
Enhancing High-Order Harmonic Generation Efficiency Through Molecular Size and Orientation Effects: A Pathway to Ultrafast Chemical Dynamics Studies


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
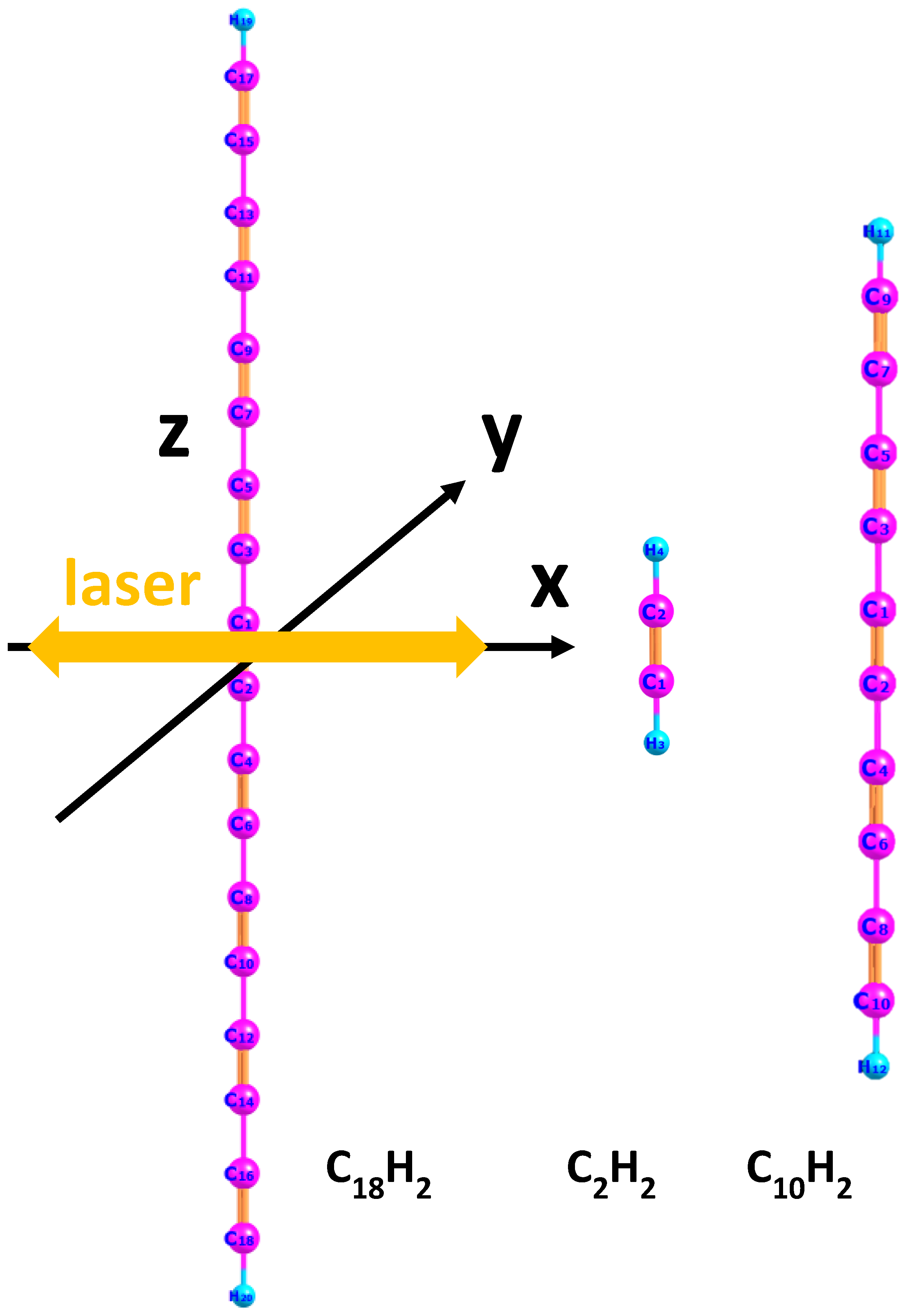
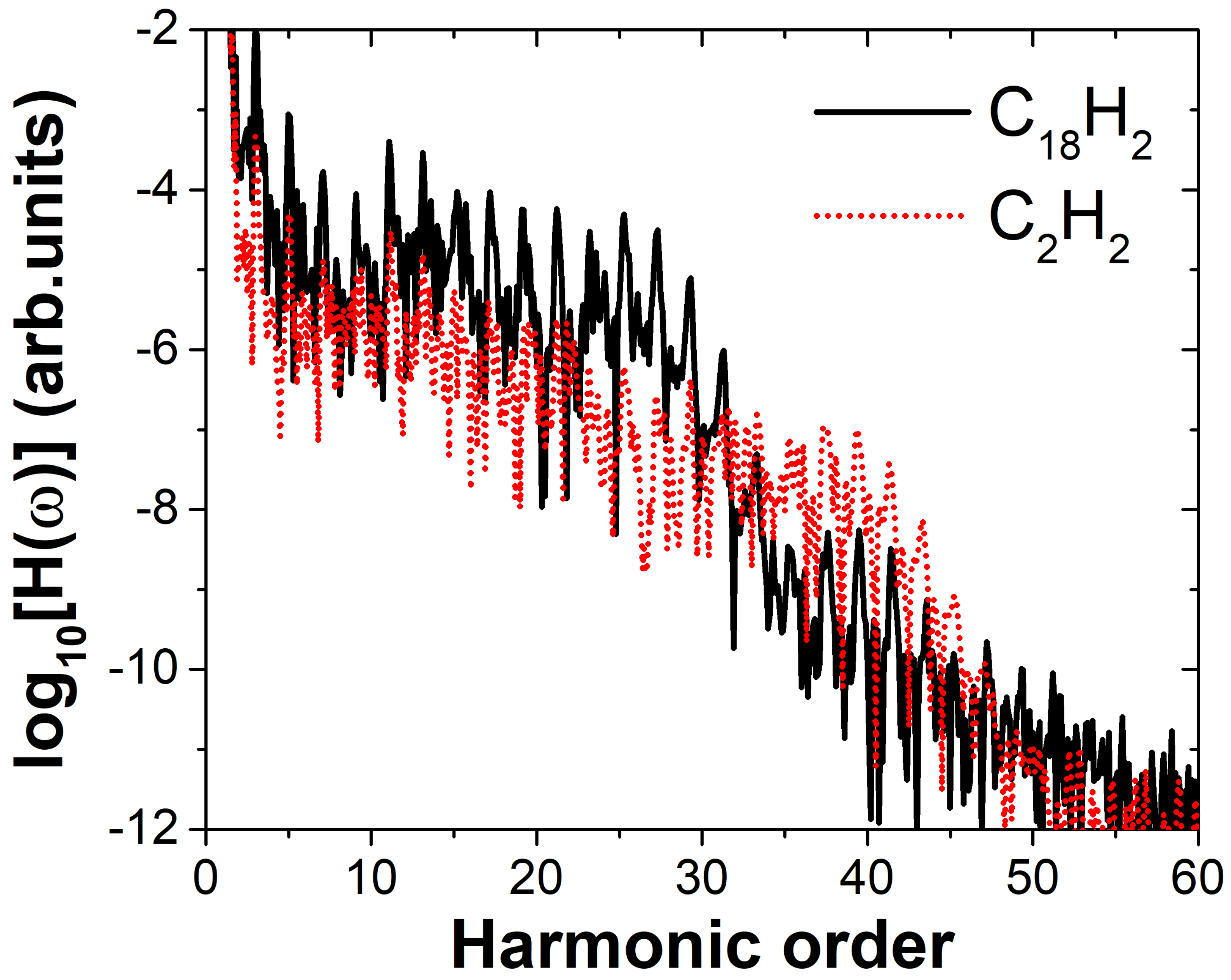
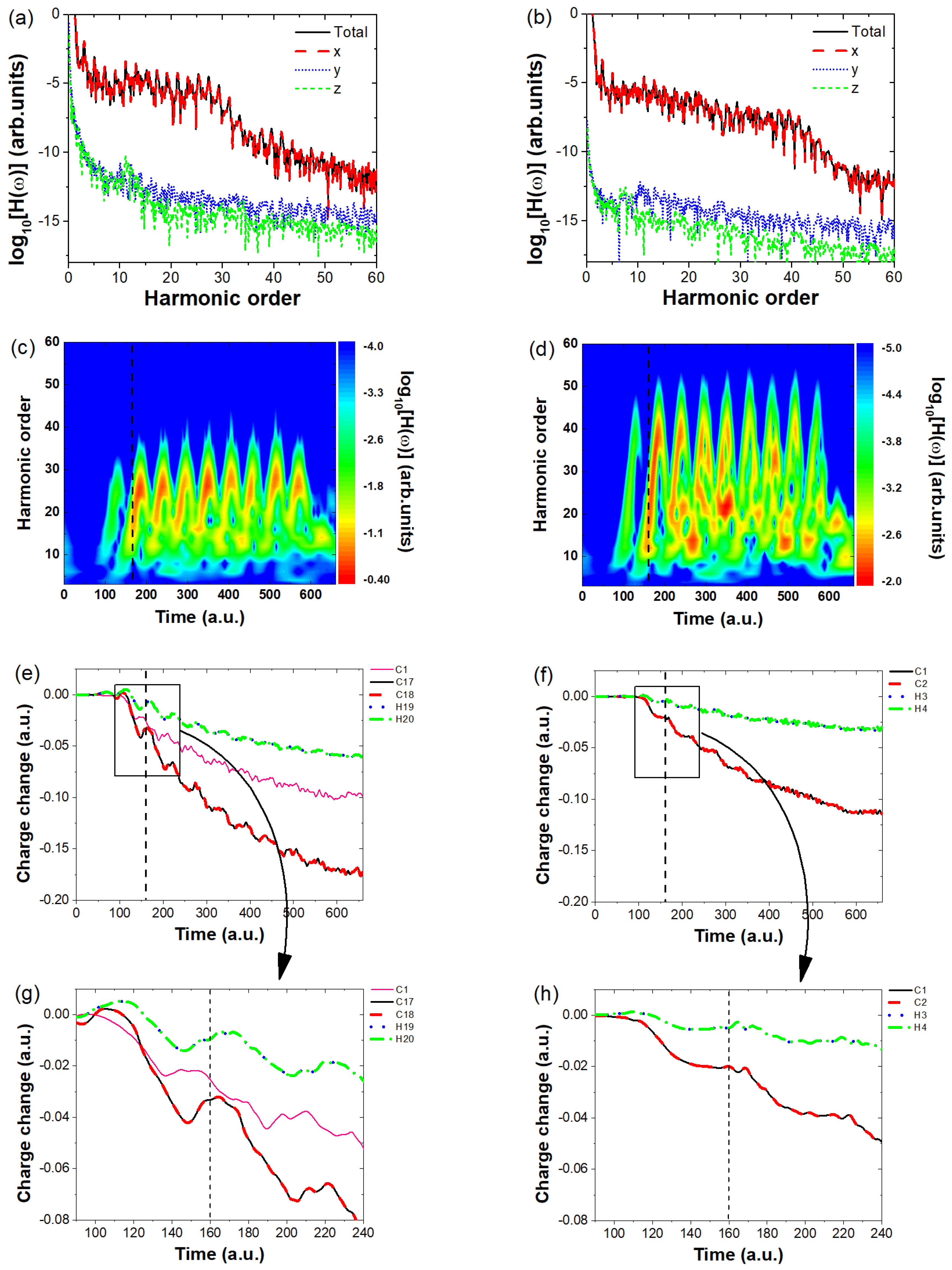
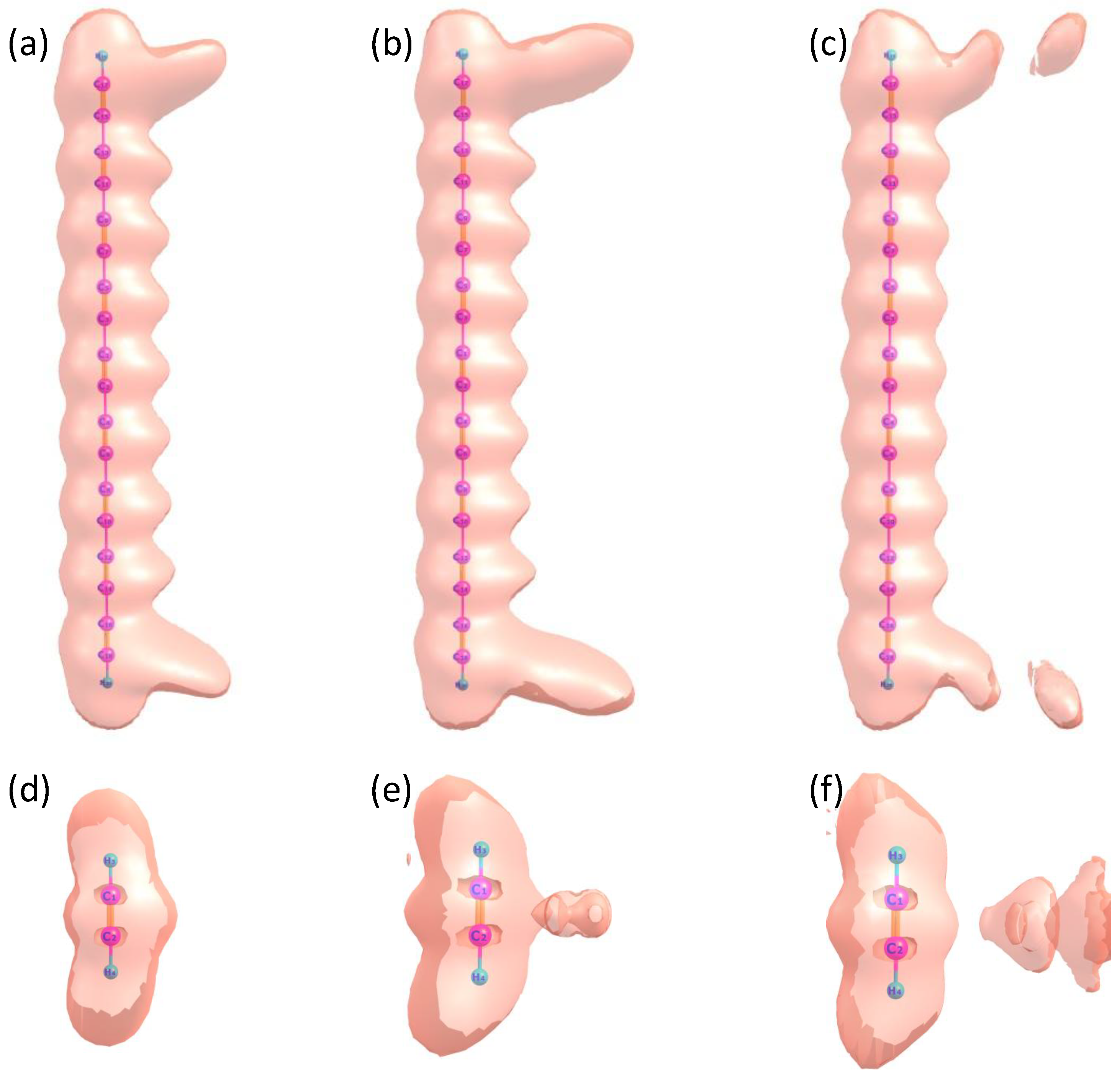
2. Results and Discussion
3. Theory and Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marcos Dantus, M.J.R.; Zewail, A.H. Real-time femtosecond probing of “transition states” in chemical reactions. J. Chem. Phys. 1987, 87, 2395. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, S.; Jiang, W.; Guo, F.; Wang, J.; Chen, J.; Yang, Y. Modulation of harmonics from solids by laser pulses with a small chirp. Phys. Rev. A 2025, 111, 013501. [Google Scholar] [CrossRef]
- Xu, N.; Zhou, S.S.; Wang, Y. Regulation of helium atom higher harmonic emission and attosecond pulse angle in inhomogeneous fields. Results Phys. 2025, 72, 108190. [Google Scholar] [CrossRef]
- Peters, M.; Dang, T.N.; Charron, E.; Keller, A.; Atabek, O. Laser-induced electron diffraction: A tool for molecular orbital imaging. Phys. Rev. A 2012, 85, 053417. [Google Scholar] [CrossRef]
- Spielmann, C.; Burnett, N.H.; Sartania, S.; Koppitsch, R.; Schnorer, M.; Kan, C.; Lenzner, M.; Wobrauschek, P.; Krausz, F. Generation of Coherent X-rays in the Water Window Using 5-Femtosecond Laser Pulses. Science 1997, 278, 661–664. [Google Scholar] [CrossRef]
