
Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Coumarin–Triazole–Isatin Hybrids as Selective Butyrylcholinesterase Inhibitors


 , , ,
, , ,
Abstract

1. Introduction
2. Results and Discussion
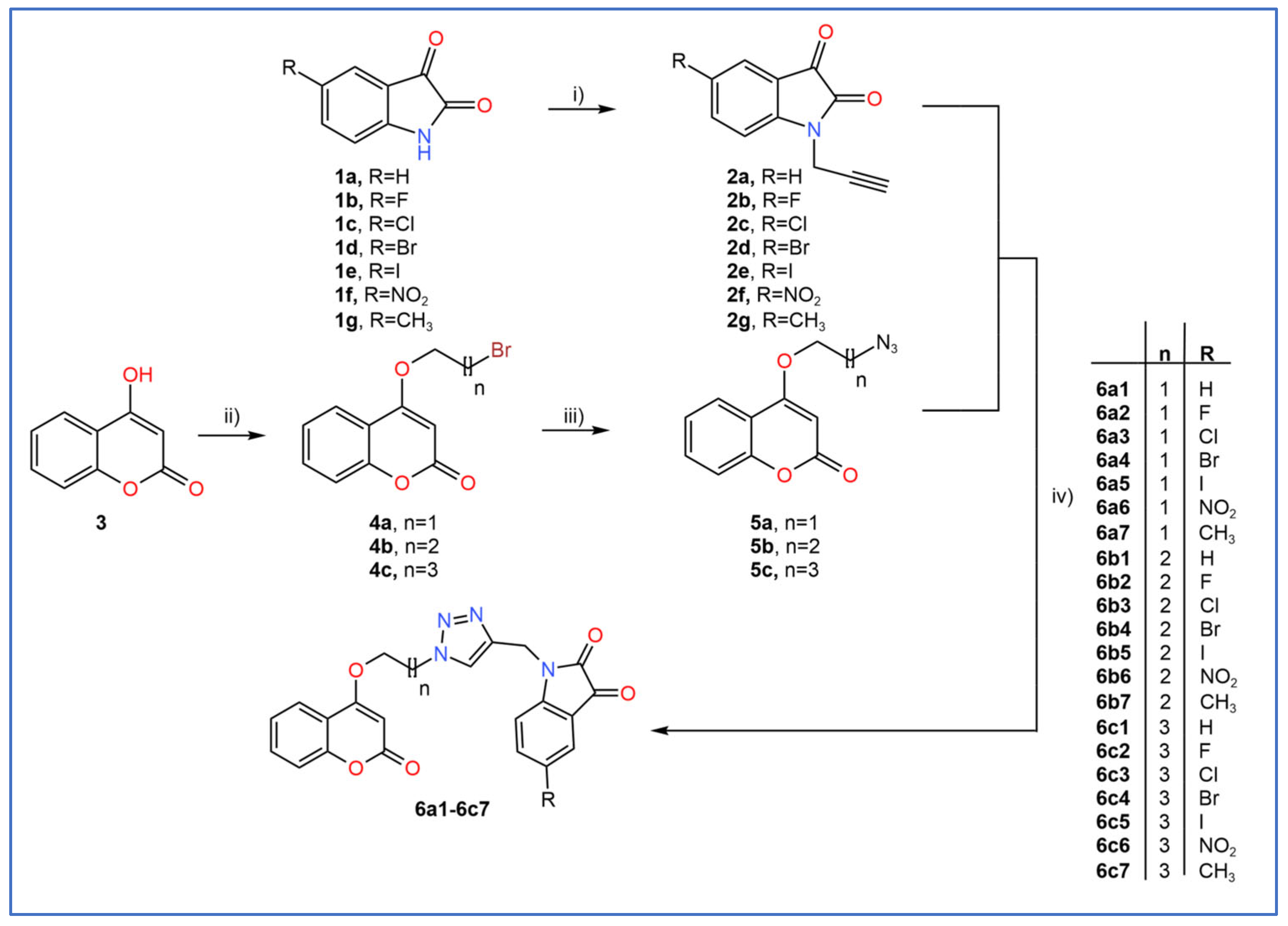
2.1. General Procedure for the Synthesis of Coumarin–Triazole–Isatin Hybrids
2.2. In Vitro Enzyme Inhibition Assays and Enzyme Kinetics
2.2.1. In Vitro Butyrylcholinesterase Inhibition Assay and SAR Analysis
