

Study on Photodeformation of Solvent Resistance in Hydrogen-Bonded Cross-Linked Main-Chain Azobenzene Films

Abstract

1. Introduction
2. Results and Discussion
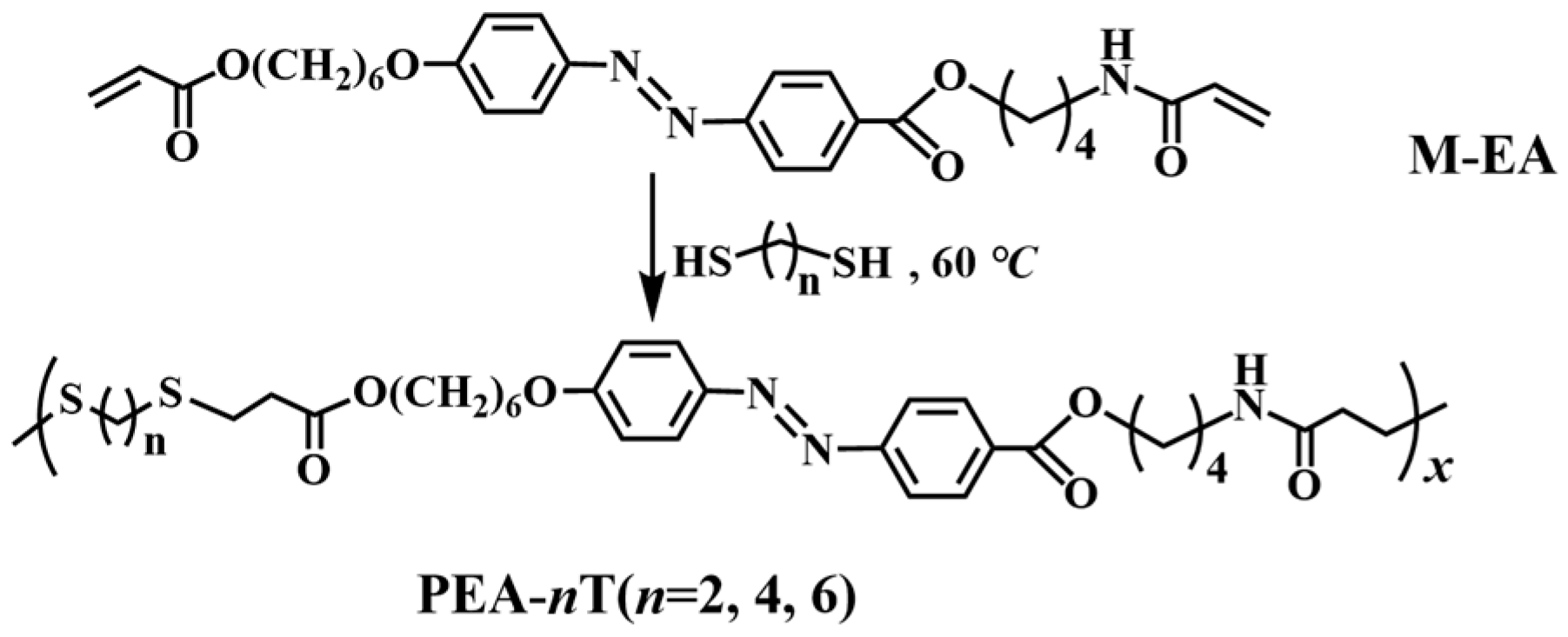
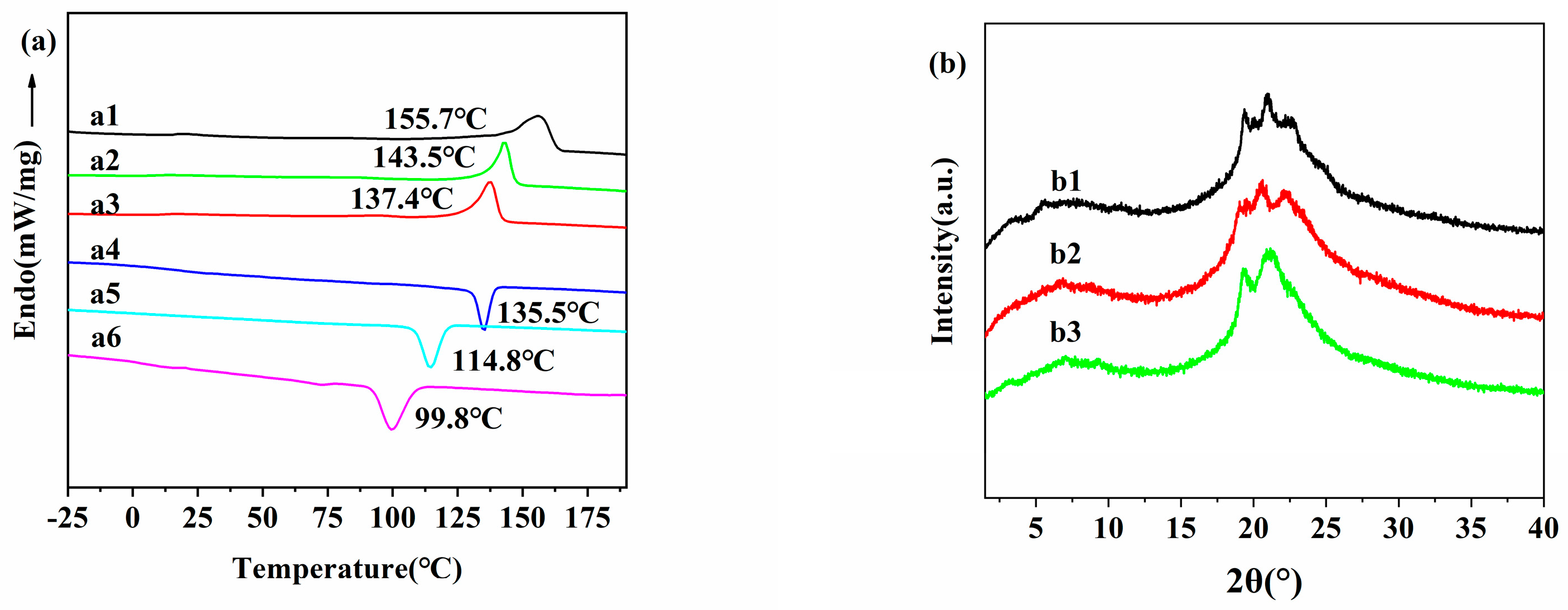
2.1. Synthesis and Characterization of the Main-Chain Azo Semi-Crystalline Poly(ester-amide) S (PEAs)
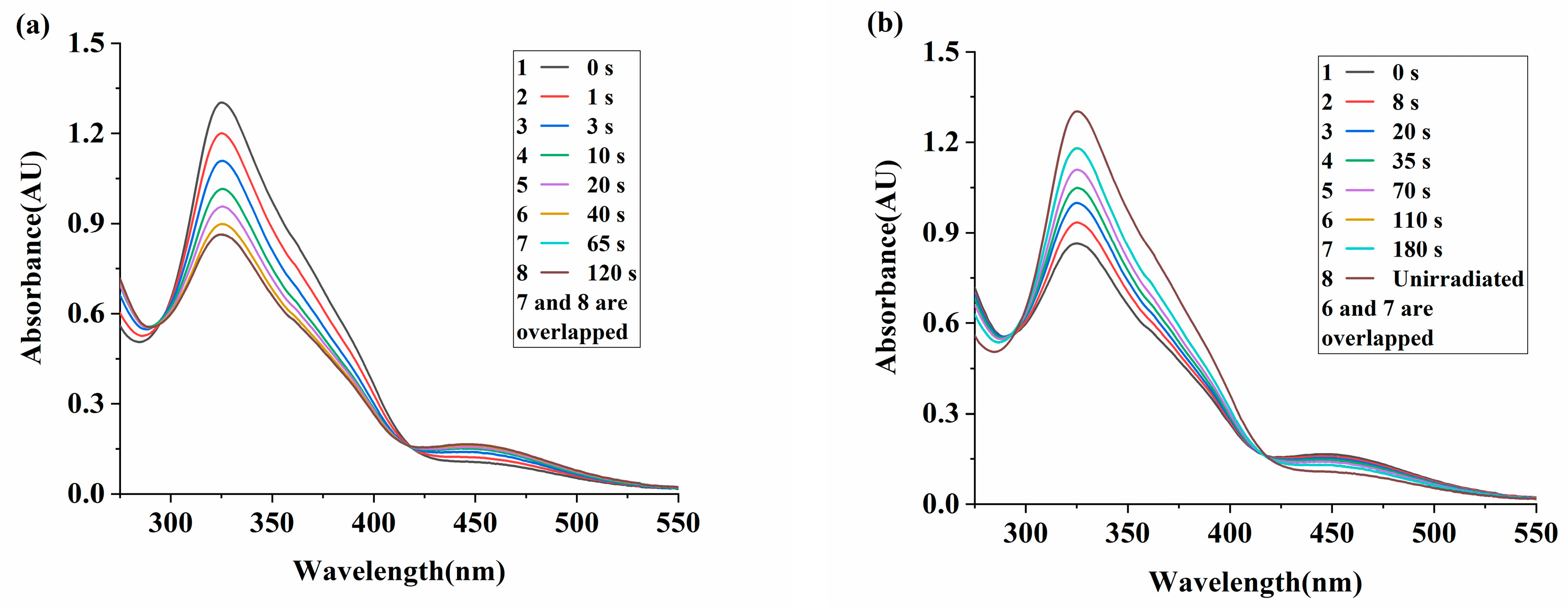
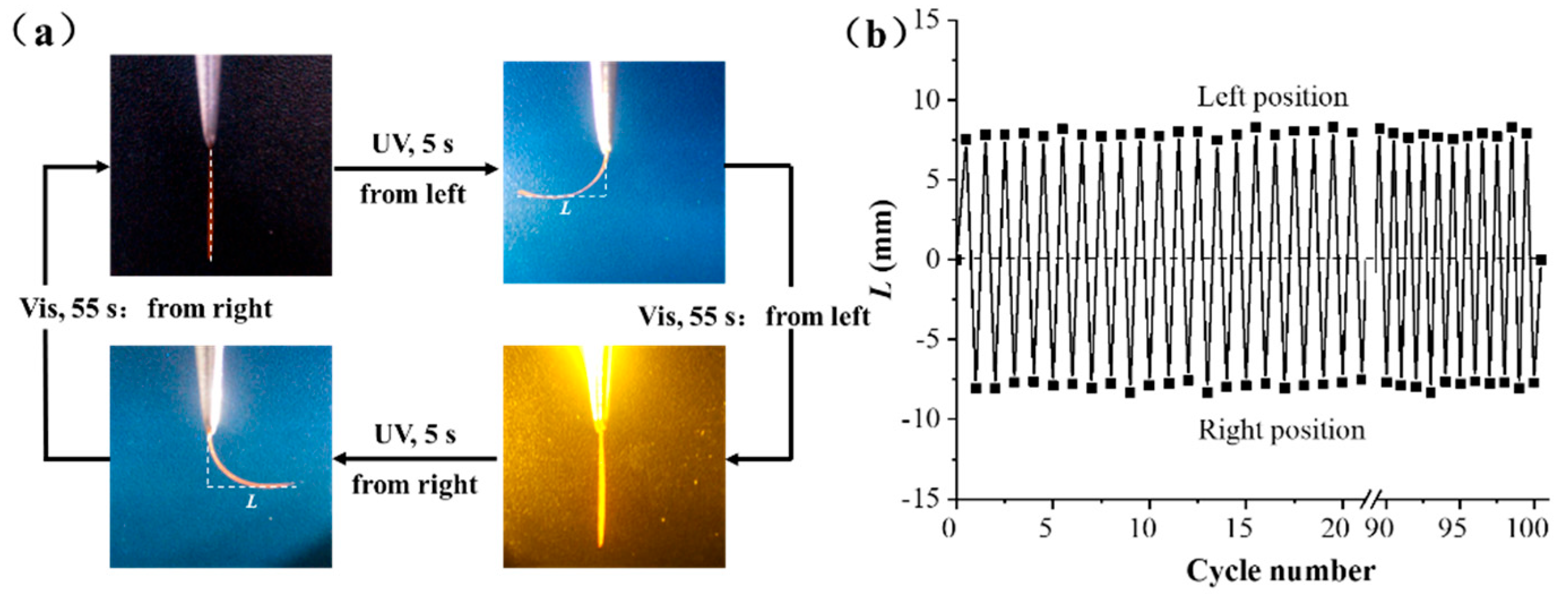
2.2. Mechanical and Photomechanical Properties of the Uniaxially Oriented Azo Films in Air
2.3. Room Temperature Three-Dimensional Shape Programmability and Recyclability of Uniaxial Oriented PEA-6T Films
2.4. Photomechanical Properties and Solvent Resistance of Uniaxially Oriented Films of Main-Chain Azo PEAs Under Common Hydrogen Bonding Solvents
3. Materials and Methods
3.1. Materials and Reagents
3.2. Synthesis of N-(4-Hydroxybutyl) Acrylamide
3.3. Synthesis of M-EA
3.4. Synthesis of PEA-nT (n = 2, 4, 6)
3.5. Preparation of the Thin PEA-nT Film for the Photoresponsivity Study
3.6. Preparation of the XRD Characterization for the Azo Polymer Powders
3.7. Preparation of Uniaxially Oriented Main-Chain Azo Polymer Films
3.8. Determination for Degree of Orientation in Uniaxially Oriented Main-Chain Azo Polymer Films
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, H.; Ikeda, T. Photocontrollable liquid-crystalline actuators. Adv. Mater. 2011, 23, 2149–2180. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Broer, D.J. Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat. Mater. 2015, 14, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Natansohn, A.; Rochon, P. Photoinduced motions in azo-containing polymers. Chem. Rev. 2002, 102, 4139–4176. [Google Scholar] [CrossRef] [PubMed]
- Mysliwiec, J.; Czajkowski, M.; Miniewicz, A.; Bartkiewicz, S.; Kochalska, A.; Polakova, L.; Sedlakova, Z.; Nespurek, S. Dynamics of photoinduced motions in azobenzene grafted polybutadienes. Opt. Mater. 2011, 33, 1398–1404. [Google Scholar] [CrossRef]
- Iamsaard, S.; Anger, E.; Asshoff, S.J.; Depauw, A.; Fletcher, S.P.; Katsonis, N. Fluorinated azobenzenes for shape-persistent liquid crystal polymer networks. Angew. Chem. Int. Ed. 2016, 55, 9908–9912. [Google Scholar] [CrossRef]
