
Preparation of Ru-Based Systems Through Metal Carbonyl Cluster Decomposition for the Base-Free 5-Hydroxymethylfurfural (HMF) Oxidation

 , , , ,
, , , ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
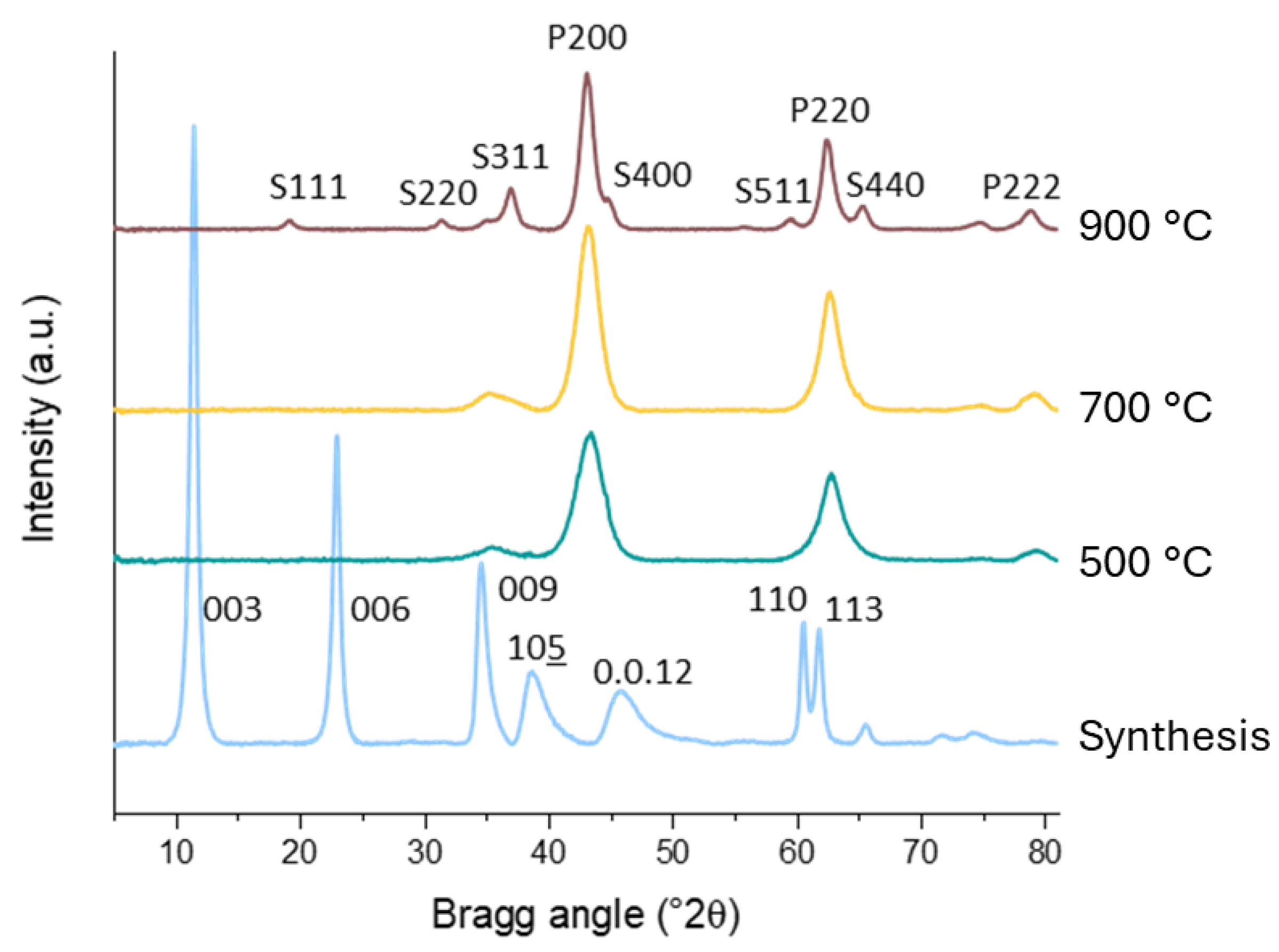
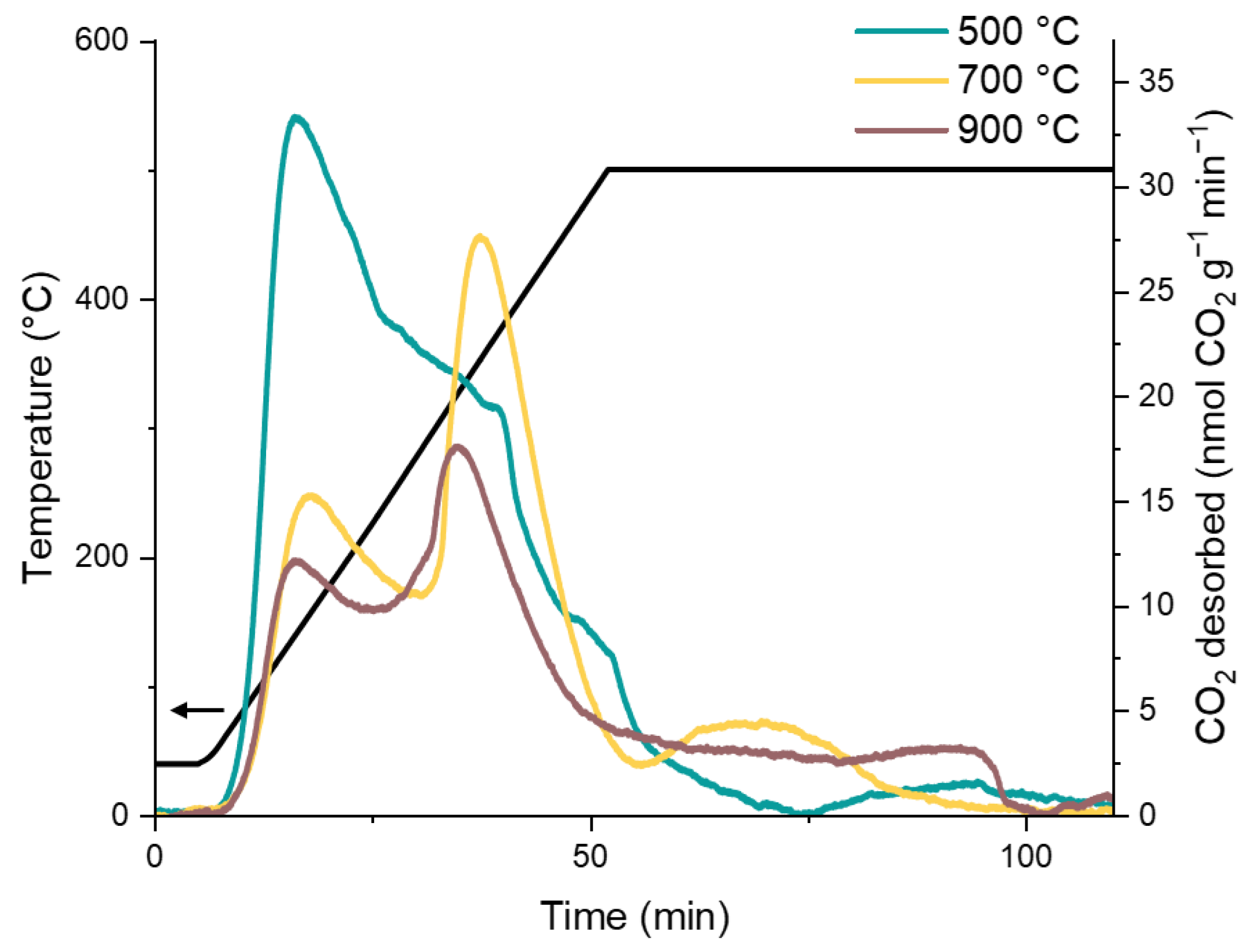
2.1. Characterisation of the Catalyst Supports
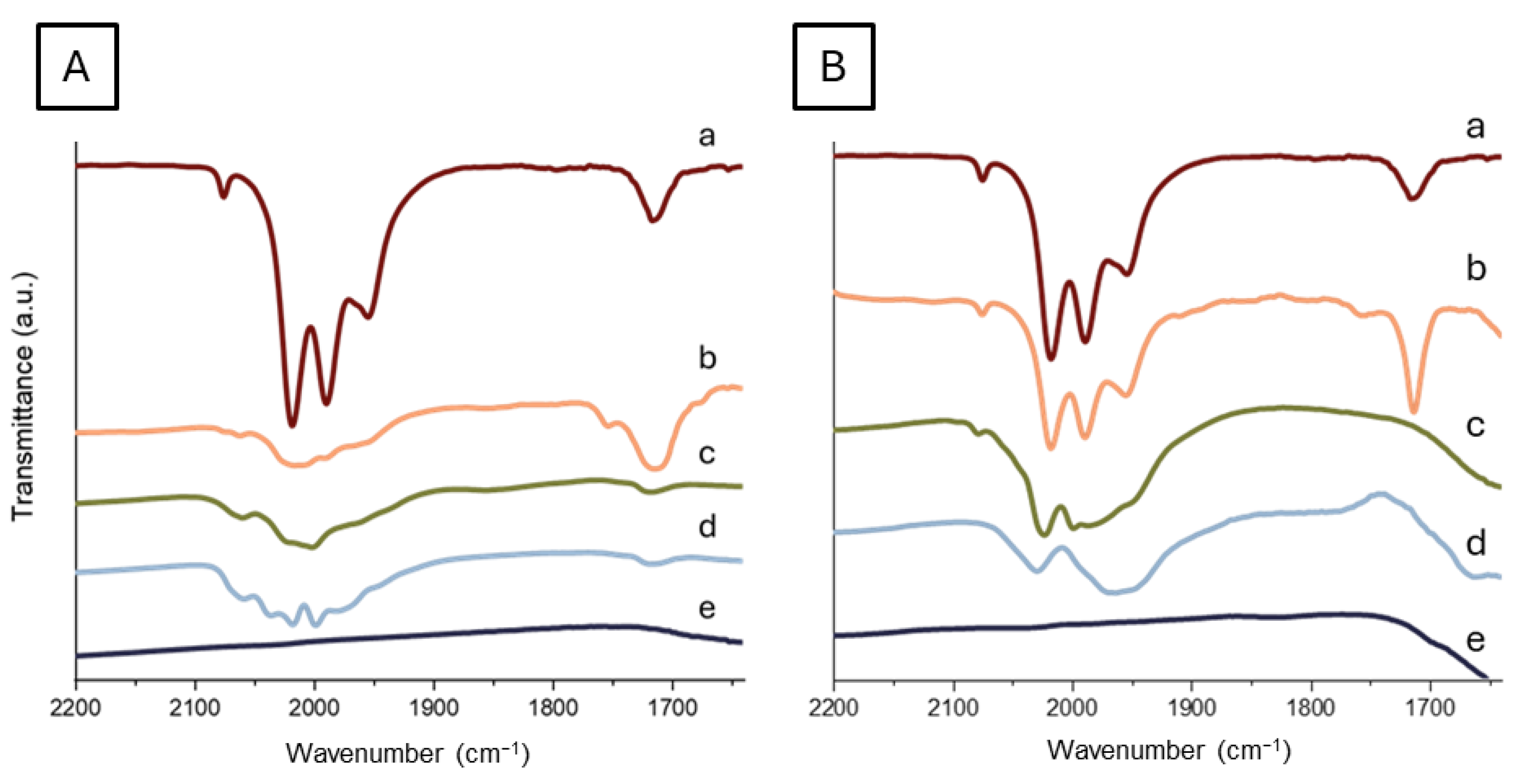
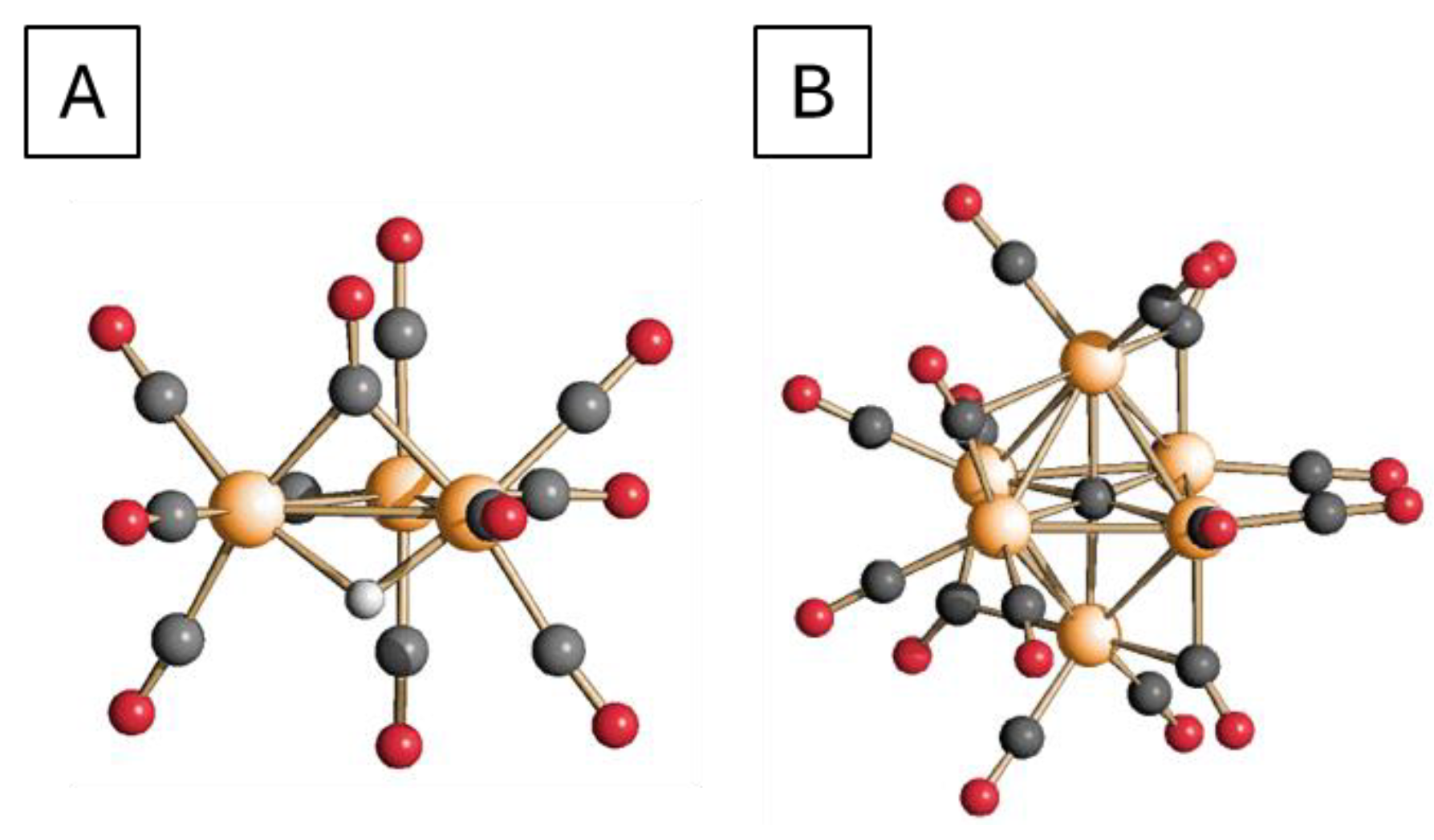
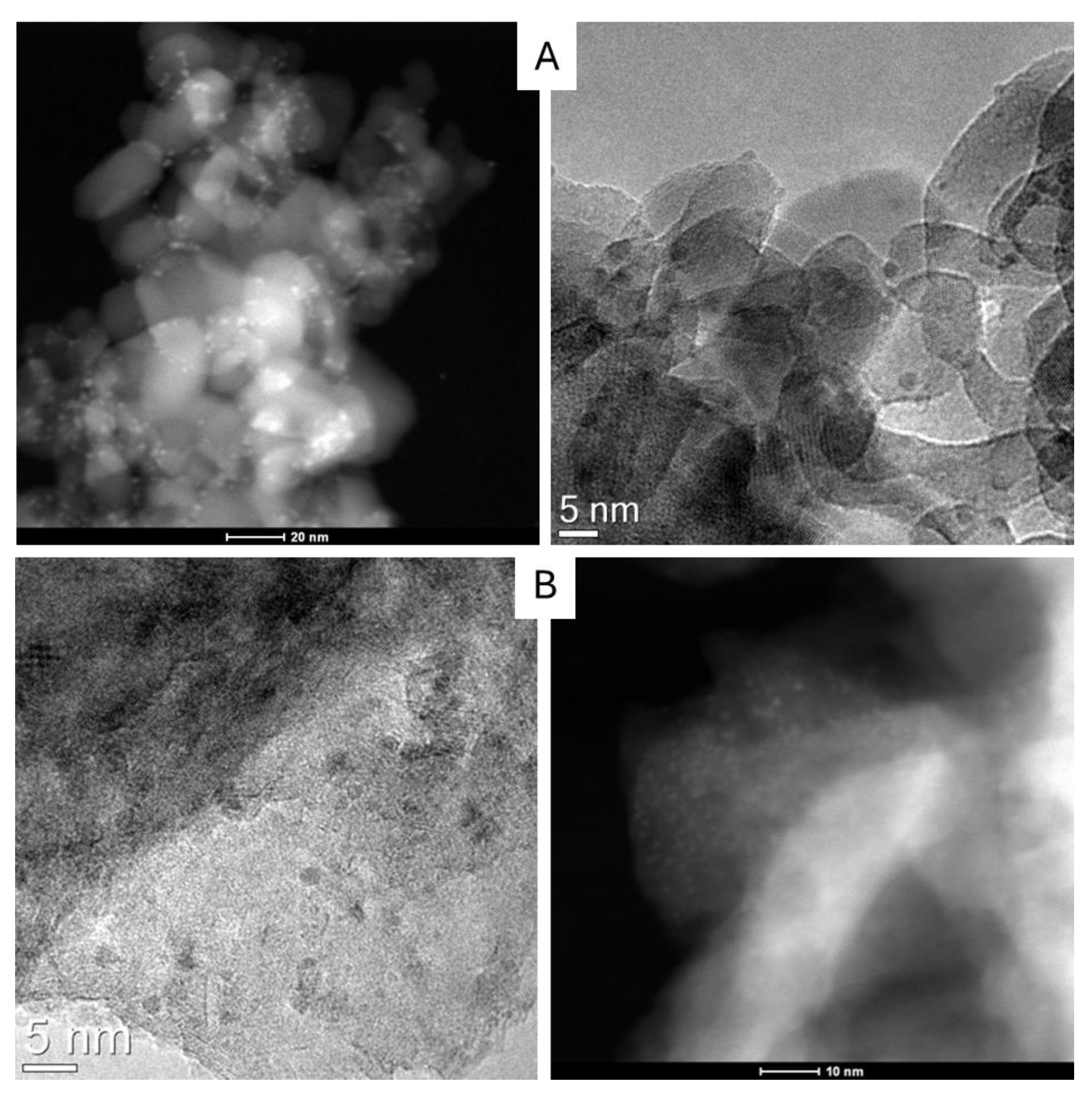
2.2. Characterization of the Ruthenium Catalysts
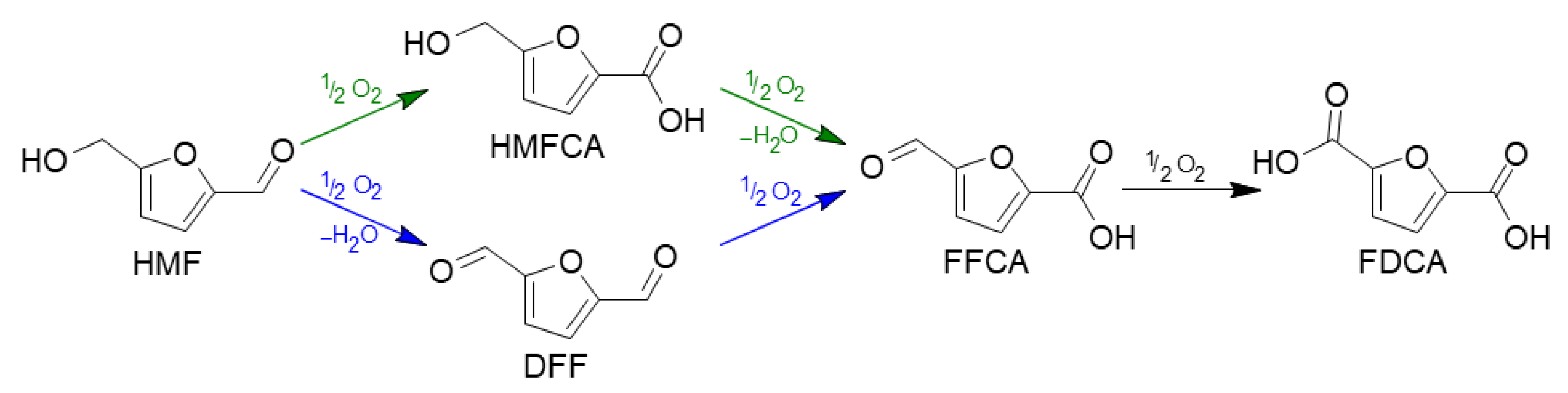
2.3. Preliminary Catalytic Tests
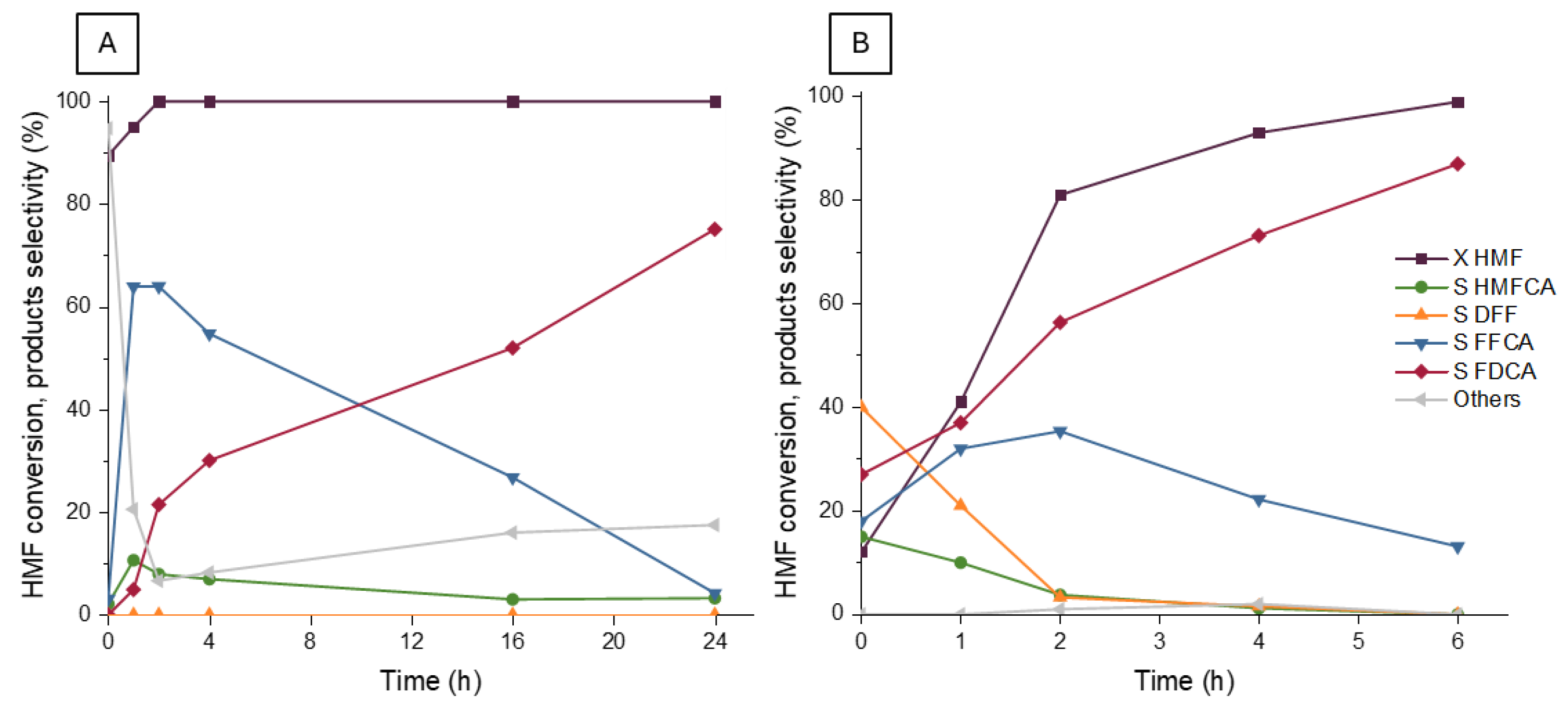
2.4. Activity of Ru/TiO2
2.5. Comparison Between TiO2- and Mg(Al)O-Based Ruthenium Catalysts
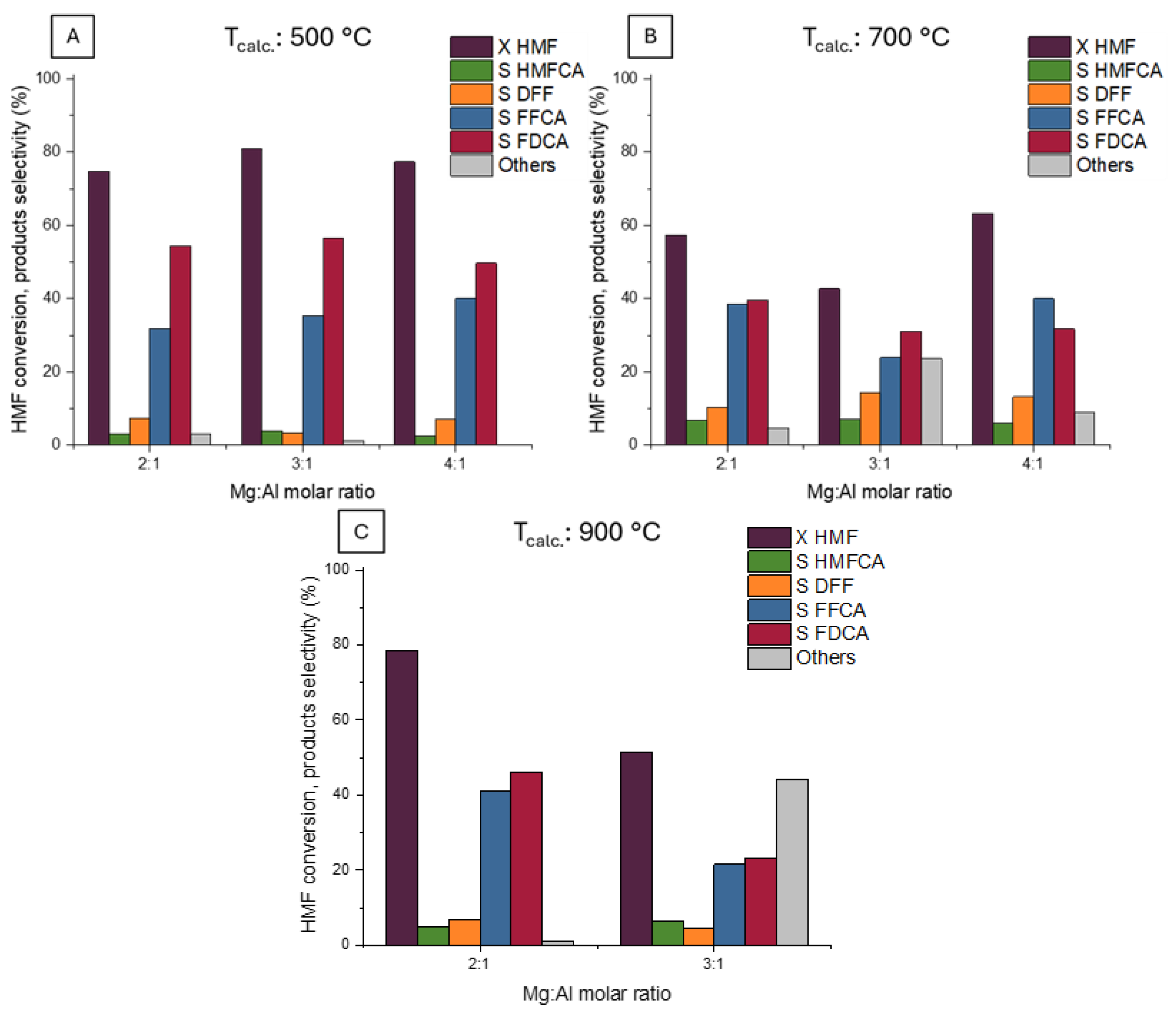
2.6. Effect of Mg/Al Ratio and Calcination Temperature on the Activity of Ru/Mg(Al)O Catalysts
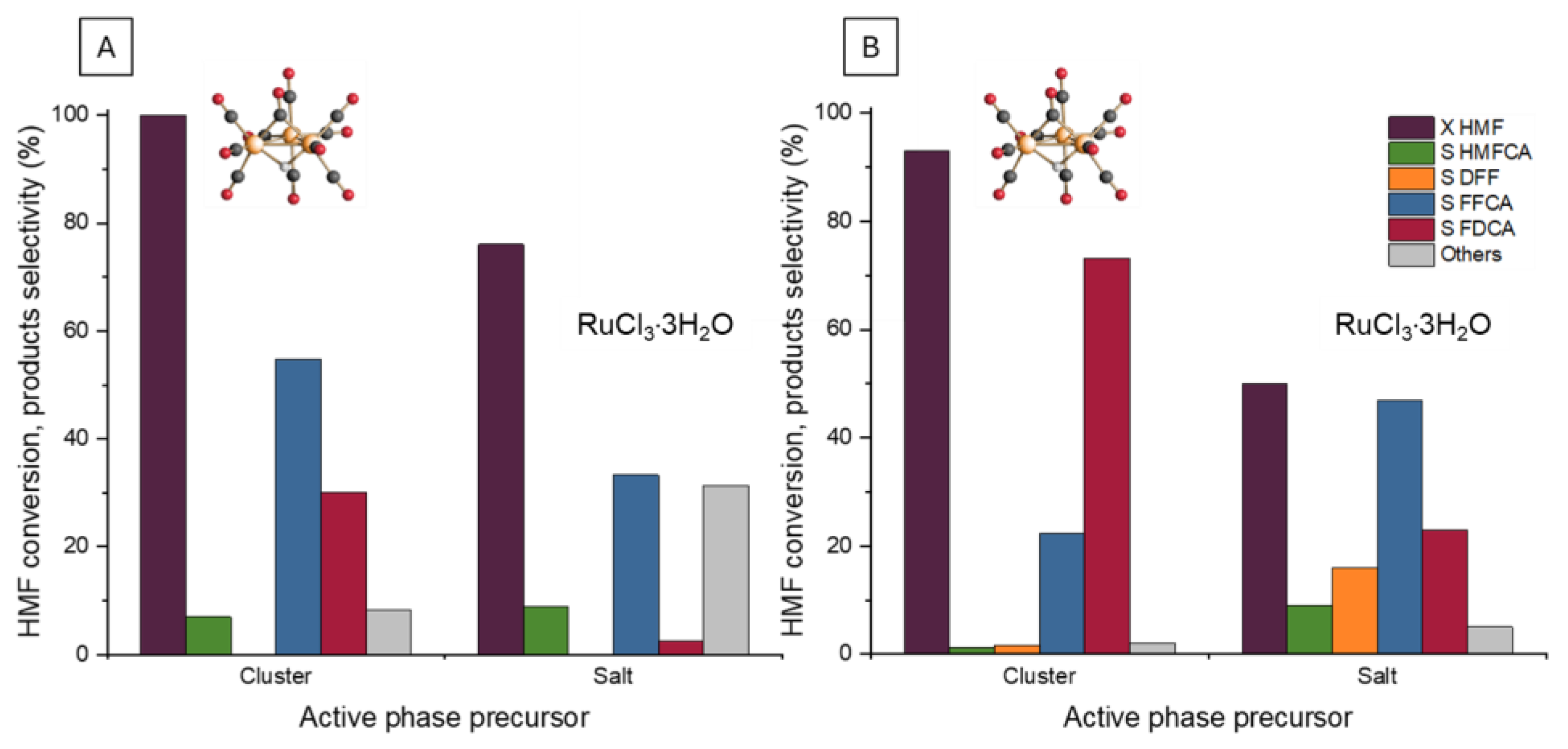
2.7. Comparison of Cluster- and Salt-Derived Ru Catalysts
