
Tuning Solvation Dynamics of Electrolytes at Their Eutectic Point Through Halide Identity

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results
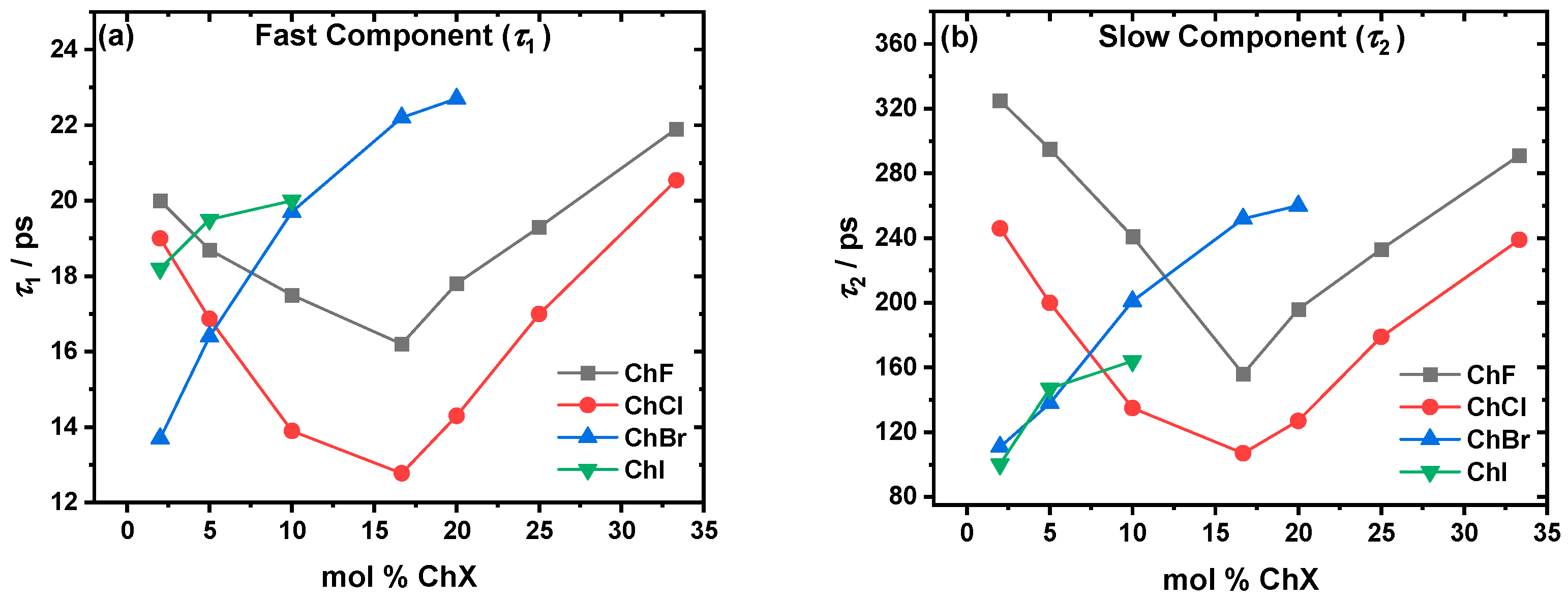
2.1. Solvent Relaxation Dynamics
2.2. Liquidus Temperatures
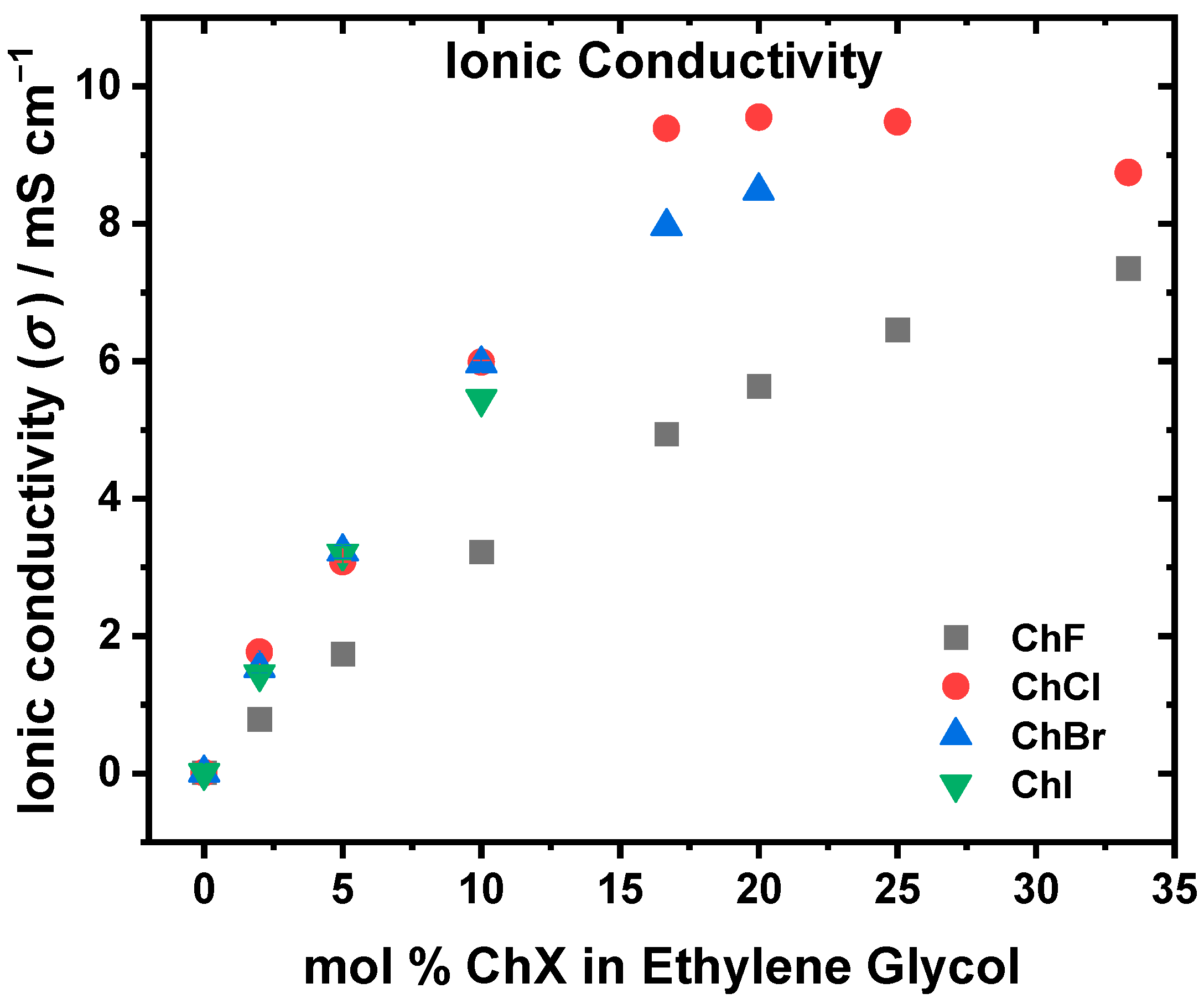
2.3. Viscosity, Ionic Conductivity, and Density
2.4. Polarity
2.5. Self-Diffusion Coefficients
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of ChF
4.3. Preparation of Solvent Mixtures
4.4. Differential Scanning Calorimetry
4.5. Femtosecond Transient Absorption Spectroscopy
4.6. Steady-State Absorption Spectroscopy and Solvent Polarity
4.7. Viscosity and Density
4.8. Ionic Conductivity
4.9. NMR Spectroscopy and Pulsed-Field Gradient (PFG) NMR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armaroli, N.; Balzani, V. The Legacy of Fossil Fuels. Chem. Asian J. 2011, 6, 768–784. [Google Scholar] [CrossRef]
- Weber, A.Z.; Mench, M.M.; Meyers, J.P.; Ross, P.N.; Gostick, J.T.; Liu, Q. Redox Flow Batteries: A Review. J. Appl. Electrochem. 2011, 41, 1137–1164. [Google Scholar] [CrossRef]
- Alotto, P.; Guarnieri, M.; Moro, F. Redox Flow Batteries for the Storage of Renewable Energy: A Review. Renew. Sustain. Energy Rev. 2014, 29, 325–335. [Google Scholar] [CrossRef]
- Mohamed, M.R.; Sharkh, S.M.; Walsh, F.C. Redox Flow Batteries for Hybrid Electric Vehicles: Progress and Challenges. In Proceedings of the 5th IEEE Vehicle Power and Propulsion Conference (VPPC ‘09), Dearborn, MI, USA, 7–10 September 2009; pp. 551–557. [Google Scholar]
- Winsberg, J.; Hagemann, T.; Janoschka, T.; Hager, M.D.; Schubert, U.S. Redox-Flow Batteries: From Metals to Organic Redox-Active Materials. Angew. Chem. Int. Ed. 2017, 129, 702–729. [Google Scholar] [CrossRef]
- Tang, L.; Leung, P.; Xu, Q.; Mohamed, M.R.; Dai, S.; Zhu, X.; Flox, C.; Shah, A.A. Future Perspective on Redox Flow Batteries: Aqueous versus Nonaqueous Electrolytes. Curr. Opin. Chem. Eng. 2022, 37, 100833. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Abbott, A.P.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep Eutectic Solvents Formed between Choline Chloride and Carboxylic Acids: Versatile Alternatives to Ionic Liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef]