- L’Huillier, A.; Balcou, P. High-order harmonic generation in rare gases with a 1-ps 1053-nm laser. Phys. Rev. Lett. 1993, 70, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Macklin, J.J.; Kmetec, J.D.; Gordon, C.L. High-order harmonic generation using intense femtosecond pulses. Phys. Rev. Lett. 1993, 70, 766–769. [Google Scholar] [CrossRef]
- Popmintchev, T.; Chen, M.C.; Popmintchev, D.; Arpin, P.; Brown, S.; Ališauskas, S.; Andriukaitis, G.; Balčiunas, T.; Mücke, O.D.; Pugzlys, A.; et al. Bright Coherent Ultrahigh Harmonics in the keV X-ray Regime from Mid-Infrared Femtosecond Lasers. Science 2012, 336, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, W.; Qiao, Y.; Yang, Y.; Chen, J. Generation of Isolated Attosecond Pulses by the Harmonic Spectrum of MgO under a Three-Color Laser Pulse. Chin. Phys. Lett. 2025, 42, 013201. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, J.; Zhou, S.; Chen, J.; Jiang, S.; Yang, Y. Modulation of High-Order Harmonic Generation from a Monolayer ZnO by Co-rotating Two-Color Circularly Polarized Laser Fields. Chin. Phys. Lett. 2024, 41, 014205. [Google Scholar] [CrossRef]
- Corkum, P.B. Plasma perspective on strong field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Wang, J.; Zhao, X.; Zhou, S. The Role of Multi-Electron and Multi-Orbital Effects in High-Order Harmonic Generation of Benzonitrile Molecules. Chin. Phys. Lett. 2025, 42, 043201. [Google Scholar] [CrossRef]
- Hentschel, M.; Kienberger, R.; Spielmann, C.; Reider, G.A.; Milosevic, N.; Brabec, T.; Corkum, P.; Heinzmann, U.; Drescher, M.; Krausz, F. Attosecond metrology. Nature 2001, 414, 509–513. [Google Scholar] [CrossRef]