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Mg-Al Hydrotalcites with Different Mg:Al Molar Ratios
3.3. Synthesis of [NEt4][HRu3(CO)11]
3.4. Catalyst Preparation
3.5. Characterization of the Materials
3.6. Catalytic Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| HMF | 5-hydroxymethylfurfural |
| HMFCA | 5-hydroxymethyl-2-furancarboxylic acid |
| DFF | 2,5-diformylfuran |
| FFCA | 5-formyl-2-furancarboxylic acid |
| FDCA | 2,5-furandicarboxylic acid |
| MCC | Metal carbonyl clusters |
| NP | Nanoparticle |
| LDH | Layer double hydroxide |
| w.b.s | weak basic sites |
| m.s.b.s. | medium and strong basic sites |
| p.a.b.s. | poorly accessible basic sites |
References
- Gates, B.C. Supported Metal Clusters: Synthesis, Structure, and Catalysis. Chem. Rev. 1995, 95, 511–522. [Google Scholar] [CrossRef]
- Guzman, J.; Gates, B.C. Supported Molecular Catalysts: Metal Complexes and Clusters on Oxides and Zeolites. Dalton Trans. 2003, 2003, 3303–3318. [Google Scholar] [CrossRef]
- Beck, A.; Horváth, A.; Sárkány, A.; Guczi, L. Building Blocks of Nanosized Supported Metals for Catalysis: Carbonyl Clusters or Colloids? Curr. Appl. Phys. 2006, 6, 200–204. [Google Scholar] [CrossRef]
- Zacchini, S. Using Metal Carbonyl Clusters to Develop a Molecular Approach towards Metal Nanoparticles. Eur. J. Inorg. Chem. 2011, 2011, 4125–4145. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Kambe, T.; Imaoka, T.; Yamamoto, K. Modern Cluster Design Based on Experiment and Theory. Nat. Rev. Chem. 2021, 5, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiao, F.-S.; Purnell, S.K.; Alexeev, O.; Kawi, S.; Deutsch, S.E.; Gates, B.C. Size-Dependent Catalytic Activity of Supported Metal Clusters. Nature 1994, 372, 346–348. [Google Scholar] [CrossRef]
- Psaro, R.; Recchia, S. Supported Metals Derived from Organometallics. Catal. Today 1998, 41, 139–147. [Google Scholar] [CrossRef]
- Heiz, U.; Bullock, E.L. Fundamental Aspects of Catalysis on Supported Metal Clusters. J. Mater. Chem. 2004, 14, 564–577. [Google Scholar] [CrossRef]