- Abbott, A.P.; Harris, R.C.; Ryder, K.S.; D’Agostino, C.; Gladden, L.F.; Mantle, M.D. Glycerol Eutectics as Sustainable Solvent Systems. Phys. Chem. Chem. Phys. 2011, 13, 82–90. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel Solvent Properties of Choline Chloride/Urea Mixtures. Chem. Commun. 2003, 1, 70–71. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- García, G.; Aparicio, S.; Ullah, R.; Atilhan, M. Deep Eutectic Solvents: Physicochemical Properties and Gas Separation Applications. Energy Fuels 2015, 29, 2616–2644. [Google Scholar] [CrossRef]
- Zhang, Q.; De Oliveira Vigier, K.; Royer, S.; Jérôme, F. Deep Eutectic Solvents: Syntheses, Properties and Applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef]
- Cao, X.; Wang, S.; Xue, X. A Zn–Ce Redox Flow Battery with Ethaline Deep Eutectic Solvent. ChemSusChem 2021, 14, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Sinclair, N.; Kellamis, C.; Gurkan, B.; Wainright, J.; Savinell, R. Effects of Alkyl Chain Length and Halide Anion on Hydrogen Bonding, Electrochemical Transport Properties and Double Layer Capacitance in Eutectic Solvents. J. Mol. Liq. 2023, 391, 123314. [Google Scholar] [CrossRef]
- Brammer, L.; Bruton, E.A.; Sherwood, P. Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Cryst. Growth Des. 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Peris, E.; Lee, J.C.J.; Rambo, J.R.; Eisenstein, O.; Crabtree, R.H. Factors Affecting the Strength of X–H···H–M Hydrogen Bonds. J. Am. Chem. Soc. 1995, 117, 3485–3491. [Google Scholar] [CrossRef]
- Prado, D.M.; Burda, C. Untapped Potential of Fluoride Ions in Maximizing the Electrochemical Stability of Deep Eutectic Solvents. J. Phys. Chem. Lett. 2024, 15, 6343–6346. [Google Scholar] [CrossRef]
- Boogaart, D.J.; Essner, J.B.; Baker, G.A. Halide effects on the performance of equimolar choline halide: Guanidinium thiocyanate deep eutectic solvents as dye-sensitized solar cell electrolytes. Green Chem. Lett. Rev. 2022, 15, 615–626. [Google Scholar] [CrossRef]
- Lane, J.N.; Klimkowski, V.J.; Hopkins, T.A. Molecular dynamics investigation of deep eutectic solvent structure and properties based on hydrogen bond acceptor variation. J. Mol. Liq. 2025, 423, 127009. [Google Scholar] [CrossRef]
- Alfurayj, I.; Pandian, R.; Springer, S.; Burda, C. Choline Fluoride–Ethylene Glycol Deep Eutectic Solvent Mixture—Synthesis and Physicochemical Properties. J. Mol. Liq. 2023, 386, 122454. [Google Scholar] [CrossRef]