- Paul, P.M.; Toma, E.S.; Breger, P.; Mullot, G.; Augé, F.; Balcou, P.; Muller, H.G.; Agostini, P. Observation of a Train of Attosecond Pulses from High Harmonic Generation. Science 2001, 292, 1689–1692. [Google Scholar] [CrossRef]
- Takahashi, E.J.; Pengfei, L.; Oliver, D.M.; Yasuo, N.; Katsumi, M. Attosecond nonlinear optics using gigawatt-scale isolated attosecond pulses. Nat. Commun. 2013, 4, 2691. [Google Scholar] [CrossRef]
- Ayuso, D.; Jiménez-Galán, A.; Morales, F.; Ivanov, M.; Smirnova, O. Attosecond control of spin polarization in electron–ion recollision driven by intense tailored fields. New J. Phys. 2017, 19, 073007. [Google Scholar] [CrossRef]
- Vozzi, C.; Negro, M.; Calegari, F.; Sansone, G.; Nisoli, M.; Silvestri, S.D.; Stagira, S. Generalized molecular orbital tomography. Nat. Phys. 2011, 7, 822–826. [Google Scholar] [CrossRef]
- Niikura, H.; Dudovich, N.; Villeneuve, D.M.; Corkum, P.B. Mapping Molecular Orbital Symmetry on High-Order Harmonic Generation Spectrum Using Two-Color Laser Fields. Phys. Rev. Lett. 2010, 105, 053003. [Google Scholar] [CrossRef]
- Itatani, J.; Levesque, J.; Zeidler, D.; Hiromichi, N.; Pépin, H.; Kieffer, J.C.; Corkum, P.B.; Villeneuve, D.M. Tomographic imaging of molecular orbitals. Nature 2004, 432, 867–871. [Google Scholar] [CrossRef]
- Sansone, G.; Benedetti, E.; Calegari, F.; Vozzi, C.; Avaldi, L.; Flammini, R.; Poletto, L.; Villoresi, P.; Altucci, C.; Velotta, R.; et al. Isolated Single-Cycle Attosecond Pulses. Science 2006, 314, 443–446. [Google Scholar] [CrossRef]