- Tyo, E.C.; Vajda, S. Catalysis by Clusters with Precise Numbers of Atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef]
- Rong, H.; Ji, S.; Zhang, J.; Wang, D.; Li, Y. Synthetic Strategies of Supported Atomic Clusters for Heterogeneous Catalysis. Nat. Commun. 2020, 11, 5884. [Google Scholar] [CrossRef]
- Li, X.; Mitchell, S.; Fang, Y.; Li, J.; Perez-Ramirez, J.; Lu, J. Advances in Heterogeneous Single-Cluster Catalysis. Nat. Rev. Chem. 2023, 7, 754–767. [Google Scholar] [CrossRef]
- Albonetti, S.; Bonelli, R.; Epoupa Mengou, J.; Femoni, C.; Tiozzo, C.; Zacchini, S.; Trifirò, F. Gold/Iron Carbonyl Clusters as Precursors for TiO2 Supported Catalysts. Catal. Today 2008, 137, 483–488. [Google Scholar] [CrossRef]
- Longo, A.; de Boed, E.J.J.; Mammen, N.; van der Linden, M.; Honkala, K.; Häkkinen, H.; de Jongh, P.E.; Donoeva, B. Towards Atomically Precise Supported Catalysts from Monolayer-Protected Clusters: The Critical Role of the Support. Chem. Eur. J. 2020, 26, 7051–7058. [Google Scholar] [CrossRef] [PubMed]
- Bonincontro, D.; Lolli, A.; Storione, A.; Gasparotto, A.; Berti, B.; Zacchini, S.; Dimitratos, N.; Albonetti, S. Pt and Pt/Sn Carbonyl Clusters as Precursors for the Synthesis of Supported Metal Catalysts for the Base-Free Oxidation of HMF. Appl. Catal. A Gen. 2019, 588, 117279. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology Development for the Production of Biobased Products from Biorefinery Carbohydrates—The US Department of Energy’s “Top 10” Revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Sousa, A.F.; Vilela, C.; Fonseca, A.C.; Matos, M.; Freire, C.S.R.; Gruter, G.-J.M.; Coelho, J.F.J.; Silvestre, A.J.D. Biobased Polyesters and Other Polymers from 2,5-Furandicarboxylic Acid: A Tribute to Furan Excellency. Polym. Chem. 2015, 6, 5961–5983. [Google Scholar] [CrossRef]
- Mineral Commodity Summaries 2025 (Version 1.2); U.S. Geological Survey: Reston, VA, USA, 2025; p. 136.