- Tkachenko, I.M.; Kurioz, Y.I.; Kravchuk, R.M.; Kobzar, Y.L.; Litoshenko, D.V.; Glushchenko, A.V.; Shevchenko, V.V.; Nazarenko, V.G. Photoinduced birefringence and liquid crystal orientation on polymers with different azobenzene content in the main chain. ACS Appl. Mater. Interfaces 2024, 16, 52945–52957. [Google Scholar] [CrossRef]
- Wang, D.H.; Lee, K.M.; Yu, Z.; Koerner, H.; Vaia, R.A.; White, T.J.; Tan, L.-S. Photomechanical response of glassy azobenzene polyimide networks. Macromolecules 2011, 44, 3840–3846. [Google Scholar] [CrossRef]
- Dong, H.; Liu, G.; Zhang, H. Preparation of photodeformable azobenzene polymer fibers by post-crosslinking strategy: Understanding the structure-property relationship. Eur. Polym. J. 2020, 135, 109863. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, Z.; Wu, F.; Wang, M.; Chen, L. Liquid-crystal elastomers based on covalent adaptable networks: From molecular design to applications. Sci. China Mater. 2023, 66, 3004–3021. [Google Scholar] [CrossRef]
- Bin Rusayyis, M.A.; Fenimore, L.M.; Purwanto, N.S.; Torkelson, J.M. Reprocessable, creep-resistant covalent adaptable networks synthesized using conventional free-radical polymerization conditions with piperidine-based and non-piperidine-based dynamic dialkylamino disulfide chemistry. Polym. Chem. 2023, 14, 3519–3534. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Nicolaÿ, R. Vitrimers: Permanently crosslinked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104, 101233. [Google Scholar] [CrossRef]
- Chakma, P.; Konkolewicz, D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58, 9682–9695. [Google Scholar] [CrossRef]
- Podgórski, M.; Fairbanks, B.D.; Kirkpatrick, B.E.; McBride, M.; Martinez, A.; Dobson, A.; Bongiardina, N.J.; Bowman, C.N. Toward stimuli-responsive dynamic thermosets through continuous development and improvements in covalent adaptable networks (CANs). Adv. Mater. 2020, 32, 1906876. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.C.; Xiao, Y.Y.; Yin, L.; Han, L.; Zhao, Y. “Self-Lockable” liquid crystalline diels-alder dynamic network actuators with room temperature programmability and solution reprocessability. Angew. Chem. Int. Ed. 2020, 59, 4925–4931. [Google Scholar] [CrossRef]