- Puthumpally-Joseph, R.; Viau-Trudel, J.; Peters, M.; Nguyen-Dang, T.T.; Atabek, O.; Charron, E. Inversion of strong-field photoelectron spectra for molecular orbital imaging. Phys. Rev. A 2016, 94, 023421. [Google Scholar] [CrossRef]
- Li, J.B.; Zhang, X.; Yue, S.J.; Wu, H.M.; Hu, B.T.; Du, H.C. Enhancement of the second plateau in solid high-order harmonic spectra by the two-color fields. Opt. Express 2017, 25, 18603–18613. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, C.; Vampa, G.; Zhang, J.L.; Sarmiento, T.; Xiao, M.; Bucksbaum, P.H.; Vučković, J.; Fan, S.; Reis, D.A. Enhanced high-harmonic generation from an all-dielectric metasurface. Nat. Phys. 2018, 14, 1006–1010. [Google Scholar] [CrossRef]
- Franz, D.; Kaassamani, S.; Gauthier, D.; Nicolas, R.; Kholodtsova, M.; Douillard, L.; Gomes, J.T.; Lavoute, L.; Gaponov, D.; Ducros, N.; et al. All semiconductor enhanced high-harmonic generation from a single nanostructured cone. Sci. Rep. 2019, 9, 5663. [Google Scholar] [CrossRef]
- Yao, D.-H.; Bo, C.; Ma, S.-Q.; Chao, Y.; Lu, R.-F. Enhancing high harmonic generation in bilayer MoS2 by interlayer atomic dislocation. Acta Phys. Sin. 2021, 70, 18–24. [Google Scholar] [CrossRef]
- Kamta-Lagmago, G.; Bandrauk, A.D. Phase dependence of enhanced ionization in asymmetric molecules. Phys. Rev. Lett. 2005, 94, 203003. [Google Scholar] [CrossRef]
- Li, Y.-P.; Yu, S.-J.; Chen, Y.-J. Wavelength-dependent perpendicular-harmonics efficiency from oriented CO2 molecule. Acta Phys. Sin. 2015, 64, 234–241. [Google Scholar]
- Shi, Y.Z.; Zhang, B.; Li, W.Y.; Yu, S.J.; Chen, Y.J. Probing degrees of orientation of polar molecules with harmonic emission in ultrashort laser pulses. Phys. Rev. A 2017, 95, 033406. [Google Scholar] [CrossRef]