2.2.2. In Vitro Acetylcholinesterase Inhibition Assay and SAR Analysis
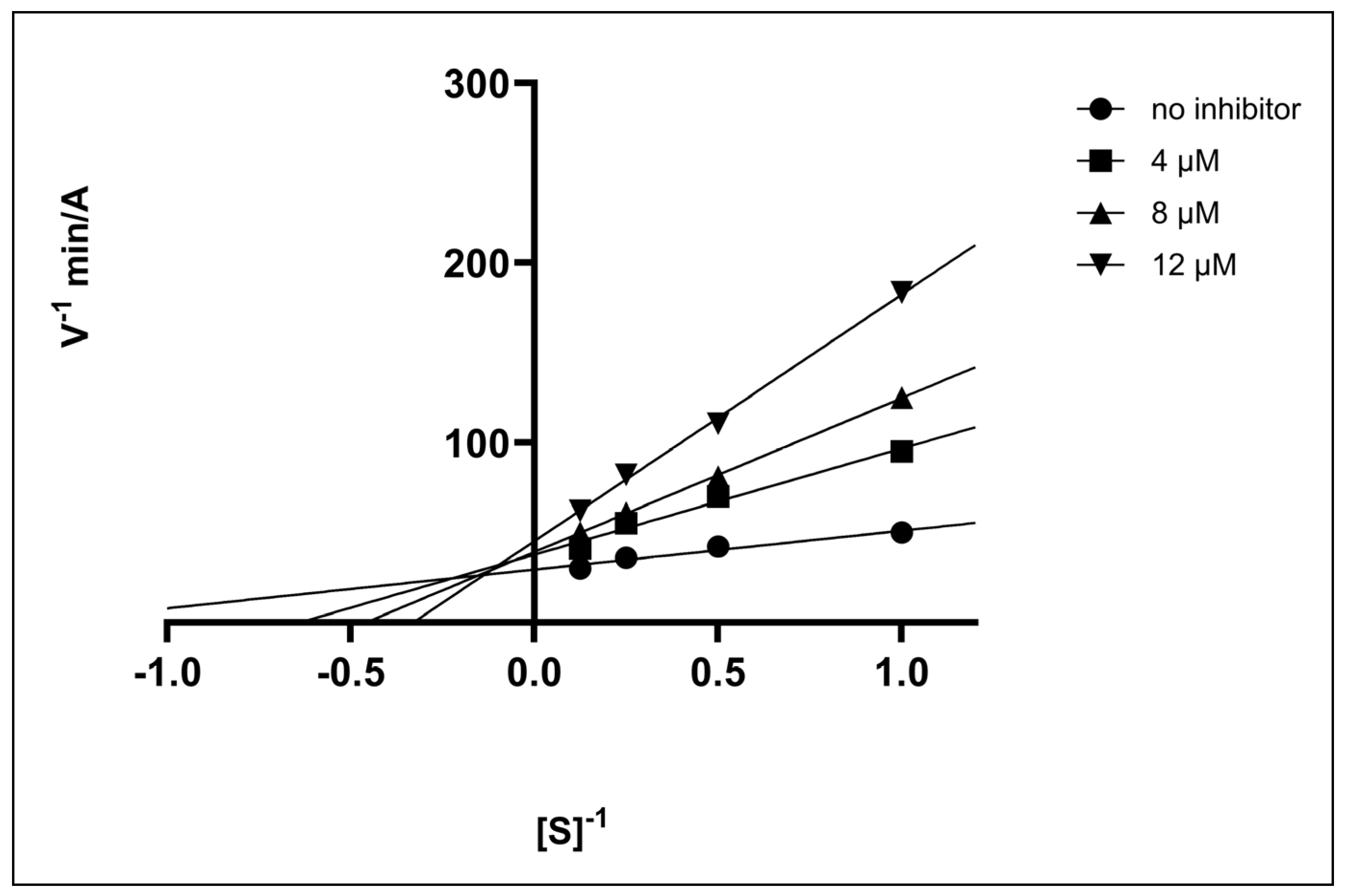
2.2.3. Enzyme Kinetics
2.3. Evaluation of Antimicrobial Activity
2.4. Molecular Docking Studies
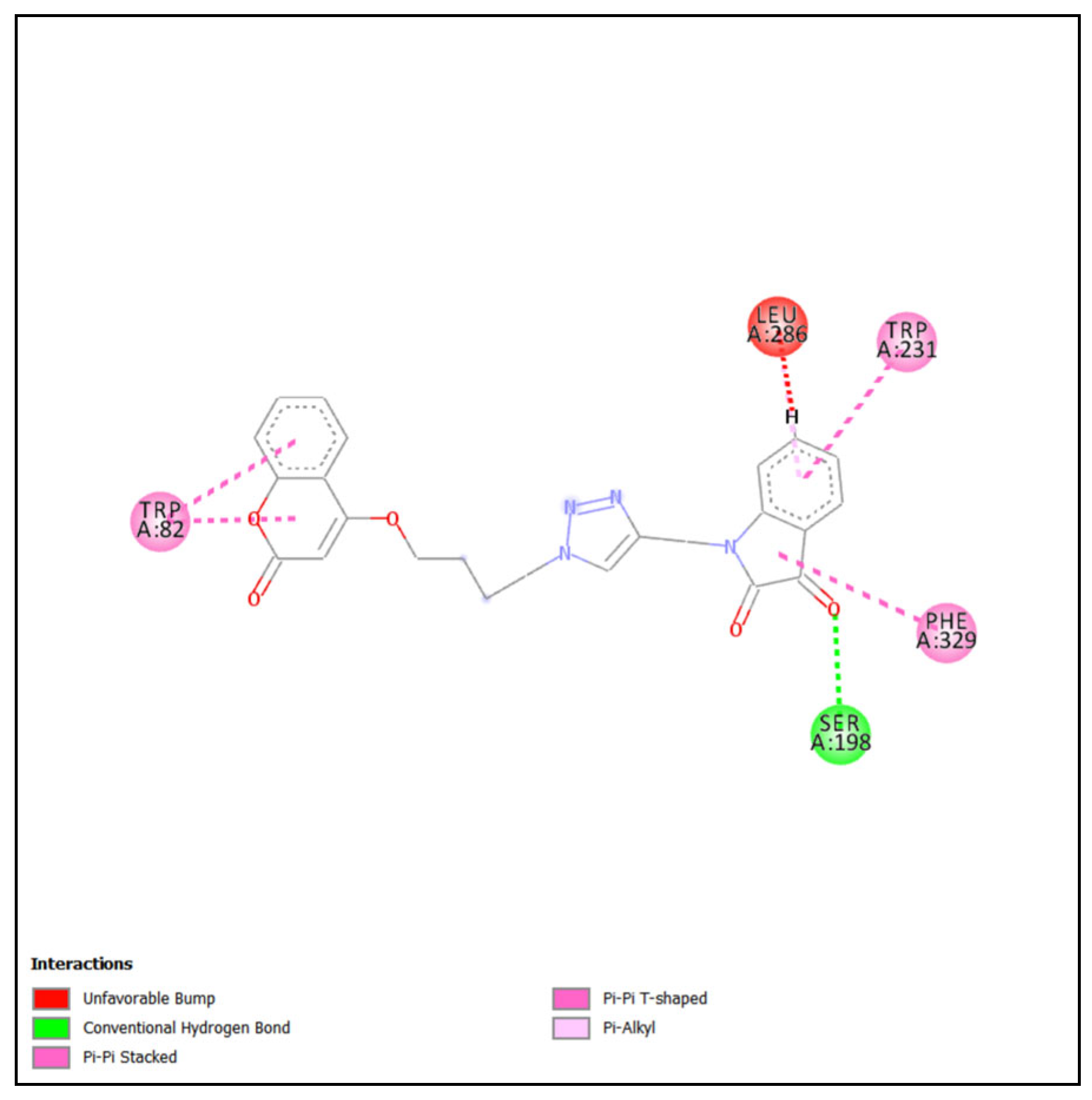
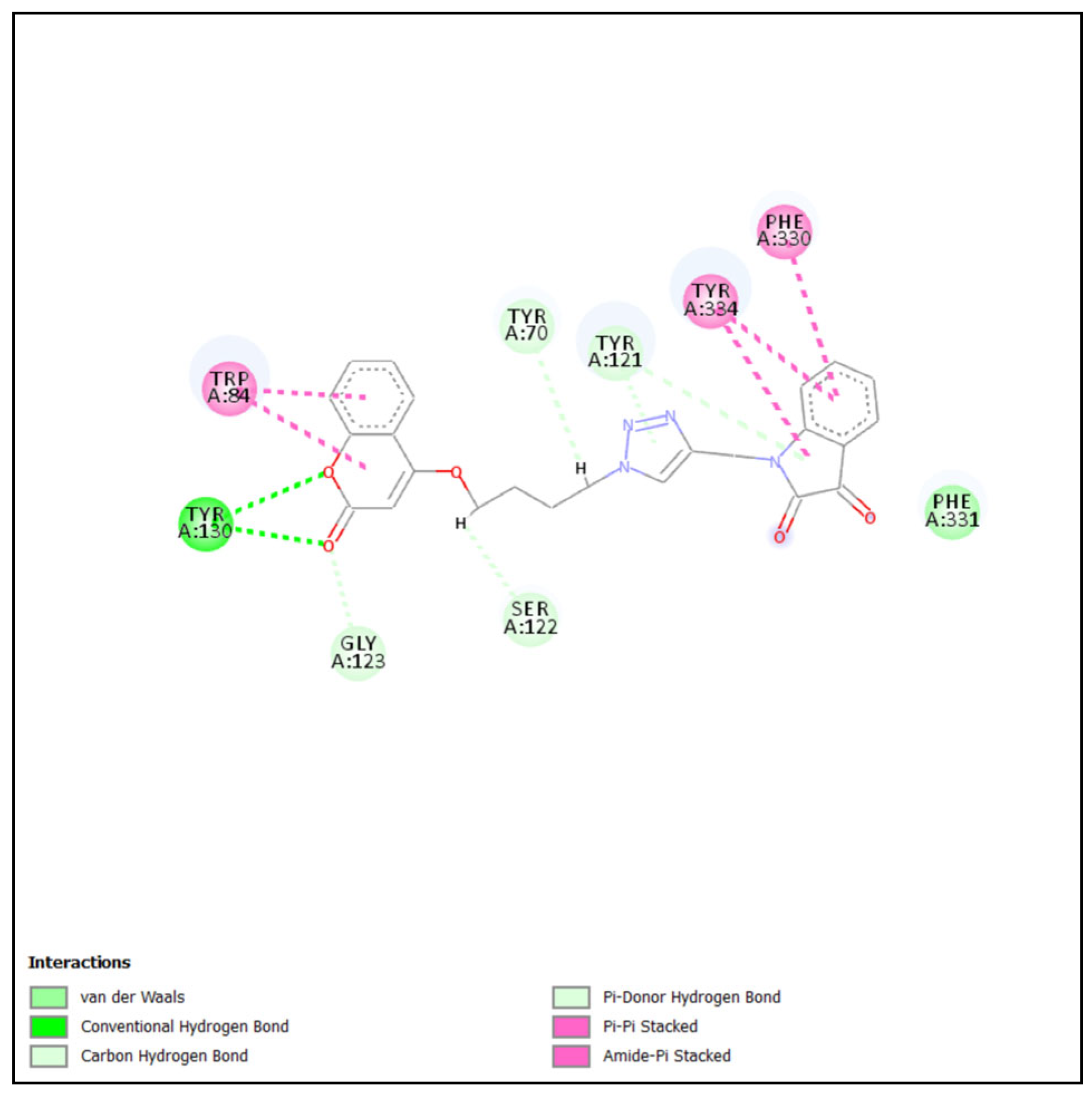
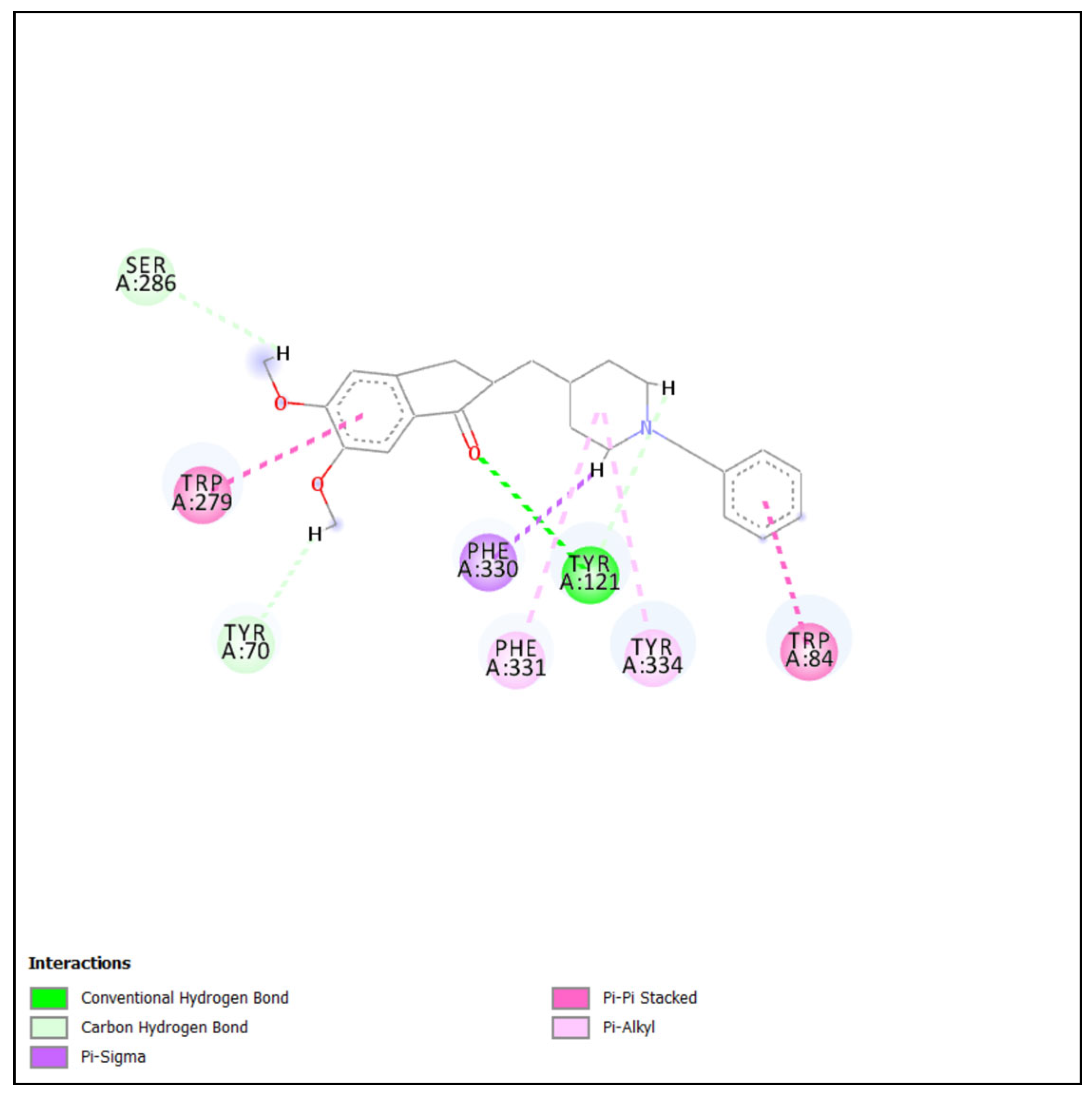
2.4.1. Molecular Docking of Most Active Compound into BChE
2.4.2. Molecular Docking of Most Active Compound into AChE
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Synthetic Procedures
3.2.1. Synthesis of 1-(Prop-2-ynyl)indoline-2,3-diones (2a–2g)
3.2.2. Synthesis of 4-(ω-bromoalkoxy)chromen-2-ones (4a–4c)
3.2.3. Synthesis of 4-(ω-azidoalkoxy)chromen-2-ones (5a–5c)
3.2.4. Synthesis of Coumarin–Triazole–Isatin Hybrids (6a1–6c7)
3.3. Biological Assays
3.3.1. Enzymatic Assays
- In Vitro Inhibitory Activity Against AChE and BChE
- Type of Enzyme Inhibition
3.3.2. Antimicrobial Screening
3.4. Molecular Docking
4. Conclusions
5. Patent
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tenchov, R.; Sasso, J.M.; Zhou, Q.A. Alzheimer’s Disease: Exploring the Landscape of Cognitive Decline. ACS Chem. Neurosci. 2024, 15, 3800–3827. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. A Blueprint for Dementia Research; WHO: Geneva, Switzerland, 2022; Available online: https://www.who.int/publications/i/item/9789240058248 (accessed on 16 February 2025).