- Alfurayj, I.; Fraenza, C.; Pandian, R.; Greenbaum, S.; Burda, C. Solvation Dynamics of Choline Fluoride in Ethylene Glycol–Water Mixtures. J. Mol. Liq. 2023, 392, 123448. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Zhao, Y.; Wang, J.; Yu, Z. Insights into the Hydrogen Bond Interactions in Deep Eutectic Solvents Composed of Choline Chloride and Polyols. ACS Sustain. Chem. Eng. 2019, 7, 7760–7767. [Google Scholar] [CrossRef]
- D’Agostino, C.H.; Harris, R.C.; Abbott, A.P.; Gladden, L.F.; Mantle, M.D. Molecular Motion and Ion Diffusion in Choline Chloride-Based Deep Eutectic Solvents Studied by ¹H Pulsed Field Gradient NMR Spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 21383–21391. [Google Scholar] [CrossRef] [PubMed]
- Dimroth, K.; Reichardt, C.; Siepmann, T.; Bohlmann, F. Über Pyridinium-N-Phenol-Betaine und Ihre Verwendung zur Charakterisierung der Polarität von Lösungsmitteln. Justus Liebigs Ann. Chem. 1963, 661, 1–37. [Google Scholar] [CrossRef]
- Beard, M.C.; Turner, G.M.; Schmuttenmaer, C.A. Measuring Intramolecular Charge Transfer via Coherent Generation of THz Radiation. J. Phys. Chem. A 2002, 106, 878–883. [Google Scholar] [CrossRef]
- Pandian, R.; Burda, H.; Alfurayj, I.; Reichardt, C.; Burda, C. 60 Years of Betaine 30─From Solvatochromic Discovery to Future Frontiers. J. Phys. Chem. B 2024, 128, 6990–7001. [Google Scholar] [CrossRef]
- Reichardt, C. Empirische Parameter der Lösungsmittelpolarität. Angew. Chem. 1965, 77, 30–40. [Google Scholar] [CrossRef]
- Reichardt, C.; Harbusch-Gornert, E.; Schafer, G. Herstellung und UV/Vis-Spektroskopische Eigenschaften Eines Wasserlöslichen Carboxylat-Substituierten Pyridinium-N-Phenolat-Betainfarbstoffs. Liebigs Ann. Chem. 1988, 1988, 839–844. [Google Scholar] [CrossRef]
- Reichardt, C. Polarity of Ionic Liquids Determined Empirically by Means of Solvatochromic Pyridinium N-Phenolate Betaine Dyes. Green Chem. 2005, 7, 339–351. [Google Scholar] [CrossRef]
- Reichardt, C. Pyridinium N-Phenolate Betaine Dyes as Empirical Indicators of Solvent Polarity: Some New Findings. Pure Appl. Chem. 2004, 76, 1903–1919. [Google Scholar] [CrossRef]