- Takahashi, E.J.; Kanai, T.; Ishikawa, K.J.; Nabekawa, Y.; Midorikawa, K. Dramatic Enhancement of High-Order Harmonic Generation. Phys. Rev. Lett. 2007, 99, 053904. [Google Scholar] [CrossRef]
- Hu, J.; Li, X. Influence of high harmonic generation of isotopic molecules. High Power Laser Part. Beams 2010, 22, 1348–1350. [Google Scholar]
- Romanov, A.A.; Silaev, A.A.; Frolov, M.V.; Vvedenskii, N.V. Influence of the polarization of a multielectron atom in a strong laser field on high-order harmonic generation. Phys. Rev. A 2020, 101, 013435. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Schirmer, J. Review of the foundations of time-dependent density-functional theory (TDDFT). Phys. Chem. Chem. Phys. 2025, 27, 4992–5005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, H.; Hu, M.; Sun, Y.; Zhao, X. Review of the Generation, Regulation, and Applications of High-Order Harmonic Generation in Gases Studied Using Time-Dependent Density Functional Theory. Symmetry 2025, 17, 359. [Google Scholar] [CrossRef]
- Castro, A.; Appel, H.; Oliveira, M.; Rozzi, C.A.; Andrade, X.; Lorenzen, F.; Marques, M.A.L.; Gross, E.K.U.; Rubio, A. octopus: A tool for the application of time-dependent density functional theory. Phys. Status Solidi B 2006, 243, 2465–2488. [Google Scholar] [CrossRef]
- Tancogne-Dejean, N.; Mücke, O.D.; Kärtner, F.X.; Rubio, A. llipticity dependence of high-harmonic generation in solids originating from coupled intraband and interband dynamics. Nat. Commun. 2017, 8, 745. [Google Scholar] [CrossRef]
