 2024, 10, e35634. [Google Scholar] [CrossRef]
- Larina, T.V.; Dovlitova, L.S.; Kaichev, V.V.; Malakhov, V.V.; Glazneva, T.S.; Paukshtis, E.A.; Bal’zhinimaev, B.S. Influence of the Surface Layer of Hydrated Silicon on the Stabilization of Co2+ Cations in Zr–Si Fiberglass Materials According to XPS, UV-Vis DRS, and Differential Dissolution Phase Analysis. RSC Adv. 2015, 5, 79898–79905. [Google Scholar] [CrossRef]
- Morgan, D.J. Photoelectron Spectroscopy of Ceria: Reduction, Quantification and the Myth of the Vacancy Peak in XPS Analysis. Surf. Interface Anal. 2023, 55, 845–850. [Google Scholar] [CrossRef]
- Bumajdad, A.; Nazeer, A.A.; Al Sagheer, F.; Nahar, S.; Zaki, M.I. Controlled Synthesis of ZrO2 Nanoparticles with Tailored Size, Morphology and Crystal Phases via Organic/Inorganic Hybrid Films. Sci. Rep. 2018, 8, 3695. [Google Scholar] [CrossRef]
- Liu, M.; Liu, W.; Liu, X.; Ouyang, Y.; Hou, H.; Lei, M.; Wei, Z. Yttrium Oxide as a Q-Switcher for the near-Infrared Erbium-Doped Fiber Laser. Nanophotonics 2020, 9, 2887–2894. [Google Scholar] [CrossRef]
- Park, I.-S.; Jung, Y.C.; Seong, S.; Ahn, J.; Kang, J.; Noh, W.; Lansalot-Matras, C. Atomic Layer Deposition of Y2O3 Films Using Heteroleptic Liquid (iPrCp)2Y(iPr-Amd) Precursor. J. Mater. Chem. C 2014, 2, 9240–9247. [Google Scholar] [CrossRef]
- Tsatsos, S.; Vakros, J.; Ladas, S.; Verykios, X.E.; Kyriakou, G. The Interplay between Acid-Base Properties and Fermi Level Pinning of a Nano Dispersed Tungsten Oxide—Titania Catalytic System. J. Colloid. Interface Sci. 2022, 614, 666–676. [Google Scholar] [CrossRef]
- Bêche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS Investigation of Cerium Oxides and Mixed Cerium Oxide (CeTiO). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Gross, T.; Ramm, M.; Sonntag, H.; Unger, W.; Weijers, H.M.; Adem, E.H. An XPS Analysis of Different SiO2 Modifications Employing a C 1s as Well as an Au 4f7/2 Static Charge Reference. Surf. Interface Anal. 1992, 18, 59–64. [Google Scholar] [CrossRef]
- Arabczyk, W.; Narkiewicz, U.; Moszyński, D. Chlorine as a Poison of the Fused Iron Catalyst for Ammonia Synthesis. Appl. Catal. Gen. 1996, 134, 331–338. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, M.; Wang, R.; Li, X.; Huang, T.; Wang, J.; Wang, Y.; Zheng, Z. Switching of CO2 Hydrogenation Selectivity via Chlorine Poisoning over Ru/TiO2 Catalyst. Nano Res. 2023, 16, 4786–4792. [Google Scholar] [CrossRef]
- de Rivas, B.; López-Fonseca, R.; Gutiérrez-Ortiz, M.A.; Gutiérrez-Ortiz, J.I. Impact of Induced Chlorine-Poisoning on the Catalytic Behaviour of Ce0.5Zr0.5O2 and Ce0.15Zr0.85O2 in the Gas-Phase Oxidation of Chlorinated VOCs. Appl. Catal. B Environ. 2011, 104, 373–381. [Google Scholar] [CrossRef]
- Crawford, J.M.; Petel, B.E.; Rasmussen, M.J.; Ludwig, T.; Miller, E.M.; Halingstad, S.; Akhade, S.A.; Pang, S.H.; Yung, M.M. Influence of Residual Chlorine on Ru/TiO2 Active Sites during CO2 Methanation. Appl. Catal. Gen. 2023, 663, 119292. [Google Scholar] [CrossRef]