- Reichardt, C. Solvation Effects in Organic Chemistry: A Short Historical Overview. J. Org. Chem. 2022, 87, 1616–1629. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromism, Thermochromism, Piezochromism, Halochromism, and Chiro-Solvatochromism of Pyridinium N-Phenoxide Betaine Dyes. Chem. Soc. Rev. 1992, 21, 147–153. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Reichardt, C.; Che, D.; Heckenkemper, G.; Schafer, G. Syntheses and UV/Vis-Spectroscopic Properties of Hydrophilic 2-, 3-, and 4-Pyridyl-Substituted Solvatochromic and Halochromic Pyridinium N-Phenolate Betaine Dyes as New Empirical Solvent Polarity Indicators. Eur. J. Org. Chem. 2001, 2001, 2343–2361. [Google Scholar] [CrossRef]
- Reichardt, C.; Harbusch-Gornert, E. Über Pyridinium-N-Phenolat-Betaine und Ihre Verwendung zur Charakterisierung der Polarität von Lösungsmitteln, X. Erweiterung, Korrektur und Neudefinition der ET-Lösungsmittelpolaritätsskala mit Hilfe Eines Lipophilen Penta-Tert-Butyl-Substituierten Pyridinium-N-Phenolat-Betainfarbstoffs. Liebigs Ann. Chem. 1983, 1983, 721–743. [Google Scholar]
- Mente, S.R.; Maroncelli, M. Computer Simulations of the Solvatochromism of Betaine-30. J. Phys. Chem. B 1999, 103, 7704–7719. [Google Scholar] [CrossRef]
- Reid, P.J.; Barbara, P.F. Dynamic Solvent Effect on Betaine-30 Electron-Transfer Kinetics in Alcohols. J. Phys. Chem. 1995, 99, 3554–3565. [Google Scholar] [CrossRef]
- Alfurayj, I.; Fraenza, C.C.; Zhang, Y.; Pandian, R.; Spittle, S.; Hansen, B.; Dean, W.; Gurkan, B.; Savinell, R.; Greenbaum, S.; et al. Solvation Dynamics of Wet Ethaline: Water Is the Magic Component. J. Phys. Chem. B 2021, 125, 8888–8901. [Google Scholar] [CrossRef]
- Pandian, R.; Kim, D.; Zhang, Y.; Alfurayj, I.; Prado, D.M.; Maginn, E.; Burda, C. Chain Length and OH-Spacing Effects on Diol-Based Deep Eutectic Solvents. J. Mol. Liq. 2024, 393, 123534. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Eilers-König, N.; Senyushkina, T.A.; Ernsting, N.P. Charge Transfer and Solvation of Betaine-30 in Polar Solvents—A Femtosecond Broadband Transient Absorption Study. J. Phys. Chem. A 2001, 105, 4834–4843. [Google Scholar] [CrossRef]
- Gajardo-Parra, N.F.; Cotroneo-Figueroa, V.P.; Aravena, P.; Vesovic, V.; Canales, R.I. Viscosity of Choline Chloride-Based Deep Eutectic Solvents: Experiments and Modeling. J. Chem. Eng. Data 2020, 65, 5581–5592. [Google Scholar] [CrossRef]
- Spittle, S.; Alfurayj, I.; Hansen, B.B.; Glynn, K.; Brackett, W.; Pandian, R.; Burda, C.; Sangoro, J. Enhanced Dynamics and Charge Transport at the Eutectic Point: A New Paradigm for the Use of Deep Eutectic Solvent Systems. JACS Au 2023, 3, 3024–3030. [Google Scholar] [CrossRef]