- Chang, Y.-F. Ruthenium-Materials Properties, Device Characterizations, and Advanced Applications; IntechOpen: London, UK, 2023; ISBN 978-1-80356-987-1. [Google Scholar]
- Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs (European Commission); Grohol, M.; Veeh, C. Study on the Critical Raw Materials for the EU 2023: Final Report; Publications Office of the European Union: Luxembourg, 2023; ISBN 978-92-68-00414-2. [Google Scholar]
- Ishida, H. Ruthenium-An Element Loved by Researchers; IntechOpen: London, UK, 2022; ISBN 978-1-83962-917-4. [Google Scholar]
- Giboulot, S.; Baldino, S.; Ballico, M.; Nedden, H.G.; Zuccaccia, D.; Baratta, W. Cyclometalated Dicarbonyl Ruthenium Catalysts for Transfer Hydrogenation and Hydrogenation of Carbonyl Compounds. Organometallics 2018, 37, 2136–2146. [Google Scholar] [CrossRef]
- Röper, M. Homogeneous Catalysis. The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes. 2. Auflage. Von G. W. Parshall Und S. D. Ittel. Wiley, New York, 1992. XII, 342 S., Geb. 39.95£.—ISBN 0-471-53829-9. Angew. Chem. 1993, 105, 1153–1154. [Google Scholar] [CrossRef]
- Hanyu, A.; Takezawa, E.; Sakaguchi, S.; Ishii, Y. Selective Aerobic Oxidation of Primary Alcohols Catalyzed by a Ru(PPh3)3Cl2/Hydroquinone System. Tetrahedron Lett. 1998, 39, 5557–5560. [Google Scholar] [CrossRef]
- Csjernyik, G.; Éll, A.H.; Fadini, L.; Pugin, B.; Bäckvall, J.-E. Efficient Ruthenium-Catalyzed Aerobic Oxidation of Alcohols Using a Biomimetic Coupled Catalytic System. J. Org. Chem. 2002, 67, 1657–1662. [Google Scholar] [CrossRef]
- Albonetti, S.; Mazzoni, R.; Cavani, F. Homogeneous, Heterogeneous and Nanocatalysis. In Transition Metal Catalysis in Aerobic Alcohol Oxidation; Royal Society of Chemistry: London, UK, 2014. [Google Scholar] [CrossRef]
- Kar, S.; Milstein, D. Oxidation of Organic Compounds Using Water as the Oxidant with H2 Liberation Catalyzed by Molecular Metal Complexes. Acc. Chem. Res. 2022, 55, 2304–2315. [Google Scholar] [CrossRef]
- Kar, S.; Zhou, Q.-Q.; Ben-David, Y.; Milstein, D. Catalytic Furfural/5-Hydroxymethyl Furfural Oxidation to Furoic Acid/Furan-2,5-Dicarboxylic Acid with H2 Production Using Alkaline Water as the Formal Oxidant. J. Am. Chem. Soc. 2022, 144, 1288–1295. [Google Scholar] [CrossRef]
- Kondo, T.; Kimura, Y.U.; Yamada, H.; Toshimitsu, A. Ruthenium-Based Catalysts for Aerobic Oxidation of Alcohols. In Transition Metal Catalysis in Aerobic Alcohol Oxidation; Royal Society of Chemistry: London, UK, 2014. [Google Scholar] [CrossRef]
- Gorbanev, Y.Y.; Kegnæs, S.; Hanning, C.W.; Hansen, T.W.; Riisager, A. Acetic Acid Formation by Selective Aerobic Oxidation of Aqueous Ethanol over Heterogeneous Ruthenium Catalysts. ACS Catal. 2012, 2, 604–612. [Google Scholar] [CrossRef]
- Thiensuwan, N.; Sankaranarayanan, S.; Yokoi, T.; Ngamcharussrivichai, C. Exfoliated Layered Metal Oxide-Supported Ruthenium Catalysts for Base-Free Oxidation of 5-Hydroxymethylfurfural into a Renewable Bioplastic Precursor. ACS Sustain. Chem. Eng. 2023, 11, 11424–11436. [Google Scholar] [CrossRef]
- Gorbanev, Y.Y.; Kegnæs, S.; Riisager, A. Effect of Support in Heterogeneous Ruthenium Catalysts Used for the Selective Aerobic Oxidation of HMF in Water. Top. Catal. 2011, 54, 1318. [Google Scholar] [CrossRef]
- Gorbanev, Y.Y.; Kegnæs, S.; Riisager, A. Selective Aerobic Oxidation of 5-Hydroxymethylfurfural in Water Over Solid Ruthenium Hydroxide Catalysts with Magnesium-Based Supports. Catal. Lett. 2011, 141, 1752–1760. [Google Scholar] [CrossRef]
- Da Fonseca Ferreira, A.D.; de Mello, M.D.; Pereira da Silva, M.A. Catalytic Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid over Ru/Al2O3 in a Trickle-Bed Reactor. Ind. Eng. Chem. Res. 2019, 58, 128–137. [Google Scholar] [CrossRef]
- Mishra, D.K.; Lee, H.J.; Kim, J.; Lee, H.-S.; Cho, J.K.; Suh, Y.-W.; Yi, Y.; Kim, Y.J. MnCo2O4 Spinel Supported Ruthenium Catalyst for Air-Oxidation of HMF to FDCA under Aqueous Phase and Base-Free Conditions. Green Chem. 2017, 19, 1619–1623. [Google Scholar] [CrossRef]
- Antonyraj, C.A.; Huynh, N.T.T.; Lee, K.W.; Kim, Y.J.; Shin, S.; Shin, J.S.; Cho, J.K. Base-Free Oxidation of 5-Hydroxymethyl-2-Furfural to 2,5-Furan Dicarboxylic Acid over Basic Metal Oxide-Supported Ruthenium Catalysts under Aqueous Conditions. J. Chem. Sci. 2018, 130, 156. [Google Scholar] [CrossRef]
- Perumal, S.K.; Lee, S.; Yu, H.; Heo, J.; Kang, M.J.; Kim, Y.; Park, M.; Lee, H.; Kim, H.S. Synergistic Interaction between Ruthenium Catalysts and Grafted Niobium on SBA-15 for 2,5-Furandicarboxylic Acid Production Using 5-Hydroxymethylfurfural. ACS Appl. Mater. Interfaces 2024, 16, 7353–7363. [Google Scholar] [CrossRef]
- Pasini, T.; Piccinini, M.; Blosi, M.; Bonelli, R.; Albonetti, S.; Dimitratos, N.; Lopez-Sanchez, J.A.; Sankar, M.; He, Q.; Kiely, C.J.; et al. Selective Oxidation of 5-Hydroxymethyl-2-Furfural Using Supported Gold–Copper Nanoparticles. Green Chem. 2011, 13, 2091–2099. [Google Scholar] [CrossRef]
- Albonetti, S.; Pasini, T.; Lolli, A.; Blosi, M.; Piccinini, M.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Hutchings, G.J.; et al. Selective Oxidation of 5-Hydroxymethyl-2-Furfural over TiO2-Supported Gold–Copper Catalysts Prepared from Preformed Nanoparticles: Effect of Au/Cu Ratio. Catal. Today 2012, 195, 120–126. [Google Scholar] [CrossRef]
- Lolli, A.; Maslova, V.; Bonincontro, D.; Basile, F.; Ortelli, S.; Albonetti, S. Selective Oxidation of HMF via Catalytic and Photocatalytic Processes Using Metal-Supported Catalysts. Molecules 2018, 23, 2792. [Google Scholar] [CrossRef]
- Cattaneo, S.; Bonincontro, D.; Bere, T.; Kiely, C.J.; Hutchings, G.J.; Dimitratos, N.; Albonetti, S. Continuous Flow Synthesis of Bimetallic AuPd Catalysts for the Selective Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid. ChemNanoMat 2020, 6, 420–426. [Google Scholar] [CrossRef]
- Allegri, A.; Maslova, V.; Blosi, M.; Costa, A.L.; Ortelli, S.; Basile, F.; Albonetti, S. Photocatalytic Oxidation of HMF under Solar Irradiation: Coupling of Microemulsion and Lyophilization to Obtain Innovative TiO2-Based Materials. Molecules 2020, 25, 5225. [Google Scholar] [CrossRef]
- Verdeguer, P.; Merat, N.; Gaset, A. Oxydation Catalytique Du HMF En Acide 2,5-Furane Dicarboxylique. J. Mol. Catal. 1993, 85, 327–344. [Google Scholar] [CrossRef]