- Islam, M.J.; Mesa, M.G.; Osatiashtiani, A.; Manayil, J.C.; Isaacs, M.A.; Taylor, M.J.; Tsatsos, S.; Kyriakou, G. PdCu Single Atom Alloys Supported on Alumina for the Selective Hydrogenation of Furfural. Appl. Catal. B Environ. 2021, 299, 120652. [Google Scholar] [CrossRef]
- Taylor, M.J.; Durndell, L.J.; Isaacs, M.A.; Parlett, C.M.A.; Wilson, K.; Lee, A.F.; Kyriakou, G. Highly Selective Hydrogenation of Furfural over Supported Pt Nanoparticles under Mild Conditions. Appl. Catal. B Environ. 2016, 180, 580–585. [Google Scholar] [CrossRef]
- Lymperi, A.; Chatzilias, C.; Xydas, F.; Martino, E.; Kyriakou, G.; Katsaounis, A. Electrochemical Promotion of CO2 Hydrogenation Using a Pt/YSZ Fuel Cell Type Reactor. Nanomaterials 2023, 13, 1930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chan, Y.S.; Zhang, R.; Yan, B. Insights into the Contribution of Oxygen Vacancies on CO2 Activation for Dry Reforming of Methane over Ceria-Based Solid Solutions. Chem. Eng. J. 2024, 481, 148360. [Google Scholar] [CrossRef]
- Pan, X.; Yang, M.-Q.; Fu, X.; Zhang, N.; Xu, Y.-J. Defective TiO2 with Oxygen Vacancies: Synthesis, Properties and Photocatalytic Applications. Nanoscale 2013, 5, 3601–3614. [Google Scholar] [CrossRef]
- Hossain, S.T.; Almesned, Y.; Zhang, K.; Zell, E.T.; Bernard, D.T.; Balaz, S.; Wang, R. Support Structure Effect on CO Oxidation: A Comparative Study on SiO2 Nanospheres and CeO2 Nanorods Supported CuOx Catalysts. Appl. Surf. Sci. 2018, 428, 598–608. [Google Scholar] [CrossRef]
- Kaneko, S.; Morimoto, T.; Ohki, Y. Cause of the Appearance of Oxygen Vacancies in Yttria-Stabilized Zirconia and Its Relation to 2.8 eV Photoluminescence. Jpn. J. Appl. Phys. 2015, 54, 06GC03. [Google Scholar] [CrossRef]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the Enhanced Photocatalytic Activity of Degussa P25 Mixed-Phase TiO2 Using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Ren, C.; Yang, R.; Li, Y.; Wang, H. Modulating of Facets-Dependent Oxygen Vacancies on Ceria and Its Catalytic Oxidation Performance. Res. Chem. Intermed. 2019, 45, 3019–3032. [Google Scholar] [CrossRef]
- Nakamura, J.; Aikawa, K.; Sato, K.; Uchijima, T. Role of Support in Reforming of CH4 with CO2 over Rh Catalysts. Catal. Lett. 1994, 25, 265–270. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Hatzisymeon, M.; Betsi-Argyropoulou, I.; Botzolaki, G.; Kousi, K.; Kondarides, D.I.; Taylor, M.J.; Parlett, C.M.A.; Osatiashtiani, A.; et al. Effect of Support Oxygen Storage Capacity on the Catalytic Performance of Rh Nanoparticles for CO2 Reforming of Methane. Appl. Catal. B Environ. 2019, 243, 490–501. [Google Scholar] [CrossRef]
- Usman, M.; Wan Daud, W.M.A.; Abbas, H.F. Dry Reforming of Methane: Influence of Process Parameters—A Review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH4 over Supported Ru Catalysts. J. Catal. 1999, 183, 69–75. [Google Scholar] [CrossRef]