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and Tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A. Neuronal and Vascular Oxidative Stress in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic System During the Progression of Alzheimer’s Disease: Therapeutic Implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase Inhibitors as Alzheimer’s Therapeutics: A Review. Mol. Med. Rep. 2019, 19, 1479–1487. [Google Scholar] [CrossRef]
- Han, S.-H.; Park, J.-C.; Byun, M.S.; Yi, D.; Lee, J.H.; Lee, D.Y.; Mook-Jung, I.; KBASE Research Group. Blood Acetylcholinesterase Level Is a Potential Biomarker for the Early Detection of Cerebral Amyloid Deposition in Cognitively Normal Individuals. Neurobiol. Aging 2019, 73, 21–29. [Google Scholar] [CrossRef]
- Jasiecki, J.; Targońska, M.; Wasąg, B. The Role of Butyrylcholinesterase and Iron in the Regulation of Cholinergic Network and Cognitive Dysfunction in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2033. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of Butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef]
- Greig, N.H.; Lahiri, D.K.; Kumar, S. Butyrylcholinesterase: An Important New Target in Alzheimer’s Disease Therapy. Int. Psychogeriatr. 2002, 14 (Suppl. 1), 77–91. [Google Scholar] [CrossRef]
- Atack, J.R.; Perry, E.K.; Bonham, J.R.; Candy, J.M.; Perry, R.H. Molecular Forms of Acetylcholinesterase and Butyrylcholinesterase in the Aged Human Central Nervous System. J. Neurochem. 1986, 47, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.R.; Maxwell, S.P.; Reid, G.A.; Cash, M.K.; DeBay, D.R.; Darvesh, S. Quantification of Butyrylcholinesterase Activity as a Sensitive and Specific Biomarker of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, H.; Yang, H.; Tan, R.; Bian, Y.; Fu, T.; Li, W.; Wu, L.; Pei, Y.; Sun, H. Discovery of New Acetylcholinesterase and Butyrylcholinesterase Inhibitors through Structure-Based Virtual Screening. RSC Adv. 2017, 7, 3429–3438. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D Structure to Function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef]
- Fang, L.; Pan, Y.; Muzyka, J.L.; Zhan, C.G. Active Site Gating and Substrate Specificity of Butyrylcholinesterase and Acetylcholinesterase: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2011, 115, 8797–8805. [Google Scholar] [CrossRef]
- De Boer, D.; Nguyen, N.; Mao, J.; Moore, J.; Sorin, E.J. A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules 2021, 11, 580. [Google Scholar] [CrossRef]
- Carney, G.; Bassett, K.; Wright, J.M.; Maclure, M.; McGuire, N.; Dormuth, C.R. Comparison of Cholinesterase Inhibitor Safety in Real-World Practice. Alzheimers Dement. Transl. Res. Clin. Interv. 2019, 5, 732–739. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Manolopoulos, A.; Ferrucci, L.; Kapogiannis, D. Critical Assessment of Anti-Amyloid-β Monoclonal Antibodies Effects in Alzheimer’s Disease: A Systematic Review and Meta-Analysis Highlighting Target Engagement and Clinical Meaningfulness. Sci. Rep. 2024, 14, 25741. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Nizamutdinov, D.; Yi, S.S.; Wu, E.; Huang, J.H. Disease Modifying Monoclonal Antibodies and Symptomatic Pharmacological Treatment for Alzheimer’s Disease. Biomedicines 2024, 12, 2636. [Google Scholar] [CrossRef]
- Jasiecki, J.; Targońska, M.; Janaszak-Jasiecka, A.; Kalinowski, L.; Waleron, K.; Wasąg, B. Butyrylcholinesterase Signal Sequence Self-Aggregates and Enhances Amyloid Fibril Formation in Vitro. Chem. Biol. Interact. 2023, 386, 110783. [Google Scholar] [CrossRef] [PubMed]
- Park-Wyllie, L.Y.; Mamdani, M.M.; Li, P.; Gill, S.S.; Laupacis, A.; Juurlink, D.N. Cholinesterase Inhibitors and Hospitalization for Bradycardia: A Population-Based Study. PLoS Med. 2009, 6, e1000157. [Google Scholar] [CrossRef]
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Sharma, A.; Bharate, S.B. Synthesis and Biological Evaluation of Coumarin Triazoles as Dual Inhibitors of Cholinesterases and β-Secretase. ACS Omega 2023, 8, 11161–11176. [Google Scholar] [CrossRef]
- Żołek, T.; Purgatorio, R.; Kłopotowski, Ł.; Catto, M.; Ostrowska, K. Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy. Int. J. Mol. Sci. 2024, 25, 12803. [Google Scholar] [CrossRef]
- Takomthong, P.; Waiwut, P.; Yenjai, C.; Sripanidkulchai, B.; Reubroycharoen, P.; Lai, R.; Kamau, P.; Boonyarat, C. Structure–Activity Analysis and Molecular Docking Studies of Coumarins from Toddalia asiatica as Multifunctional Agents for Alzheimer’s Disease. Biomedicines 2020, 8, 107. [Google Scholar] [CrossRef]
- Hasan, A.; Amran, S.; Hussain, F.; Jaff, A.; Jamalis, J. Molecular Docking and Recent Advances in the Design and Development of Cholinesterase Inhibitor Scaffolds: Coumarin Hybrids. ChemistrySelect 2019, 4, 14140–14156. [Google Scholar] [CrossRef]
- Benazzouz-Touami, A.; Chouh, A.; Halit, S.; Terrachet-Bouaziz, S.; Makhloufi-Chebli, M.; Ighil-Ahriz, K.; Silva, A.M.S. New Coumarin-Pyrazole hybrids: Synthesis, Docking studies and Biological evaluation as potential cholinesterase inhibitors. J. Mol. Struct. 2022, 1249, 131591. [Google Scholar] [CrossRef]