- Huang, X.; Qin, L.; Wang, J.; Zhang, X.; Peng, B.; Yu, Y. Multiple Shape Manipulation of Liquid Crystal Polymers Containing Diels-Alder Network. Adv. Funct. Mater. 2022, 32, 2208312. [Google Scholar] [CrossRef]
- Han, G.; Nie, J.; Zhang, H. Facile preparation of recyclable photodeformable azobenzene polymer fibers with chemically crosslinked networks. Polym. Chem. 2016, 7, 5088–5092. [Google Scholar] [CrossRef]
- Qin, C.; Feng, Y.; Luo, W.; Cao, C.; Hu, W.; Feng, W. A supramolecular assembly of cross-linked azobenzene/polymers for a high-performance light-driven actuator. J. Mater. Chem. A 2015, 3, 16453–16460. [Google Scholar] [CrossRef]
- Qin, C.; Feng, Y.; An, H.; Han, J.; Cao, C.; Feng, W. Tetracarboxylated azobenzene/polymer supramolecular assemblies as high-performance multiresponsive actuators. ACS Appl. Mater. Interfaces 2017, 9, 4066–4073. [Google Scholar] [CrossRef]
- Vapaavuori, J.; Bazuin, C.G.; Priimagi, A. Supramolecular design principles for efficient photoresponsive polymer–azobenzene complexes. J. Mater. Chem. C 2018, 6, 2168–2188. [Google Scholar] [CrossRef]
- Yu, H.-T.; Tang, J.-W.; Feng, Y.-Y.; Feng, W. Structural design and application of azo-based supramolecular polymer systems. Chin. J. Polym. Sci. 2019, 37, 1183–1199. [Google Scholar] [CrossRef]
- Houben, S.J.A.; Lugger, S.J.D.; van Raak, R.J.H.; Schenning, A.P.H.J. A ph-responsive liquid crystal hydrogel actuator with calcium-induced reprogrammable shape fixing. ACS Appl. Polym. Mater. 2022, 4, 1298–1304. [Google Scholar] [CrossRef]
- Zhong, H.-Y.; Chen, L.; Yang, R.; Meng, Z.-Y.; Ding, X.-M.; Liu, X.-F.; Wang, Y.-Z. Azobenzene-containing liquid crystalline polyester with π–π interactions: Diverse thermo- and photo-responsive behaviours. J. Mater. Chem. C 2017, 5, 3306–3314. [Google Scholar] [CrossRef]
- Zhong, H.-Y.; Chen, L.; Liu, X.-F.; Yang, R.; Wang, Y.-Z. Novel liquid crystalline copolyester containing amphi-mesogenic units toward multiple stimuli-response behaviors. J. Mater. Chem. C 2017, 5, 9702–9711. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Ma, S.; Zhang, H. Fully room-temperature reprogrammable, reprocessable, and photomobile soft actuators from a high-molecular-weight main-chain azobenzene crystalline poly(ester-amide). ACS Appl. Mater. Interfaces 2022, 14, 3264–3273. [Google Scholar] [CrossRef]