- Keldysh, L.V. Ionization in the Field of a Strong Electromagnetic Wave. J. Exp. Theor. Phys. 1965, 20, 1307–1314. [Google Scholar]
- Krause, J.L.; Schafer, K.J.; Kulander, K.C. High-order harmonic generation from atoms and ions in the high intensity regime. Phys. Rev. Lett. 1992, 68, 3535–3538. [Google Scholar] [CrossRef]
- Graves, C.E.; Reid, A.H.; Wang, T.; Wu, B.; de Jong, S.; Vahaplar, K.; Radu, I.; Bernstein, D.P.; Messerschmidt, M.; Müller, L.; et al. Nanoscale spin reversal by non-local angular momentum transfer following ultrafast laser excitation in ferrimagnetic GdFeCo. Nat. Mater. 2013, 12, 293–298. [Google Scholar] [CrossRef]
- Ofer, N.; Zahra, N.; Nicolas, T.; Angel, R. Ab Initio Cluster Approach for High Harmonic Generation in Liquids. J. Chem. Theory Comput. 2022, 18, 4117–4126. [Google Scholar]
- Angana, M.; Ofer, N.; Zhong, Y.; Zahra, N.; Vít, S.; Angel, R.; Nicolas, T.D.; Jakob, W.H. High-harmonic spectroscopy of low-energy electron-scattering dynamics in liquids. Nat. Phys. 2023, 19, 1813–1820. [Google Scholar]
- Kepceoglu, A.; Gundogdo, Y.; Dereli, O.; Kilic, H.S. Molecular Structure and TD-DFT Study of the Xylene Isomers. Gazi Univ. J. Sci. 2019, 32, 300–308. [Google Scholar]
- Khan, M.F.S.; Wu, J.; Liu, B.; Cheng, C.; Akbar, M.; Chai, Y.; Memon, A. Fluorescence and photophysical properties of xylene isomers in water: With experimental and theoretical approaches. R. Soc. 2018, 5, 171719. [Google Scholar] [CrossRef]