- Alcorn, K.N.; Oberhauser, I.A.; Politeski, M.D.; Eckroat, T.J. Evaluation of N-alkyl isatins and indoles as acetylcholinesterase and butyrylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2023, 39, 2286935. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Eckroat, T.J. Isatin-linked 4,4-dimethyl-5-methylene-4,5-dihydrothiazole-2-thiols for inhibition of acetylcholinesterase. Med. Chem. Res. 2021, 30, 2289–2300. [Google Scholar] [CrossRef]
- Shaik, B.B.; Katari, N.K.; Seboletswe, P.; Gundla, R.; Kushwaha, N.D.; Kumar, V.; Singh, P.; Karpoormath, R.; Bala, M.D. Recent Literature Review on Coumarin Hybrids as Potential Anticancer Agents. Anticancer Agents Med. Chem. 2023, 23, 142–163. [Google Scholar] [CrossRef] [PubMed]
- Rohman, N.; Ardiansah, B.; Wukirsari, T.; Judeh, Z. Recent Trends in the Synthesis and Bioactivity of Coumarin, Coumarin–Chalcone, and Coumarin–Triazole Molecular Hybrids. Molecules 2024, 29, 1026. [Google Scholar] [CrossRef]
- Cheke, R.S.; Patil, V.M.; Firke, S.D.; Ambhore, J.P.; Ansari, I.A.; Patel, H.M.; Shinde, S.D.; Pasupuleti, V.R.; Hassan, M.I.; Adnan, M.; et al. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals 2022, 15, 272. [Google Scholar] [CrossRef]
- Puerta, A.; González-Bakker, A.; Brandão, P.; Pineiro, M.; Burke, A.J.; Giovannetti, E.; Fernandes, M.X.; Padrón, J.M. Early Pharmacological Profiling of Isatin Derivatives as Potent and Selective Cytotoxic Agents. Biochem. Pharmacol. 2024, 222, 116059. [Google Scholar] [CrossRef]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A Review on Pharmacological Properties of Coumarins. Mini Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef]
- de Souza, L.G.; Rennã, M.N.; Figueroa-Villar, J.D. Coumarins as cholinesterase inhibitors: A review. Chem. Biol. Interact. 2016, 254, 11–23. [Google Scholar] [CrossRef]
- Yildirim, M.; Ersatır, M.; Yalın, S.; Giray, E.S. Coumarin Hybrids as Cholinesterase Inhibitors. Russ. J. Bioorg. Chem. 2023, 49, 970–975. [Google Scholar] [CrossRef]
- Sharma, A.; Nuthakki, V.K.; Gairola, S.; Singh, B.; Bharate, S.B. A Coumarin−Donepezil Hybrid as a Blood−Brain Barrier Permeable Dual Cholinesterase Inhibitor: Isolation, Synthetic Modifications, and Biological Evaluation of Natural Coumarins. ChemMedChem 2022, 17, 1535–1545. [Google Scholar] [CrossRef]
- Hamulakova, S.; Janovec, L.; Hrabinova, M.; Spilovska, K.; Korabecny, J.; Kristian, P.; Kuca, K.; Imrich, J. Synthesis and Biological Evaluation of Novel Tacrine Derivatives and Tacrine–Coumarin Hybrids as Cholinesterase Inhibitors. J. Med. Chem. 2014, 57, 6785–6796. [Google Scholar] [CrossRef] [PubMed]
- Baruah, P.; Basumatary, G.; Yesylevskyy, S.O.; Aguan, K.; Bez, G.; Mitra, S. Novel Coumarin Derivatives as Potent Acetylcholinesterase Inhibitors: Insight into Efficacy, Mode and Site of Inhibition. J. Biomol. Struct. Dyn. 2018, 36, 1750–1765. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Hussain, R.; Khan, Y.; Khan, M.U.; Sattar, A.; Saleem, M. Synthesis, in vitro bio-evaluation, and molecular docking study of thiosemicarbazone-based isatin/bis-Schiff base hybrid analogues as effective cholinesterase inhibitors. J. Mol. Struct. 2023, 1284, 135351. [Google Scholar] [CrossRef]
- Vaishnani, M.J.; Bijani, S.; Rahamathulla, M.; Baldaniya, L.; Jain, V.; Thajudeen, K.Y. Biological importance and synthesis of 1,2,3-triazole derivatives: A review. J. Chem. Technol. Biotechnol. 2024, 99, 2307989. [Google Scholar] [CrossRef]
- Raman, A.P.S.; Aslam, M.; Awasthi, A.; Ansari, A.; Jain, P.; Lal, K.; Bahadur, I.; Singh, P.; Kumari, K. An updated review on 1,2,3-/1,2,4-triazoles: Synthesis and diverse range of biological potential. Mol. Divers. 2025, 29, 899–964. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9530. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Peng, Y.; Zhu, L.; Wang, S.; Ji, J.; Rakesh, K.P. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019, 180, 656–672. [Google Scholar] [CrossRef]
- Cheke, R.S.; Patel, H.M.; Patil, V.M.; Ansari, I.A.; Ambhore, J.P.; Shinde, S.D.; Kadri, A.; Snoussi, M.; Adnan, M.; Kharkar, P.S.; et al. Molecular Insights into Coumarin Analogues as Antimicrobial Agents: Recent Developments in Drug Discovery. Antibiotics 2022, 11, 566. [Google Scholar] [CrossRef]
- Guo, H. Isatin Derivatives and Their Anti-Bacterial Activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef]
- Strzelecka, M.; Świątek, P. 1,2,4-Triazoles as Important Antibacterial Agents. Pharmaceuticals. 2021, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-Q.; Min, W.; Wang, J.; Si, X.-X.; Wang, X.-J.; Liu, Y.-W.; Shi, D.-H. Design, Synthesis and Biological Evaluation of New Carbazole–Coumarin Hybrids as Dual Binding Site Inhibitors of Acetylcholinesterase. J. Mol. Struct. 2021, 1229, 129784. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Timokhina, E.N.; Serkov, I.V.; Proshin, A.N.; Soldatova, Y.V.; et al. Combining Experimental and Computational Methods to Produce Conjugates of Anticholinesterase and Antioxidant Pharmacophores with Linker Chemistries Affecting Biological Activities Related to Treatment of Alzheimer’s Disease. Molecules 2024, 29, 321. [Google Scholar] [CrossRef] [PubMed]