- Lee, W.; Kwak, S.-Y.; Chung, J.W. Arm-length-dependent phase transformation and dual dynamic healing behavior of supramolecular networks consisting of ureidopyrimidinone-end-functionalized semi-crystalline star polymers. Eur. Polym. J. 2020, 138, 109976. [Google Scholar] [CrossRef]
- Ma, S.; Zhou, Y.; Wang, L.; Zhang, H. Multifunctional UV-NIR dual light-responsive soft actuators from a main-chain azobenzene semi-crystalline poly(ester-amide) doped with polydopamine nanoparticles. Chem. Eur. J. 2024, 30, e202303306. [Google Scholar] [CrossRef]
- Li, G.; Xu, M.; Zhang, S.; Yang, G.; Li, W. Reversible controlling the supramolecular chirality of side chain azobenzene polymers: Chiral induction and modulation. Macromol. Rapid Commun. 2022, 43, 2100904. [Google Scholar] [CrossRef]
- Zhang, H. Reprocessable photodeformable azobenzene polymers. Molecules 2021, 26, 4455. [Google Scholar] [CrossRef]
- Fang, L.; Han, G.; Zhang, J.; Zhang, H.; Zhang, H. Synthesis of well-defined easily crosslinkable azobenzene side-chain liquid crystalline polymers via reversible addition–fragmentation chain transfer polymerization and photomechanical properties of their post-crosslinked fibers. Eur. Polym. J. 2015, 69, 592–604. [Google Scholar] [CrossRef]
- Han, G.; Zhang, H.; Chen, J.; Sun, Q.; Zhang, Y.; Zhang, H. Easily crosslinkable side-chain azobenzene polymers for fast and persistent fixation of surface relief gratings. New J. Chem. 2015, 39, 1410–1420. [Google Scholar] [CrossRef]
- Li, X.; Fang, L.; Hou, L.; Zhu, L.; Zhang, Y.; Zhang, B.; Zhang, H. Photoresponsive side-chain liquid crystalline polymers with amide group-substituted azobenzene mesogens: Effects of hydrogen bonding, flexible spacers, and terminal tails. Soft Matter 2012, 8, 5532–5542. [Google Scholar] [CrossRef]