- Reichardt, C. Empirical Parameters of the Polarity of Solvents. Angew. Chem. Int. Ed. Engl. 1965, 4, 29–40. [Google Scholar] [CrossRef]
- Migliorati, V.; D’Angelo, P. Deep Eutectic Solvents: A Structural Point of View on the Role of the Anion. Chem. Phys. Lett. 2021, 777, 138702. [Google Scholar] [CrossRef]
- Harmon, K.M.; Madeura, S.L.; Jacks, M.J.; Avci, G.F.; Thiel, A.C. Part 18. The Nature of the CHF Hydrogen Bond in Choline Fluoride. J. Mol. Struct. 1885, 128, 305–314. [Google Scholar] [CrossRef]
- Alfurayj, I.; Prado, D.M.; Prado, R.C.; Samia, A.C.; Burda, C. Unusual Hydration Properties of Choline Fluoride-Based Deep Eutectic Solvents. J. Phys. Chem. B 2024, 128, 2762–2772. [Google Scholar] [CrossRef]
- Gaur, A.; Avula, N.V.S.; Balasubramanian, S. Insights into the Stabilization of Fluoride Ions in Ionic Liquids: Pointers to Better Fluorinating Agents. J. Phys. Chem. B 2020, 124, 8844–8856. [Google Scholar] [CrossRef]
- Das, L.; Mukherjee, S.; Kumar Maity, D.; Adhikari, S. Synthesis and Characterization of a Computationally Predicted Redox and Radiation Stable Deep Eutectic Solvent. J. Mol. Liq. 2022, 360, 119377. [Google Scholar] [CrossRef]
- van den Bruinhorst, A.; Kollau, L.J.B.M.; Vis, M.; Hendrix, M.M.R.M.; Meuldijk, J.; Tuinier, R.; Esteves, A.C.C. From a eutectic mixture to a deep eutectic system via anion selection: Glutaric acid + tetraethylammonium halides. J. Chem. Phys. 2021, 155, 014502. [Google Scholar] [CrossRef]
- Agieienko, V.; Neklyudov, V.; Buchner, R. Why Does Ethaline Apparently Behave as an Ideal Binary Mixture? J. Phys. Chem. Lett. 2022, 13, 10805–10809. [Google Scholar] [CrossRef]
- Kadhim, M.J.; Gamaj, M.I. Estimation of the Diffusion Coefficient and Hydrodynamic Radius (Stokes Radius) for Inorganic Ions in Solution Depending on Molar Conductivity as Electro-Analytical Technique—A Review. J. Chem. Rev. 2020, 2, 182–188. [Google Scholar]
- Curnow, O.J.; MacFarlene, D.R.; Walst, K.J. Fluoride Ionic Liquids in Salts of Ethylmethylimidazolium and Substituted Cyclopropenium Cation Families. Front. Chem. 2018, 6, 603. [Google Scholar] [CrossRef]
- Radomski, R.; Radomska, M. Determination of Solidus and Liquidus Temperatures by Means of a Perkin-Elmer 1B Differential Scanning Calorimeter. J. Therm. Anal. 1982, 24, 101–109. [Google Scholar] [CrossRef]