- Shu, V.A.; Eni, D.B.; Ntie-Kang, F. A Survey of Isatin Hybrids and Their Biological Properties. Mol. Divers. 2025, 29, 1737–1760. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, T.A.; van Beek, B.; Wolters, L.P.; Bickelhaupt, F.M. Nucleophilic Substitution in Solution: Activation Strain Analysis of Weak and Strong Solvent Effects. Chem. Eur. J. 2018, 24, 5927–5938. [Google Scholar] [CrossRef]
- Tri, N.M.; Thanh, N.D.; Ha, L.N.; Anh, D.T.T.; Toan, V.N.; Giang, N.T.K. Study on Synthesis of Some Substituted N-Propargyl Isatins by Propargylation Reaction of Corresponding Isatins Using Potassium Carbonate as Base under Ultrasound- and Microwave-Assisted Conditions. Chem. Pap. 2021, 75, 4793–4801. [Google Scholar] [CrossRef]
- Derr, J.B.; Clark, J.A.; Morales, M.; Espinoza, E.M.; Vadhin, S.; Vullev, V.I. Solvent-Induced Selectivity of Williamson Etherification in the Pursuit of Amides Resistant against Oxidative Degradation. RSC Adv. 2020, 10, 24419–24424. [Google Scholar] [CrossRef]
- Mandal, S.; Mandal, S.; Ghosh, S.K.; Sar, P.; Ghosh, A.; Saha, R.; Saha, B. A Review on the Advancement of Ether Synthesis from Organic Solvent to Water. RSC Adv. 2016, 6, 69605–69614. [Google Scholar] [CrossRef]
- Bräse, S.; Banert, K. Organic Azides: Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Praveena Devi, C.H.B.; Vijay, K.; Hari Babu, B.; Adil, S.F.; Alam, M.M.; Vijjulatha, M.; Ansari, M.B. CuSO4/Sodium Ascorbate Catalysed Synthesis of Benzosuberone and 1,2,3-Triazole Conjugates: Design, Synthesis and In Vitro Anti-Proliferative Activity. J. Saudi Chem. Soc. 2019, 23, 980–991. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Ganeshpurkar, A.; Singh, R.; Shivhare, S.; Divya; Kumar, D.; Gutti, G.; Singh, R.; Kumar, A.; Singh, S.K. Improved Machine Learning Scoring Functions for Identification of Electrophorus electricus’s Acetylcholinesterase Inhibitors. Mol. Divers. 2022, 26, 1455–1479. [Google Scholar] [CrossRef]
- Teralı, Y.; Dalmizrak, O.; Uzairu, S.M.; Ozer, N. New Insights into the Interaction between Mammalian Butyrylcholinesterase and Amitriptyline: A Combined Experimental and Computational Approach. Turk. J. Biochem. 2019, 44, 55–61. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, S.; Sussman, J.L.; Silman, I.; Jiang, H. Computational Studies on Acetylcholinesterases. Molecules 2017, 22, 1324. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Colletier, J.-P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase: X-ray versus Molecular Dynamics. Biophys. J. 2008, 95, 2500–2511. [Google Scholar] [CrossRef]
- Khaw, K.Y.; Choi, S.B.; Tan, S.C.; Wahab, H.A.; Chan, K.L.; Murugaiyah, V. Prenylated Xanthones from Mangosteen as Promising Cholinesterase Inhibitors and Their Molecular Docking Studies. Phytomedicine 2014, 21, 1303–1309. [Google Scholar] [CrossRef]
- Maher, C.; Hassan, K.A. The Gram-Negative Permeability Barrier: Tipping the Balance of the In and the Out. mBio 2023, 14, e01205-23. [Google Scholar] [CrossRef]
- Rohde, M. The Gram-Positive Bacterial Cell Wall. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- OpenEye Scientific Software. OEDocking 4.0.0.4: FRED, ChemGauss4 Scoring Function; OpenEye Scientific Software: Santa Fe, NM, USA, 2024; Available online: https://docs.eyesopen.com/applications/oedocking (accessed on 11 April 2025).
- Miličević, A.; Šinko, G. Evaluation of the Key Structural Features of Various Butyrylcholinesterase Inhibitors Using Simple Molecular Descriptors. Molecules 2022, 27, 6894. [Google Scholar] [CrossRef]
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive Review of Mechanisms of Pathogenesis Involved in Alzheimer’s Disease and Potential Therapeutic Strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Barron, M.G. Development of 3D-QSAR Model for Acetylcholinesterase Inhibitors Using a Combination of Fingerprint, Molecular Docking, and Structure-Based Pharmacophore Approaches. Toxicol. Sci. 2015, 148, 60–70. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 27 December 2024).
- van der Westhuizen, C.J.; Stander, A.; Riley, D.L.; Panayides, J.-L. Discovery of Novel Acetylcholinesterase Inhibitors by Virtual Screening, In Vitro Screening, and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Deng, S.; Li, G.; Zhang, Y.; Wang, L.; Wu, C.; Deng, Y. Butenolide Derivatives from Aspergillus terreus Selectively Inhibit Butyrylcholinesterase. Front. Chem. 2022, 10, 1063284. [Google Scholar] [CrossRef] [PubMed]
- MAKE Receptor 3.2.0.2: OpenEye Scientific Software, Santa Fe, USA. Available online: https://docs.eyesopen.com/applications/oedocking/make_receptor/make_receptor_setup.html (accessed on 29 December 2024).
- FRED 3.2.0.2: OpenEye Scientific Software, Santa Fe, NM. Available online: https://www.eyesopen.com/ (accessed on 29 December 2024).