- Li, X.; Wen, R.; Zhang, Y.; Zhu, L.; Zhang, B.; Zhang, H. Photoresponsive side-chain liquid crystalline polymers with an easily cross-linkable azobenzene mesogen. J. Mater. Chem. 2009, 19, 236–245. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Zhu, L.; Shen, T.; Zhang, H. Efficient synthesis of photoresponsive azobenzene-containing side-chain liquid crystalline polymers with high molecular weights by click chemistry. Polym. Chem. 2010, 1, 1501–1511. [Google Scholar] [CrossRef]
- Wani, O.M.; Zeng, H.; Priimagi, A. A light-driven artificial flytrap. Nat. Commun. 2017, 8, 15546. [Google Scholar] [CrossRef]
- Guo, C.; Gao, J.; Ma, S.; Zhang, H. Efficient preparation of chemically crosslinked recyclable photodeformable azobenzene polymer fibers with high processability and reconstruction ability via a facile post-crosslinking method. Eur. Polym. J. 2020, 139, 109998. [Google Scholar] [CrossRef]
- Nie, J.; Liu, X.; Yan, Y.; Zhang, H. Supramolecular hydrogen-bonded photodriven actuators based on an azobenzene-containing main-chain liquid crystalline poly(ester-amide). J. Mater. Chem. C 2017, 5, 10391–10398. [Google Scholar] [CrossRef]
- Tan, S.; Sha, Y.; Zhu, T.; Rahman, M.A.; Tang, C. Photoresponsive supramolecular polymers based on quadruple hydrogen-bonding and a photochromic azobenzene motif. Polym. Chem. 2018, 9, 5395–5401. [Google Scholar] [CrossRef]
- Ube, T.; Nakayama, R.; Ikeda, T. Photoinduced motions of thermoplastic polyurethanes containing azobenzene moieties in main chains. Macromolecules 2022, 55, 413–420. [Google Scholar] [CrossRef]
- Ma, S.; Wang, L.; Zhou, Y.; Zhang, H. Fully room temperature reprogrammable, recyclable, and photomobile soft actuators from physically cross-linked main-chain azobenzene liquid crystalline polymers. Molecules 2023, 28, 4174. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Zhang, H. Enhancing the performances of physically cross-linked photodeformable main-chain azobenzene poly(ester-amide)s via chemical structure engineering. Polym. Chem. 2022, 13, 3713–3725. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.; Ma, S.; Zhang, H. Reprocessable and healable room temperature photoactuators based on a main-chain azobenzene liquid crystalline poly(ester-urea). J. Mater. Chem. C 2021, 9, 13255–13265. [Google Scholar] [CrossRef]