- Romeo-Gella, F.; Corral, I.; Faraji, S. Theoretical investigation of a novel xylene-based light-driven unidirectional molecular motor. J. Chem. Phys. 2021, 154, 064111. [Google Scholar] [CrossRef]
- Zeng, A.W.; Bian, X.B. Impact of Statistical Fluctuations on High Harmonic Generation in Liquids. Phys. Rev. Lett. 2020, 124, 203901. [Google Scholar] [CrossRef] [PubMed]
- Vampa, G.; McDonald, C.; Orlando, G.; Corkum, P.B.; Brabec, T. Semiclassical analysis of high harmonic generation in bulk crystals. Phys. Rev. B Condens. Matter Mater. Phys. 2015, 91, 064302. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Ma, S.Y.; Jiang, S.C.; Yang, Y.J.; Zhao, X.; Chen, J.G. All-optical reconstruction of k-dependent transition dipole moment by solid harmonic spectra from ultrashort laser pulses. Opt. Express 2019, 27, 34392–34404. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, J.; Dong, W.; Liu, J.; Huang, Y.; Zhao, Z. Spatial coherence in high-order-harmonic generation from periodic solid structures. Phys. Rev. A 2017, 96, 053403. [Google Scholar] [CrossRef]
- Jin, J.Z.; Xiao, X.R.; Liang, H.; Wang, M.X.; Chen, S.G.; Gong, Q.; Peng, L.Y. High-order harmonic generation from a two-dimensional band structure. Phys. Rev. A 2018, 97, 043420. [Google Scholar] [CrossRef]
- Ghimire, S.; DiChiara, A.D.; Sistrunk, E.; Agostini, P.; DiMauro, L.F.; Reis, D.A. Observation of high-order harmonic generation in a bulk crystal. Nat. Phys. 2011, 7, 138–141. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, J.; Li, Z.; Liu, Y.; Jiang, S.; Liu, W.; Yang, Y.; Chen, J. Analysis on the minimum structure of harmonic spectra from MgO crystals. Opt. Lett. 2024, 49, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Klemke, N.; Tancogne-Dejean, N.; Rossi, G.; Yang, Y.; Scheiba, F.; Mainz, R.; Sciacca, G.D.; Rubio, A.; Kaertner, F.; Muecke, O. Polarization-state-resolved high-harmonic spectroscopy of solids. Nat. Commun. 2019, 10, 1319. [Google Scholar] [CrossRef] [PubMed]
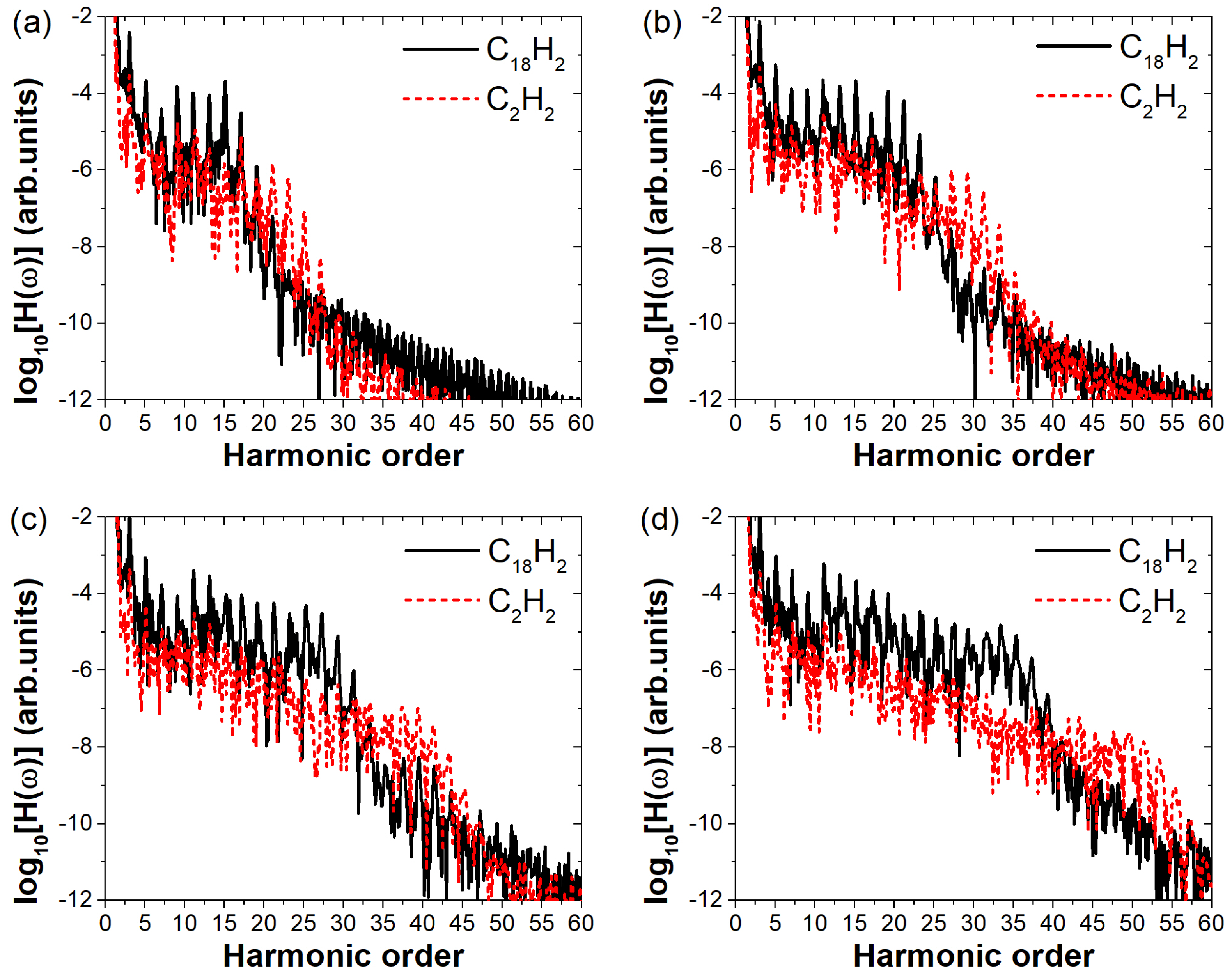
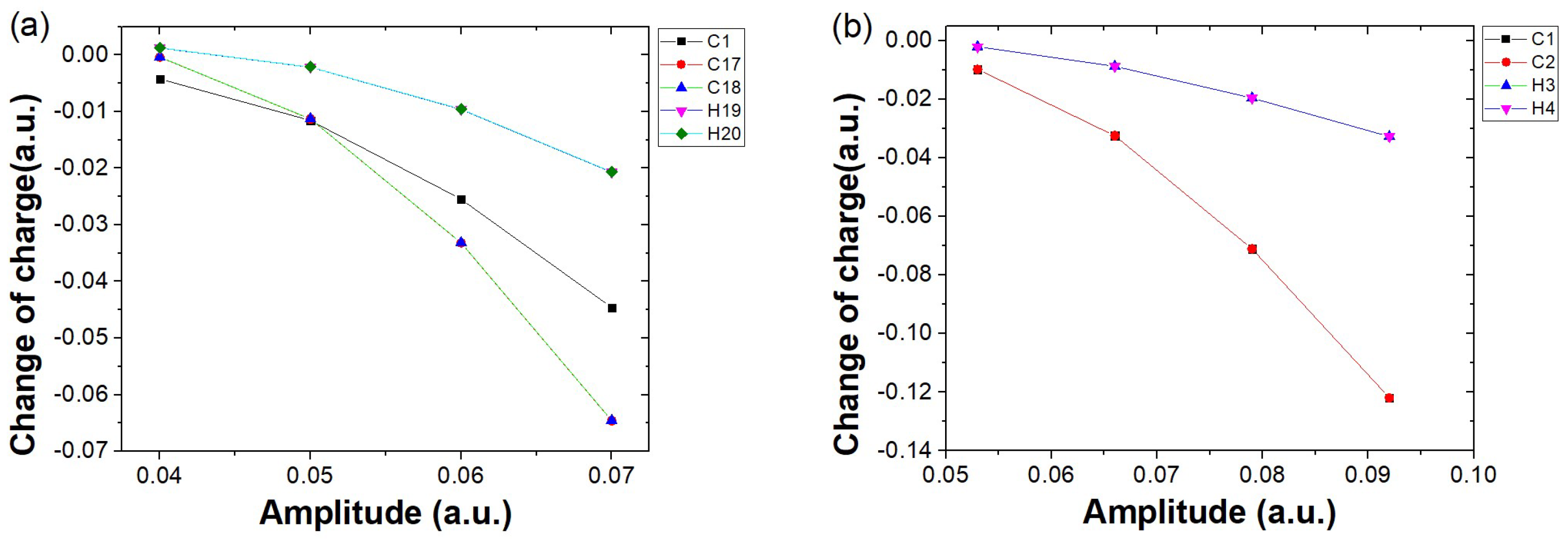
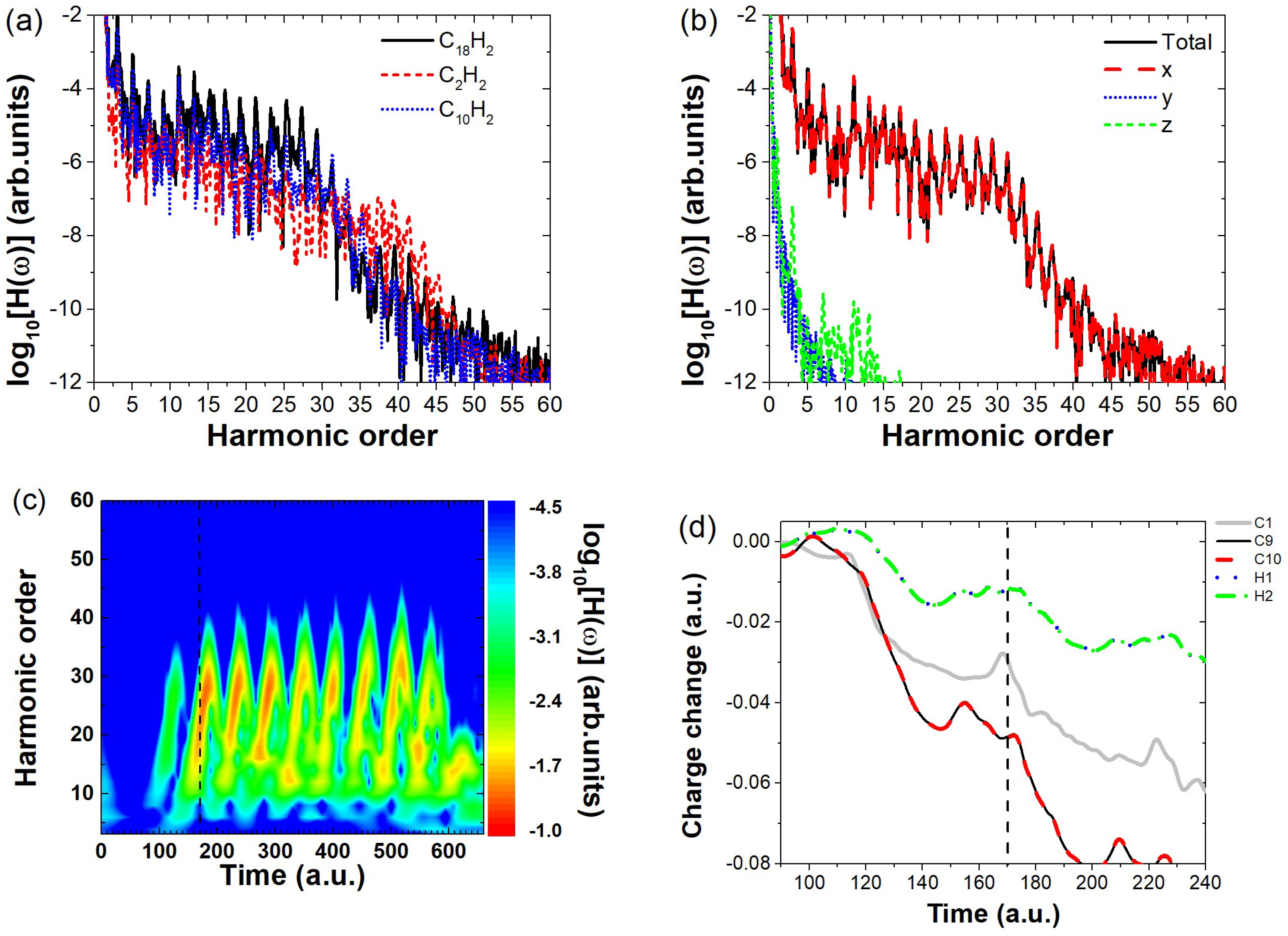
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Wang, H.; Yu, D.; Xu, N.; Hu, M. Enhancing High-Order Harmonic Generation Efficiency Through Molecular Size and Orientation Effects: A Pathway to Ultrafast Chemical Dynamics Studies. Molecules 2025, 30, 2133. https://doi.org/10.3390/molecules30102133
Zhou S, Wang H, Yu D, Xu N, Hu M. Enhancing High-Order Harmonic Generation Efficiency Through Molecular Size and Orientation Effects: A Pathway to Ultrafast Chemical Dynamics Studies. Molecules. 2025; 30(10):2133. https://doi.org/10.3390/molecules30102133
Chicago/Turabian StyleZhou, Shushan, Hao Wang, Dongming Yu, Nan Xu, and Muhong Hu. 2025. "Enhancing High-Order Harmonic Generation Efficiency Through Molecular Size and Orientation Effects: A Pathway to Ultrafast Chemical Dynamics Studies" Molecules 30, no. 10: 2133. https://doi.org/10.3390/molecules30102133
APA StyleZhou, S., Wang, H., Yu, D., Xu, N., & Hu, M. (2025). Enhancing High-Order Harmonic Generation Efficiency Through Molecular Size and Orientation Effects: A Pathway to Ultrafast Chemical Dynamics Studies. Molecules, 30(10), 2133. https://doi.org/10.3390/molecules30102133