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- OMEGA 2.5.1.4: OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com/ (accessed on 29 December 2024).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Shamsian, S.; Sokouti, B.; Dastmalchi, S. Benchmarking Different Docking Protocols for Predicting the Binding Poses of Ligands Complexed with Cyclooxygenase Enzymes and Screening Chemical Libraries. Bioimpacts 2023, 14, 29955. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BChE | AChE | Selectivity BChE/AChE | ||
|---|---|---|---|---|---|
| % of Inhibition (Conc. 20 μM) | IC50 μM | % of Inhibition (Conc. 20 μM) | IC50 μM | ||
| 6a1 | 72.81 ± 2.17 | 6.08 ± 1.19 | 29.27 ± 2.14 | - | - |
| 6a2 | 66.15 ± 5.18 | 9.16 ± 1.69 | 11.78 ± 1.18 | - | - |
| 6a3 | 62.65 ± 6.19 | 10.58 ± 1.13 | 5.57 ± 1.19 | - | - |
| 6a4 | 22.70 ± 3.87 | - | 4.57 ± 0.87 | - | - |
| 6a5 | 40.89 ± 6.26 | - | 3.17 ± 0.26 | - | - |
| 6a6 | 4.89 ± 0.74 | - | 3.11 ± 0.44 | - | - |
| 6a7 | 2.91 ± 0.15 | - | 22.76 ± 3.66 | - | - |
| 6b1 | 69.23 ± 5.27 | 8.52 ± 1.28 | 69.23 ± 4.27 | 18.16 ± 1.19 | 2.13 |
| 6b2 | 75.51 ± 5.69 | 5.90 ± 1.09 | 44.27 ± 5.69 | - | - |
| 6b3 | 61.64 ± 7.24 | 11.25 ± 1.16 | 31.50 ± 3.24 | - | - |
| 6b4 | 66.22 ± 8.11 | 13.28 ± 2.19 | 22.91 ± 2.11 | - | - |
| 6b5 | 61.65 ± 7.49 | 14.84 ± 1.44 | 27.58 ± 1.49 | - | - |
| 6b6 | 5.89 ± 1.91 | - | 17.88 ± 1.91 | - | - |
| 6b7 | 4.88 ± 0.27 | - | 24.41 ± 1.27 | - | - |
| 6c1 | 84.52 ± 8.46 | 1.74 ± 0.29 | 67.12 ± 3.27 | 15.28 ± 1.22 | 8.78 |
| 6c2 | 79.13 ± 5.49 | 5.51 ± 1.38 | 60.98 ± 3.49 | 17.13 ± 1.65 | 3.11 |
| 6c3 | 5.19 ± 1.14 | - | 44.23 ± 3.14 | - | - |
| 6c4 | 27.10 ± 2.19 | - | 9.84 ± 1.19 | - | - |
| 6c5 | 22.93 ± 1.64 | - | 11.21 ± 1.64 | - | - |
| 6c6 | 4.15 ± 0.54 | - | 18.98 ± 1.96 | - | - |
| 6c7 | 10.32 ± 1.53 | - | 12.12 ± 1.26 | - | - |
| Donepezil | 74.22 ± 6.17 | 5.24 ± 1.16 | ~100 | 0.08 ± 0.02 | - |
| Compound | Staphylococcus aureus ATCC 25923 ZOI (mm ± SD) | Staphylococcus epidermidis ATCC 12228 ZOI (mm ± SD) |
|---|---|---|
| 6a1 | * | 6.08 ± 1.19 |
| 6a2 | * | 9.16 ± 1.69 |
| 6a3 | * | 10.58 ± 1.13 |
| 6a4 | 12.67 ± 2.31 | * |
| 6a5 | * | * |
| 6a6 | * | * |
| 6a7 | * | * |
| 6b1 | * | 8.52 ± 1.28 |
| 6b2 | 9.33 ± 4.04 | 5.90 ± 1.09 |
| 6b3 | 10.33 ± 1.15 | 11.25 ± 1.16 |
| 6b4 | 20.00 ± 3.61 | 13.28 ± 2.19 |
| 6b5 | * | 14.84 ± 1.44 |
| 6b6 | * | * |
| 6b7 | * | * |
| 6c1 | 8.67 ± 1.15 | 1.74 ± 0.29 |
| 6c2 | 12.00 ± 2.65 | 5.51 ± 1.38 |
| 6c3 | 10.67 ± 3.06 | * |
| 6c4 | 12.67 ± 0.58 | * |
| 6c5 | * | * |
| 6c6 | * | * |
| 6c7 | * | * |
| Ciprofloxacine | 27.00 ± 1.62 | 5.24 ± 1.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimkovski, A.; Dobričić, V.; Simić, M.R.; Jurhar Pavlova, M.; Mihajloska, E.; Sterjev, Z.; Poceva Panovska, A. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Coumarin–Triazole–Isatin Hybrids as Selective Butyrylcholinesterase Inhibitors. Molecules 2025, 30, 2121. https://doi.org/10.3390/molecules30102121
Dimkovski A, Dobričić V, Simić MR, Jurhar Pavlova M, Mihajloska E, Sterjev Z, Poceva Panovska A. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Coumarin–Triazole–Isatin Hybrids as Selective Butyrylcholinesterase Inhibitors. Molecules. 2025; 30(10):2121. https://doi.org/10.3390/molecules30102121
Chicago/Turabian StyleDimkovski, Aleksandar, Vladimir Dobričić, Milena R. Simić, Maja Jurhar Pavlova, Evgenija Mihajloska, Zoran Sterjev, and Ana Poceva Panovska. 2025. "Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Coumarin–Triazole–Isatin Hybrids as Selective Butyrylcholinesterase Inhibitors" Molecules 30, no. 10: 2121. https://doi.org/10.3390/molecules30102121
APA StyleDimkovski, A., Dobričić, V., Simić, M. R., Jurhar Pavlova, M., Mihajloska, E., Sterjev, Z., & Poceva Panovska, A. (2025). Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Coumarin–Triazole–Isatin Hybrids as Selective Butyrylcholinesterase Inhibitors. Molecules, 30(10), 2121. https://doi.org/10.3390/molecules30102121