- Pilz da Cunha, M.; van Thoor, E.A.J.; Debije, M.G.; Broer, D.J.; Schenning, A.P.H.J. Unravelling the photothermal and photomechanical contributions to actuation of azobenzene-doped liquid crystal polymers in air and water. J. Mater. Chem. C 2019, 7, 13502–13509. [Google Scholar] [CrossRef]
- Zhang, P.; Lan, Z.; Wei, J.; Yu, Y. Photodeformable azobenzene-containing polyimide with flexible linkers and molecular alignment. ACS Macro Lett. 2021, 10, 469–475. [Google Scholar] [CrossRef]
- Kuenstler, A.S.; Clark, K.D.; de Alaniz, J.R.; Hayward, R.C. Reversible actuation via photoisomerization-induced melting of a semicrystal line poly(azobenzene). ACS Macro Lett. 2020, 9, 902–909. [Google Scholar] [CrossRef]
- Niemann, M.; Ritter, H. Comb-like methacrylamide polymers containing condensates of amino acids and azobenzene moieties in the side chains. Makromol. Chem. 1993, 194, 1169–1181. [Google Scholar] [CrossRef]
- Wang, G.-J.; Tong, X.; Zhao, Y.J.M. Preparation of azobenzene-containing amphiphilic diblock copolymers for light-responsive micellar aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Akiyama, H.; Tamaoki, N. Synthesis and photoinduced phase transitions of poly(N-isopropylacrylamide) derivative functionalized with terminal azobenzene units. Macromolecules 2007, 40, 5129–5132. [Google Scholar] [CrossRef]
- Wei, R.; He, Y.; Wang, X. Diblock copolymers composed of a liquid crystalline azo block and a poly(dimethylsiloxane) block: Synthesis, morphology and photoresponsive properties. RSC Adv. 2014, 4, 58386–58396. [Google Scholar] [CrossRef]
- Wie, J.J.; Wang, D.H.; Lee, K.M.; White, T.J.; Tan, L.S. The contribution of hydrogen bonding to the photomechanical response of azobenzene-functionalized polyamides. J. Mater. Chem. C 2018, 6, 5964–5974. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, H.; Hsu, C.; Wang, X. Azo molecular glass patterning from chiral submicron pillar array to self-organized topographic transition via irradiation with circularly polarized light. Adv. Opt. Mater. 2021, 9, 2100922. [Google Scholar] [CrossRef]
- Corrado, F.; Bruno, U.; Prato, M.; Carella, A.; Criscuolo, V.; Massaro, A.; Pavone, M.; Muñoz-García, A.B.; Forti, S.; Coletti, C.; et al. Azobenzene-based optoelectronic transistors for neurohybrid building blocks. Nat. Commun. 2023, 14, 6760. [Google Scholar] [CrossRef] [PubMed]