- Quiroga, M.M.B.; Luna, A.E.C. Kinetic Analysis of Rate Data for Dry Reforming of Methane. Ind. Eng. Chem. Res. 2007, 46, 5265–5270. [Google Scholar] [CrossRef]
- Triviño, M.L.T.; Arriola, N.C.; Kang, Y.S.; Seo, J.G. Transforming CO2 to Valuable Feedstocks: Emerging Catalytic and Technological Advances for the Reverse Water Gas Shift Reaction. Chem. Eng. J. 2024, 487, 150369. [Google Scholar] [CrossRef]
- Spencer, M.S. On the Activation Energies of the Forward and Reverse Water-Gas Shift Reaction. Catal. Lett. 1995, 32, 9–13. [Google Scholar] [CrossRef]
- Baudouin, D.; Rodemerck, U.; Krumeich, F.; de Mallmann, A.; Szeto, K.C.; Ménard, H.; Veyre, L.; Candy, J.-P.; Webb, P.B.; Thieuleux, C.; et al. Particle Size Effect in the Low Temperature Reforming of Methane by Carbon Dioxide on Silica-Supported Ni Nanoparticles. J. Catal. 2013, 297, 27–34. [Google Scholar] [CrossRef]
- Özdemir, H.; Öksüzömer, M.A.F.; Gürkaynak, M.A. Preparation and Characterization of Ni Based Catalysts for the Catalytic Partial Oxidation of Methane: Effect of Support Basicity on H2/CO Ratio and Carbon Deposition. Int. J. Hydrogen Energy 2010, 35, 12147–12160. [Google Scholar] [CrossRef]
- Lucrédio, A.F.; Assaf, J.M.; Assaf, E.M. Methane Conversion Reactions on Ni Catalysts Promoted with Rh: Influence of Support. Appl. Catal. Gen. 2011, 400, 156–165. [Google Scholar] [CrossRef]
- Manan, W.N.; Isahak, W.N.R.W.; Yaakob, Z. CeO2-Based Heterogeneous Catalysts in Dry Reforming Methane and Steam Reforming Methane: A Short Review. Catalysts 2022, 12, 452. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.; Breen, J.P.; Burch, R.; Petch, M.I.; Faur Ghenciu, A. Study of the Origin of the Deactivation of a Pt/CeO2 Catalyst during Reverse Water Gas Shift (RWGS) Reaction. J. Catal. 2004, 226, 382–392. [Google Scholar] [CrossRef]
- He, L.; Ren, Y.; Fu, Y.; Yue, B.; Tsang, S.C.E.; He, H. Morphology-Dependent Catalytic Activity of Ru/CeO2 in Dry Reforming of Methane. Molecules 2019, 24, 526. [Google Scholar] [CrossRef]
- Zhou, R.; Ren, Y.; Lu, Q.; Mahinpey, N. Microwave-Assisted Hydrothermal Synthesis of Ru/CeO2 Catalyst for Efficient and Stable Low-Temperature Dry Reforming of Methane. Fuel 2023, 346, 128366. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, F.; Rui, N.; Li, X.; Lin, L.; Betancourt, L.E.; Su, D.; Xu, W.; Cen, J.; Attenkofer, K.; et al. Highly Active Ceria-Supported Ru Catalyst for the Dry Reforming of Methane: In Situ Identification of Ruδ+–Ce3+ Interactions for Enhanced Conversion. ACS Catal. 2019, 9, 3349–3359. [Google Scholar] [CrossRef]
- Yong, X.; Tse, J.S.; Chen, J. Mechanism of Chemical Reactions between SiO2 and CO2 under Mantle Conditions. ACS Earth Space Chem. 2018, 2, 548–555. [Google Scholar] [CrossRef]
- Ueno, A.; Bennett, C.O. Infrared Study of CO2 Adsorption on SiO2. J. Catal. 1978, 54, 31–41. [Google Scholar] [CrossRef]