- Sels, B.F.; De Vos, D.E.; Jacobs, P.A. Hydrotalcite-like Anionic Clays in Catalytic Organic Reactions. Catal. Rev. 2001, 43, 443–488. [Google Scholar] [CrossRef]
- Ardemani, L.; Cibin, G.; Dent, A.J.; Isaacs, M.A.; Kyriakou, G.; Lee, A.F.; Parlett, C.M.A.; Parry, S.A.; Wilson, K. Solid Base Catalysed 5-HMF Oxidation to 2,5-FDCA over Au/Hydrotalcites: Fact or Fiction? Chem. Sci. 2015, 6, 4940–4945. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, K.; Lei, D.; Si, W.; Feng, Y.; Lou, L.-L.; Liu, S. Basicity-Tuned Hydrotalcite-Supported Pd Catalysts for Aerobic Oxidation of 5-Hydroxymethyl-2-Furfural under Mild Conditions. ACS Sustain. Chem. Eng. 2016, 4, 4752–4761. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Effective Ionic Radii in Oxides and Fluorides. Acta Cryst. B 1969, 25, 925–946. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-Type Anionic Clays: Preparation, Properties and Applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Hazen, R.M. Effects of Temperature and Pressure on the Cell Dimension and X-Ray Temperature Factors of Periclase. Am. Mineral. 1976, 61, 266–271. [Google Scholar]
- Saraiva, M.; Georgieva, V.; Mahieu, S.; Van Aeken, K.; Bogaerts, A.; Depla, D. Compositional Effects on the Growth of Mg(M)O Films. J. Appl. Phys. 2010, 107, 034902. [Google Scholar] [CrossRef]
- Hiremath, V.; Kwon, H.J.; Jung, I.-S.; Kwon, S.; Kwon, S.H.; Lee, S.G.; Lee, H.C.; Seo, J.G. Mg-Ion Inversion in MgO@MgO−Al2O3 Oxides: The Origin of Basic Sites. ChemSusChem 2019, 12, 2810–2818. [Google Scholar] [CrossRef]
- Sato, T.; Kato, K.; Endo, T.; Shimada, M. Preparation and Chemical Properties of Magnesium Aluminium Oxide Solid Solutions. React. Solids 1986, 2, 253–260. [Google Scholar] [CrossRef]
- Vyas, S.; Grimes, R.W.; Binks, D.J.; Rey, F. Metastable Solid Solutions of Alumina in Magnesia. J. Phys. Chem. Solids 1997, 58, 1619–1624. [Google Scholar] [CrossRef]
- Hobbs, C.; Jaskaniec, S.; McCarthy, E.K.; Downing, C.; Opelt, K.; Güth, K.; Shmeliov, A.; Mourad, M.C.D.; Mandel, K.; Nicolosi, V. Structural Transformation of Layered Double Hydroxides: An In Situ TEM Analysis. npj 2D Mater. Appl. 2018, 2, 4. [Google Scholar] [CrossRef]
- Rebours, B.; d’Espinose De La Caillerie, J.-B.; Clause, O. Decoration of Nickel and Magnesium Oxide Crystallites with Spinel-Type Phases. J. Am. Chem. Soc. 1994, 116, 1707–1717. [Google Scholar] [CrossRef]
- Fornasari, G.; Gazzano, M.; Matteuzzi, D.; Trifirò, F.; Vaccari, A. Structure and Reactivity of High-Surface-Area Ni/Mg/Al Mixed Oxides. Appl. Clay Sci. 1995, 10, 69–82. [Google Scholar] [CrossRef]
- Gazzano, M.; Kagunya, W.; Matteuzzi, D.; Vaccari, A. Neutron Diffraction Studies of Polycrystalline Ni/Mg/Al Mixed Oxides Obtained from Hydrotalcite-like Precursors. J. Phys. Chem. B 1997, 101, 4514–4519. [Google Scholar] [CrossRef]
- Lavina, B.; Salviulo, G.; Giusta, A.D. Cation Distribution and Structure Modelling of Spinel Solid Solutions. Phys. Chem. Miner. 2002, 29, 10–18. [Google Scholar] [CrossRef]
- Hill, R.J.; Craig, J.R.; Gibbs, G.V. Systematics of the Spinel Structure Type. Phys. Chem. Miner. 1979, 4, 317–339. [Google Scholar] [CrossRef]
- Andreozzi, G.B.; Princivalle, F. Kinetics of Cation Ordering in Synthetic MgAl2O4 Spinel. Am. Mineral. 2002, 87, 838–844. [Google Scholar] [CrossRef]
- Shukla, P.; Chernatynskiy, A.; Nino, J.C.; Sinnott, S.B.; Phillpot, S.R. Effect of Inversion on Thermoelastic and Thermal Transport Properties of MgAl2O4 Spinel by Atomistic Simulation. J. Mater. Sci. 2011, 46, 55–62. [Google Scholar] [CrossRef]
- Warringham, R.; Mitchell, S.; Murty, R.; Schäublin, R.; Crivelli, P.; Kenvin, J.; Pérez-Ramírez, J. Mapping the Birth and Evolution of Pores upon Thermal Activation of Layered Hydroxides. Chem. Mater. 2017, 29, 4052–4062. [Google Scholar] [CrossRef]
- Menezes, A.O.; Silva, P.S.; Padrón Hernández, E.; Borges, L.E.P.; Fraga, M.A. Tuning Surface Basic Properties of Nanocrystalline MgO by Controlling the Preparation Conditions. Langmuir 2010, 26, 3382–3387. [Google Scholar] [CrossRef]
- Gorte, R.J. Design Parameters for Temperature Programmed Desorption from Porous Catalysts. J. Catal. 1982, 75, 164–174. [Google Scholar] [CrossRef]
- Tronconi, E.; Lietti, L.; Forzatti, P. Diffusion and Readsorption Effects during TPD from Porous Materials. Thermochim. Acta 1988, 135, 147–153. [Google Scholar] [CrossRef]
- Evans, J.V.; Whateley, T.L. Infra-Red Study of Adsorption of Carbon Dioxide and Water on Magnesium Oxide. Trans. Faraday Soc. 1967, 63, 2769–2777. [Google Scholar] [CrossRef]
- Morterra, C.; Ghiotti, G.; Boccuzi, F.; Coluccia, S. An Infrared Spectroscopic Investigation of the Surface Properties of Magnesium Aluminate Spinel. J. Catal. 1978, 51, 299–313. [Google Scholar] [CrossRef]
- Philipp, R.; Fujimoto, K. FTIR Spectroscopic Study of Carbon Dioxide Adsorption/Desorption on Magnesia/Calcium Oxide Catalysts. J. Phys. Chem. 1992, 96, 9035–9038. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Takaoka, K.; Yamabe, S.; Ito, T. Interaction of CO2 with Magnesium Oxide Surfaces: A TPD, FTIR, and Cluster-Model Calculation Study. J. Phys. Chem. 1995, 99, 3704–3710. [Google Scholar] [CrossRef]
- Ungermann, C.; Landis, V.; Moya, S.A.; Cohen, H.; Walker, H.; Pearson, R.G.; Rinker, R.G.; Ford, P.C. Homogeneous Catalysis of the Water Gas Shift Reaction by Ruthenium and Other Metal Carbonyls. Studies in Alkaline Solutions. J. Am. Chem. Soc. 1979, 101, 5922–5929. [Google Scholar] [CrossRef]
- Liu, N.; Guo, L.; Cao, Z.; Li, W.; Zheng, X.; Shi, Y.; Guo, J.; Xi, Y. Mechanisms of the Water–Gas Shift Reaction Catalyzed by Ruthenium Carbonyl Complexes. J. Phys. Chem. A 2016, 120, 2408–2419. [Google Scholar] [CrossRef]
- Bergmeister, J.J.; Hanson, B.E. In Situ Synthesis of [HRu3(CO)11]− on the Surface of Hydroxylated Aluminum Oxide. J. Organomet. Chem. 1988, 352, 367–372. [Google Scholar] [CrossRef]