- Kizhakidathazhath, R.; Geng, Y.; Jampani, V.S.R.; Charni, C.; Sharma, A.; Lagerwall, J.P.F. Facile anisotropic deswelling method for realizing large-area cholesteric liquid crystal elastomers with uniform structural color and broad-range mechanochromic response. Adv. Funct. Mater. 2019, 30, 1909537. [Google Scholar] [CrossRef]
- Kaniyoor, A.; Gspann, T.S.; Mizen, J.E.; Elliott, J.A. Quantifying alignment in carbon nanotube yarns and similar two-dimensional anisotropic systems. J. Appl. Polym. Sci. 2021, 138, 50939. [Google Scholar] [CrossRef]
- Patil, N.; Balzano, L.; Portale, G.; Rastogi, S. Influence of nanoparticles on the rheological behaviour and initial stages of crystal growth in linear polyethylene. Macromol. Chem. Phys. 2009, 210, 2174–2187. [Google Scholar] [CrossRef]
- Cinader, D.K.; Burghardt, W.R. X-ray scattering studies of orientation in channel flows of a lyotropic liquid crystalline polymer. Polymer 1999, 40, 4169–4180. [Google Scholar] [CrossRef]
- Ube, T.; Miyamoto, K.; Kurihara, S.; Ikeda, T. Sunlight-Driven Photomobile Polymer Materials Containing Push–Pull Azobenzene Moieties. ACS Appl. Mater. Interfaces 2025, 17, 16010–16015. [Google Scholar] [CrossRef]
- Pang, X.; Xu, B.; Qing, X.; Wei, J.; Yu, Y. Photo-Induced Bending Behavior of Post-Crosslinked Liquid Crystalline Polymer/Polyurethane Blend Films. Macromol. Rapid Commun. 2018, 39, 1700237. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sample a | Yield (%) | Degree of Polymerization (DP) b | Mn, NMR (g·mol−1) c | Thermal Transition T (°C) d | ΔHsi (J·g−1) g | Td (°C) h |
|---|---|---|---|---|---|---|---|
| 1 | PEA-2T | 90 | 47 | 28,400 | G 16.3 Cr 155.7 I e I 135.5 Cr 10.5 G f | 31.3 −26.4 | 288 |
| 2 | PEA-4T | 91 | 48 | 30,900 | G 12.6 Cr 143.5 I e I 114.8 Cr 4.5 G f | 29.4 −24.4 | 292 |
| 3 | PEA-6T | 90 | 49 | 32,900 | G 8.3 Cr 137.4 I e I 99.8 Cr 1.4 G f | 28.6 −25.5 | 294 |
| Entry | Sample | Elastic Modulus (MPa) | Yield Strength (MPa) | Rupture Strength (MPa) | Elongation at Break (%) | Toughness (MJ/m3) | Photoinduced Stress (kPa) |