- Tahari, M.N.A.; Yarmo, M.A. Adsorption of CO2 on Silica Dioxide Catalyst Impregnated with Various Alkylamine. AIP Conf. Proc. 2014, 1614, 334–341. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Comparative Study at Low and Medium Reaction Temperatures of Syngas Production by Methane Reforming with Carbon Dioxide over Silica and Alumina Supported Catalysts. Appl. Catal. Gen. 1998, 170, 177–187. [Google Scholar] [CrossRef]
- Han, J.W.; Park, J.S.; Choi, M.S.; Lee, H. Uncoupling the Size and Support Effects of Ni Catalysts for Dry Reforming of Methane. Appl. Catal. B Environ. 2017, 203, 625–632. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Deo, G. Influence of Particle Size and Metal-Support Interaction on the Catalytic Performance of Ni-Al2O3 Catalysts for the Dry and Oxidative-Dry Reforming of Methane. Colloids Surf. Physicochem. Eng. Asp. 2022, 646, 128973. [Google Scholar] [CrossRef]
- Awad, M.M.; Kotob, E.; Taialla, O.A.; Hussain, I.; Ganiyu, S.A.; Alhooshani, K. Recent Developments and Current Trends on Catalytic Dry Reforming of Methane: Hydrogen Production, Thermodynamics Analysis, Techno Feasibility, and Machine Learning. Energy Convers. Manag. 2024, 304, 118252. [Google Scholar] [CrossRef]
- Andraos, S.; Abbas-Ghaleb, R.; Chlala, D.; Vita, A.; Italiano, C.; Laganà, M.; Pino, L.; Nakhl, M.; Specchia, S. Production of Hydrogen by Methane Dry Reforming over Ruthenium-Nickel Based Catalysts Deposited on Al2O3, MgAl2O4, and YSZ. Int. J. Hydrogen Energy 2019, 44, 25706–25716. [Google Scholar] [CrossRef]
- Tsatsos, S.; Kyriakou, G. Copper Growth on a Stepped Nickel Surface: Electronic and Geometric Effects on CO Reactivity. J. Phys. Chem. C 2023, 127, 6337–6346. [Google Scholar] [CrossRef]
- Drivas, C.; Kennou, S.; Kyriakou, G. Phthalocyanine Interfaces with MoS2(0001): The Effect of the Metal Centre on the Charge Transfer. Appl. Surf. Sci. 2025, 682, 161672. [Google Scholar] [CrossRef]
- Kerkhof, F.P.J.M.; Moulijn, J.A. Quantitative Analysis of XPS Intensities for Supported Catalysts. J. Phys. Chem. 1979, 83, 1612–1619. [Google Scholar] [CrossRef]
- Yin, A.; Guo, X.; Dai, W.; Fan, K. High Activity and Selectivity of Ag/SiO2 Catalyst for Hydrogenation of Dimethyl Oxalate. Chem. Commun. 2010, 46, 4348–4350. [Google Scholar] [CrossRef]
- Vargas-Hernández, D.; Rubio-Caballero, J.M.; Santamaría-González, J.; Moreno-Tost, R.; Mérida-Robles, J.M.; Pérez-Cruz, M.A.; Jiménez-López, A.; Hernández-Huesca, R.; Maireles-Torres, P. Furfuryl Alcohol from Furfural Hydrogenation over Copper Supported on SBA-15 Silica Catalysts. J. Mol. Catal. Chem. 2014, 383–384, 106–113. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, B.; Lin, Y.; Wang, Q.; Zhang, Q.; Su, D.S. Enhanced Chemoselective Hydrogenation through Tuning the Interaction between Pt Nanoparticles and Carbon Supports: Insights from Identical Location Transmission Electron Microscopy and X-Ray Photoelectron Spectroscopy. ACS Catal. 