- Crichton, S.N.; Moynihan, C.T. Dependence of the Glass Transition Temperature on Heating Rate. J. Non-Cryst. Solids 1988, 99, 413–417. [Google Scholar] [CrossRef]
- Kremer, F.; Schönhals, A. Broadband Dielectric Spectroscopy; Springer Science & Business Media: Berlin, Germany, 2002. [Google Scholar]
- Reid, P.J.; Alex, S.; Jarzeba, W.; Schlief, R.E.; Johnson, A.E.; Barbara, P.F. Evidence for Intermolecular Hydrogen-Bond Rearrangement in the Electron Transfer Dynamics of Betaine-30 in n-Butanol. Chem. Phys. Lett. 1994, 229, 93–100. [Google Scholar] [CrossRef]
- Åkesson, E.; Walker, G.C.; Barbara, P.F. Dynamic Solvent Effects on Electron Transfer Rates in the Inverted Regime: Ultrafast Studies on the Betaines. J. Chem. Phys. 1991, 95, 4188–4194. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Self-Diffusion Coefficient (×10−11 m2 s−1) | ||
|---|---|---|---|
| Ch+ | EG | 19F | |
| ChF:EG (10 mol %) | 4.87 | 7.71 | 3.90 (downfield peak); 7.75 (upfield peak) |
| ChCl:EG (10 mol %) | 5.77 | 8.59 | - |
| ChBr:EG (10 mol %) | 5.86 | 9.07 | - |
| ChI:EG (10 mol %) | 6.15 | 9.42 | - |
| ChCl:EG (20 mol %) | 4.39 | 7.14 | - |
| ChBr:EG (20 mol %) | 4.58 | 6.04 | - |
| ChF:EG (33.33 mol %) [22] | 3.09 | 4.64 | 2.20 (downfield peak); 5.29 (upfield peak) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandian, R.; Hansen, B.B.; de Araujo Lima e Souza, G.; Sangoro, J.R.; Greenbaum, S.; Burda, C. Tuning Solvation Dynamics of Electrolytes at Their Eutectic Point Through Halide Identity. Molecules 2025, 30, 2113. https://doi.org/10.3390/molecules30102113
Pandian R, Hansen BB, de Araujo Lima e Souza G, Sangoro JR, Greenbaum S, Burda C. Tuning Solvation Dynamics of Electrolytes at Their Eutectic Point Through Halide Identity. Molecules. 2025; 30(10):2113. https://doi.org/10.3390/molecules30102113
Chicago/Turabian StylePandian, Rathiesh, Benworth B. Hansen, Giselle de Araujo Lima e Souza, Joshua R. Sangoro, Steven Greenbaum, and Clemens Burda. 2025. "Tuning Solvation Dynamics of Electrolytes at Their Eutectic Point Through Halide Identity" Molecules 30, no. 10: 2113. https://doi.org/10.3390/molecules30102113
APA StylePandian, R., Hansen, B. B., de Araujo Lima e Souza, G., Sangoro, J. R., Greenbaum, S., & Burda, C. (2025). Tuning Solvation Dynamics of Electrolytes at Their Eutectic Point Through Halide Identity. Molecules, 30(10), 2113. https://doi.org/10.3390/molecules30102113