|---|---|---|---|---|---|---|---|
| 1 | 1,2-film | 179.2 ± 4.2 | 12.6 ± 1.1 | 12.6 ± 0.7 | 472.9 ± 28.7 | 53.7 ± 8.4 | 582.7 ± 43.1 |
| 2 | 1,4-film | 197.5 ± 3.2 | 14.8 ± 0.9 | 16.1 ± 1.1 | 547.6 ± 22.7 | 72.7 ± 5.2 | 722.5 ± 48.6 |
| 3 | 1,6-film | 210.3 ± 4.0 | 16.9 ± 1.1 | 22.1 ± 2.1 | 632.9 ± 15.4 | 108.1 ± 2.3 | 835.9 ± 48.2 |
| Entry | Sample | Environment | Temperature (°C) | Induced Bending Time (s) a | Bending Amplitude L (mm) b | Cycling Number (times) c | |
|---|---|---|---|---|---|---|---|
| UV | Vis | ||||||
| 1 | PEA-2T | Air | 25 | 12 | 110 | 6.8 ± 0.2 | >100 |
| 2 | PEA-4T | Air | 25 | 10 | 90 | 7.0 ± 0.3 | >100 |
| 3 | PEA-6T | Air | 25 | 5 | 55 | 7.9 ± 0.4 | >100 |
| 4 | PEA-6T | Water | 25 | 6 | 60 | 7.9 ± 0.3 | >100 |
| 30 | 6 | 55 | 7.6 ± 0.2 | >100 | |||
| 40 | 5.5 | 51 | 7.6 ± 0.3 | >100 | |||
| 50 | 4 | 46 | 7.5 ± 0.5 | ~40 | |||
| 5 | PEA-6T | Methanol | 25 | 6 | 65 | 7.8 ± 0.2 | >100 |
| 6 | PEA-6T | Ethanol | 25 | 6.5 | 73 | 7.8 ± 0.2 | >100 |
| 7 | PEA-6T | N-butanol | 25 | 8 | 90 | 7.6 ± 0.2 | ~40 |
| 8 | PEA-6T | (0.9 wt%) NaCl | 25 | 5.5 | 55 | 7.6 ± 0.2 | >100 |
| 50 | 3.5 | 42 | 7.1 ± 0.6 | ~55 | |||
| 9 | PEA-6T | (3.5 wt%) NaCl | 25 | 6 | 65 | 7.7 ± 0.2 | >100 |
| 50 | 4 | 33 | 6.8 ± 0.7 | ~60 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Ma, S.; Gao, J. Study on Photodeformation of Solvent Resistance in Hydrogen-Bonded Cross-Linked Main-Chain Azobenzene Films. Molecules 2025, 30, 2106. https://doi.org/10.3390/molecules30102106
Zhang Z, Ma S, Gao J. Study on Photodeformation of Solvent Resistance in Hydrogen-Bonded Cross-Linked Main-Chain Azobenzene Films. Molecules. 2025; 30(10):2106. https://doi.org/10.3390/molecules30102106
Chicago/Turabian StyleZhang, Zhaoyang, Shengkui Ma, and Jianfeng Gao. 2025. "Study on Photodeformation of Solvent Resistance in Hydrogen-Bonded Cross-Linked Main-Chain Azobenzene Films" Molecules 30, no. 10: 2106. https://doi.org/10.3390/molecules30102106
APA StyleZhang, Z., Ma, S., & Gao, J. (2025). Study on Photodeformation of Solvent Resistance in Hydrogen-Bonded Cross-Linked Main-Chain Azobenzene Films. Molecules, 30(10), 2106. https://doi.org/10.3390/molecules30102106