2016, 6, 7844–7854. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater Subshell Photoionization Cross-Sections at 1254 and 1487 eV. J. Electron. Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Shinotsuka, H.; Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of Electron Inelastic Mean Free Paths. X. Data for 41 Elemental Solids over the 50 eV to 200 keV Range with the Relativistic Full Penn Algorithm. Surf. Interface Anal. 2015, 47, 871–888. [Google Scholar] [CrossRef]
- Pietraszek, A.; Koubaissy, B.; Roger, A.-C.; Kiennemann, A. The Influence of the Support Modification over Ni-Based Catalysts for Dry Reforming of Methane Reaction. Catal. Today 2011, 176, 267–271. [Google Scholar] [CrossRef]
- Rotaru, C.G.; Postole, G.; Florea, M.; Matei-Rutkovska, F.; Pârvulescu, V.I.; Gelin, P. Dry Reforming of Methane on Ceria Prepared by Modified Precipitation Route. Appl. Catal. Gen. 2015, 494, 29–40. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ru wt% 1 | SSA 2 (m2 g−1) | Ru Particle Size 3 (nm) | Ru Particle Size 4 (nm) | Ru Dispersion 5 % | Ru Particle Size 6 (nm) |
|---|---|---|---|---|---|---|
| Ru/YSZ | 1.3 | 14.4 | 7.4 ± 4.1 | 14.9 | 40.0 | 2.7 |
| Ru/CeO2 | 2.3 | 48.9 | 10.1 ± 3.8 | 12.3 | 16.7 | 6.2 |
| Ru/TiO2 | 3.7 | 46.8 | 5.5 ± 2.7 | n.a. 7 | 19.2 | 5.5 |
| Ru/SiO2 | 1.3 | 281.7 | 6.3 ± 1.7 | 9.1 | 81.7 | 1.2 |
| Catalyst | Reaction | Reducing | Stoichiometric | Oxidizing |
|---|---|---|---|---|
| Ru/YSZ | DRM | 33 ± 2 | 30 ± 1 | 29 ± 1 |
| RWGS | 24 ± 3 | 24 ± 2 | 24 ± 2 | |
| Ru/CeO2 | DRM | 28 ± 2 | 28 ± 1 | 30 ± 2 |
| RWGS | 30 ± 3 | 24 ± 2 | 20 ± 2 | |
| Ru/TiO2 | DRM | 24 ± 1 | 23 ± 2 | 26 ± 1 |
| RWGS | 21 ± 1 | 19 ± 1 | 21 ± 1 | |
| Ru/SiO2 | DRM | 25 ± 2 | 24 ± 1 | 24 ± 1 |
| RWGS | 22 ± 2 | 21 ± 1 | 22 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bikogiannakis, A.K.; Lymperi, A.; Dimitropoulos, P.; Bourikas, K.; Katsaounis, A.; Kyriakou, G. CO2 Reforming of Methane over Ru Supported Catalysts Under Mild Conditions. Molecules 2025, 30, 2135. https://doi.org/10.3390/molecules30102135
Bikogiannakis AK, Lymperi A, Dimitropoulos P, Bourikas K, Katsaounis A, Kyriakou G. CO2 Reforming of Methane over Ru Supported Catalysts Under Mild Conditions. Molecules. 2025; 30(10):2135. https://doi.org/10.3390/molecules30102135
Chicago/Turabian StyleBikogiannakis, Alexandros K., Andriana Lymperi, Paraskevas Dimitropoulos, Kyriakos Bourikas, Alexandros Katsaounis, and Georgios Kyriakou. 2025. "CO2 Reforming of Methane over Ru Supported Catalysts Under Mild Conditions" Molecules 30, no. 10: 2135. https://doi.org/10.3390/molecules30102135
APA StyleBikogiannakis, A. K., Lymperi, A., Dimitropoulos, P., Bourikas, K., Katsaounis, A., & Kyriakou, G. (2025). CO2 Reforming of Methane over Ru Supported Catalysts Under Mild Conditions. Molecules, 30(10), 2135. https://doi.org/10.3390/molecules30102135