- Cesari, C.; Bortoluzzi, M.; Funaioli, T.; Femoni, C.; Iapalucci, M.C.; Zacchini, S. Highly Reduced Ruthenium Carbide Carbonyl Clusters: Synthesis, Molecular Structure, Reactivity, Electrochemistry, and Computational Investigation of [Ru6C(CO)15]4–. Inorg. Chem. 2023, 62, 14590–14603. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Yeh, W.-Y.; Lee, G.-H.; Peng, S.-M. One-Step Synthesis of [HM3(CO)11]− from M3(CO)12 (M = Fe, Ru, Os) via Unusual Hydride Transfer from 1,3,5-Trimethyl-1,3,5-Triazacyclohexane. Organometallics 2003, 22, 4163–4166. [Google Scholar] [CrossRef]
- Li, C.; Xu, J.; Zhao, J.; Tian, D.; King, R.B. The Maximum Number of Carbonyl Groups around an Ru6C Polyhedral Cluster: Hexanuclear Ruthenium Carbonyl Carbides. Dalton Trans. 2010, 39, 10697–10701. [Google Scholar] [CrossRef]
- Asakura, K.; Iwasawa, Y. Surface Structure and Catalysis for CO Hydrogenation of the Supported Ru Species Derived from the Ru3(CO)12 Inorganic Oxides. J. Chem. Soc. Faraday Trans. 1990, 86, 2657–2662. [Google Scholar] [CrossRef]
- Chen, P.; Khetan, A.; Yang, F.; Migunov, V.; Weide, P.; Stürmer, S.P.; Guo, P.; Kähler, K.; Xia, W.; Mayer, J.; et al. Experimental and Theoretical Understanding of Nitrogen-Doping-Induced Strong Metal–Support Interactions in Pd/TiO2 Catalysts for Nitrobenzene Hydrogenation. ACS Catal. 2017, 7, 1197–1206. [Google Scholar] [CrossRef]
- Bonincontro, D.; Lolli, A.; Villa, A.; Prati, L.; Dimitratos, N.; Veith, G.M.; Chinchilla, L.E.; Botton, G.A.; Cavani, F.; Albonetti, S. AuPd-nNiO as an Effective Catalyst for the Base-Free Oxidation of HMF under Mild Reaction Conditions. Green Chem. 2019, 21, 4090–4099. [Google Scholar] [CrossRef]
- Davis, S.E.; Benavidez, A.D.; Gosselink, R.W.; Bitter, J.H.; de Jong, K.P.; Datye, A.K.; Davis, R.J. Kinetics and Mechanism of 5-Hydroxymethylfurfural Oxidation and Their Implications for Catalyst Development. J. Mol. Catal. A Chem. 2014, 388–389, 123–132. [Google Scholar] [CrossRef]
- Vuyyuru, K.R.; Strasser, P. Oxidation of Biomass Derived 5-Hydroxymethylfurfural Using Heterogeneous and Electrochemical Catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Yi, G.; Teong, S.P.; Zhang, Y. Base-Free Conversion of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid over a Ru/C Catalyst. Green Chem. 2016, 18, 979–983. [Google Scholar] [CrossRef]
- Grillo, F.; Soethoudt, J.; Marques, E.A.; de Martín, L.; Van Dongen, K.; van Ommen, J.R.; Delabie, A. Area-Selective Deposition of Ruthenium by Area-Dependent Surface Diffusion. Chem. Mater. 2020, 32, 9560–9572. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Jiao, Y.; Wen, X.; Jiao, H. Surface Hydroxyl Dependent Adsorption of Ruthenium on SiO2(0 0 1)–Understanding Metal–Support Interaction. Appl. Surf. Sci. 2022, 593, 153396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LDH a (Å) | LDH c (Å) | Mg(Al)O a (Å) | Spinel w/w Fraction | |||
|---|---|---|---|---|---|---|
| Calcination Temperature (°C) | ||||||
| 500 | 700 | 900 | 900 | |||
| Mg4Al | 3.072 | 23.461 | 4.177 | 4.177 | - | - |
| Mg3Al | 3.061 | 23.297 | 4.177 | 4.180 | 4.199 | 0.179 |
| Mg2Al | 3.045 | 22.840 | 4.178 | 4.182 | 4.194 | 0.267 |
| Surface Area (m2 g−1) | Mg(Al)O Size (nm) | Spinel Size nm | |||||
|---|---|---|---|---|---|---|---|
| Calcination Temperature (°C) | 500 | 700 | 900 | 500 | 700 | 900 | 900 |
| Mg4Al | 115 | 157 | - | 5.0 | 5.7 | - | - |
| Mg3Al | 111 | 195 | 122 | 4.0 | 4.8 | 7.2 | 9.8 |
| Mg2Al | 91 | 141 | 101 | 3.8 | 4.1 | 7.6 | 9.2 |
| Sample | Calcination Temperature | Total CO2 | CO2 Fraction | |||
|---|---|---|---|---|---|---|
| °C | μmol g−1 | μmol m−2 | w.b.s. * | m.s.b.s. * | p.a.b.s. * | |
| TiO2 | 500 | 15 | 0.19 | 0.64 | 0.34 | 0.02 |
| Mg2Al | 500 | 290 | 3.19 | 0.475 | 0.402 | 0.123 |
| Mg3Al | 320 | 2.88 | 0.522 | 0.441 | 0.037 | |
| Mg4Al | 240 | 2.09 | 0.426 | 0.553 | 0.021 | |
| Mg2Al | 700 | 130 | 0.92 | 0.409 | 0.585 | 0.006 |
| Mg3Al | 250 | 1.28 | 0.278 | 0.559 | 0.163 | |
| Mg4Al | 120 | 0.76 | 0.373 | 0.430 | 0.198 | |
| Mg2Al | 900 | 130 | 1.29 | 0.522 | 0.353 | 0.125 |
| Mg3Al | 210 | 1.72 | 0.303 | 0.440 | 0.256 | |
| Catalyst | Mean Particle Size (nm) | Experimental Metal Loading (%) | Total Basicity (mmol CO2/g) |
|---|---|---|---|
| Ru/TiO2-C a | 1.4 ± 0.4 | 1.5 | 0.02 |
| Ru/TiO2-S b | 1.8 ± 0.4 | 1.4 | 0.02 |
| Ru/Mg3Al-500-C | 1.2 ± 0.3 | 1.5 | 0.32 |
| Catalyst | HMF Conversion (%) | HMFCA Yield (%) | DFF Yield (%) | FFCA Yield (%) | FDCA Yield (%) | Others Yield (%) |
|---|---|---|---|---|---|---|
| No support, base-free | 4 | - | - | - | - | 4 |
| No support with base | 26 | 2 | - | 5 | 1 | 18 |
| TiO2, base-free | 8 | - | - | - | - | 8 |
| TiO2 with base | 35 | 5 | - | 7 | 2 | 21 |
| Mg3Al-500 | 26 | 2 | 5 | 5 | 2 | 12 |
| Mg3Al-700 | 27 | 2 | 4 | 3 | 1 | 17 |
| Mg3Al-900 | 28 | 2 | 4 | 4 | 0 | 18 |
| Entry | Reagent | Conversion (%) | Yield FFCA (%) | Yield FDCA (%) | Others (%) |
|---|---|---|---|---|---|
| 1 | HMFCA | 39 | 25 | 8 | 6 |
| 2 | DFF | 100 | 78 | 19 | 3 |
| 3 | FFCA | 26 | - | 22 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liuzzi, F.; Di Renzo, F.; Cesari, C.; Mammi, A.; Monti, L.; Allegri, A.; Zacchini, S.; Fornasari, G.; Dimitratos, N.; Albonetti, S. Preparation of Ru-Based Systems Through Metal Carbonyl Cluster Decomposition for the Base-Free 5-Hydroxymethylfurfural (HMF) Oxidation. Molecules 2025, 30, 2120. https://doi.org/10.3390/molecules30102120
Liuzzi F, Di Renzo F, Cesari C, Mammi A, Monti L, Allegri A, Zacchini S, Fornasari G, Dimitratos N, Albonetti S. Preparation of Ru-Based Systems Through Metal Carbonyl Cluster Decomposition for the Base-Free 5-Hydroxymethylfurfural (HMF) Oxidation. Molecules. 2025; 30(10):2120. https://doi.org/10.3390/molecules30102120
Chicago/Turabian StyleLiuzzi, Francesca, Francesco Di Renzo, Cristiana Cesari, Alice Mammi, Lorenzo Monti, Alessandro Allegri, Stefano Zacchini, Giuseppe Fornasari, Nikolaos Dimitratos, and Stefania Albonetti. 2025. "Preparation of Ru-Based Systems Through Metal Carbonyl Cluster Decomposition for the Base-Free 5-Hydroxymethylfurfural (HMF) Oxidation" Molecules 30, no. 10: 2120. https://doi.org/10.3390/molecules30102120
APA StyleLiuzzi, F., Di Renzo, F., Cesari, C., Mammi, A., Monti, L., Allegri, A., Zacchini, S., Fornasari, G., Dimitratos, N., & Albonetti, S. (2025). Preparation of Ru-Based Systems Through Metal Carbonyl Cluster Decomposition for the Base-Free 5-Hydroxymethylfurfural (HMF) Oxidation. Molecules, 30(10), 2120. https://doi.org/10.3390/molecules30102120