
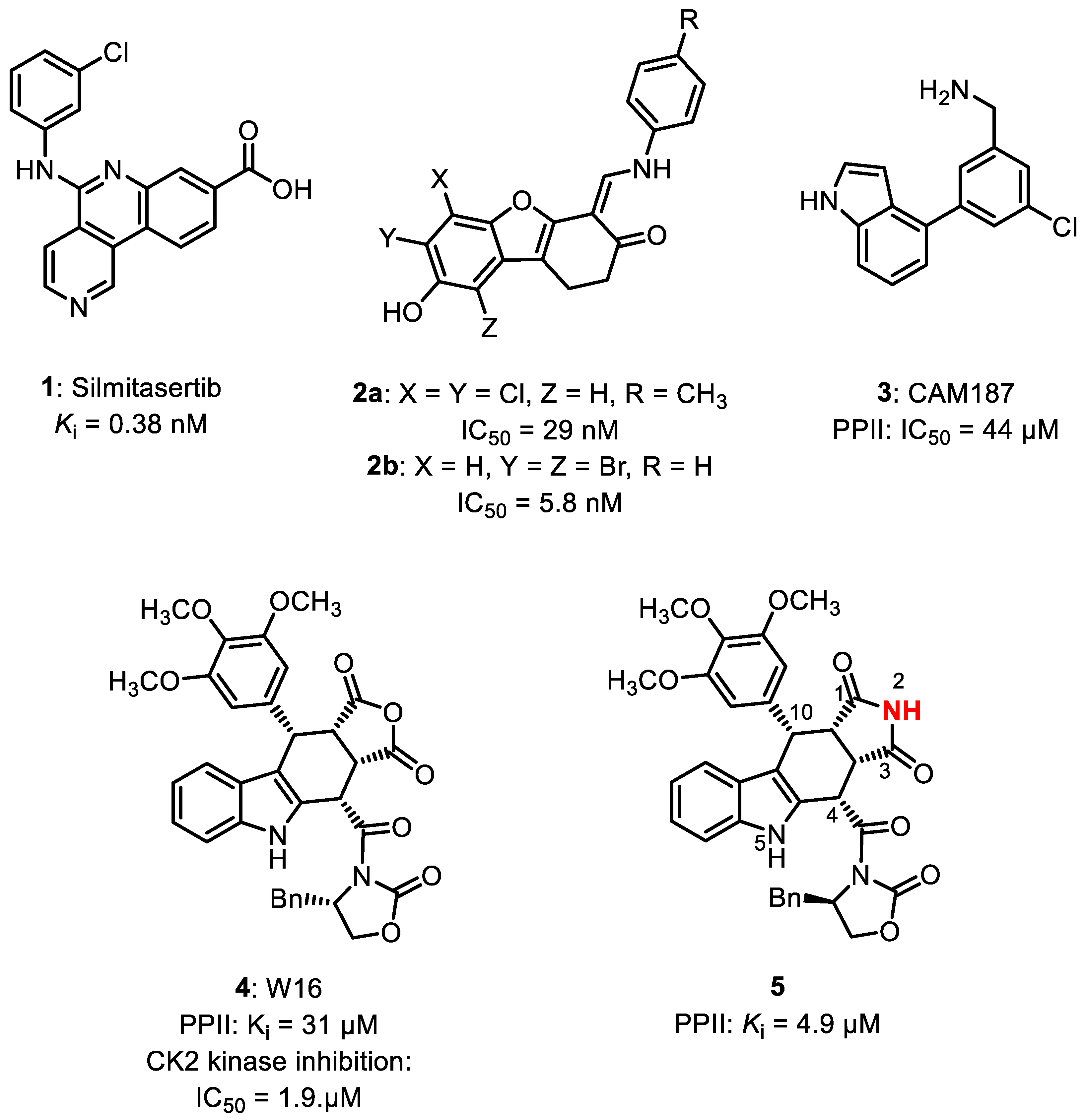
Structure–Activity Relationship Studies of Tetracyclic Pyrrolocarbazoles Inhibiting Heterotetrameric Protein Kinase CK2

 , ,
, ,  , and
, and
Abstract

1. Introduction
2. Results and Discussion
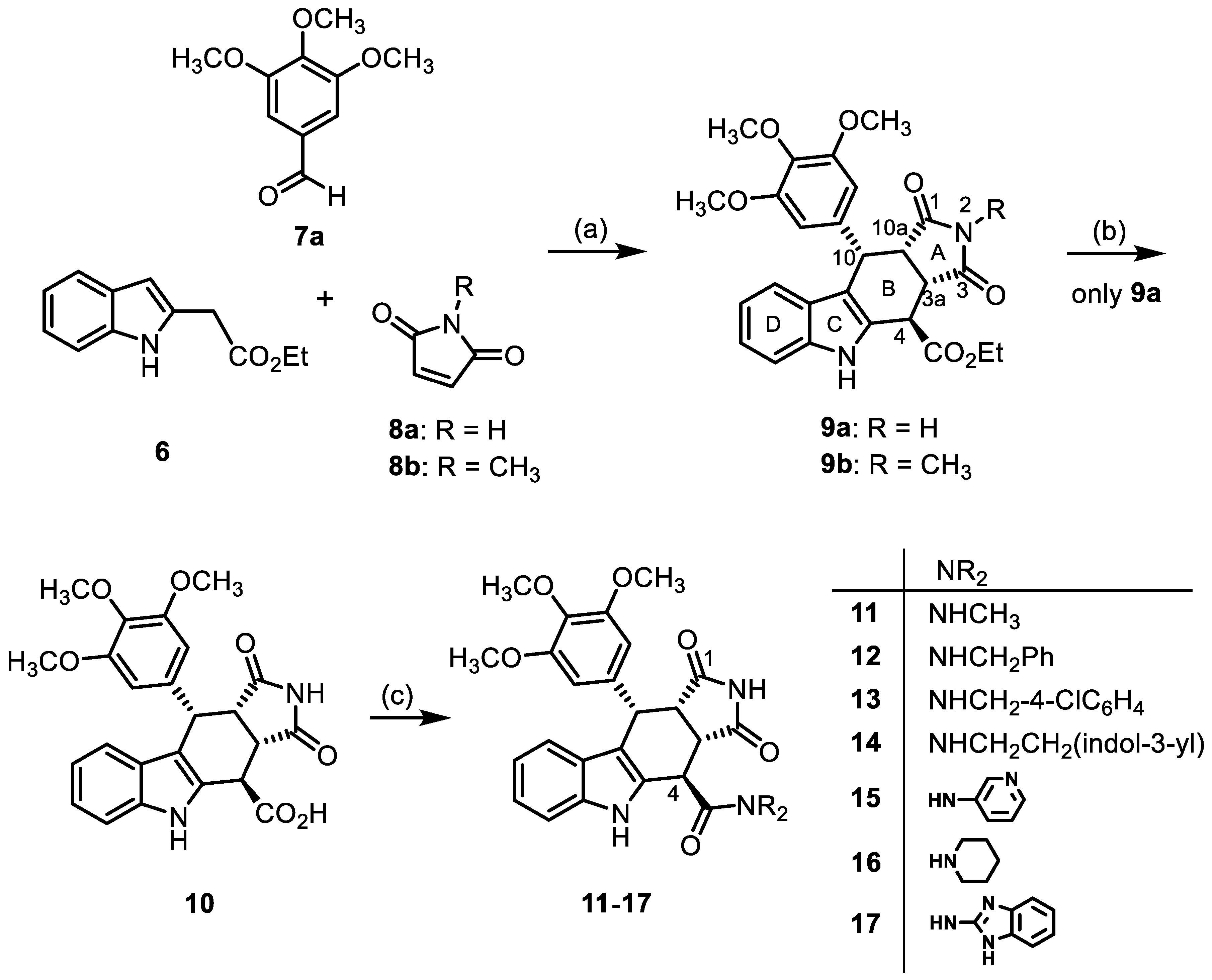
2.1. Synthesis of Tetracyclic Pyrrolocarbazoles
2.2. Pharmacological Evaluation
2.2.1. Inhibition of the CK2α/CK2β Interaction
2.2.2. Inhibition of the Enzymatic Activity
3. Conclusions
4. Experimental Part
4.1. Chemistry, General Methods
4.2. HPLC Method for the Determination of the Purity
4.3. Synthetic Procedures
- Ethyl (3aRS,4SR,10RS,10aRS)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl9a)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate () [40]
![Molecules 30 00063 i007]()
- Under N2, indolylacetate 6 (203 mg, 1.00 mmol), maleimide (8a, 292 mg, 3.01 mmol) and 3,4,5-trimethoxybenzaldehyde (7a, 296 mg, 1.51 mmol) were dissolved in dry o-xylene (10 mL) in a pressure-resistant Schlenk tube. Crushed CuSO4 ∙ 5 H2O (25.2 mg, 0.10 mmol) was added to the solution and the mixture was heated to reflux for 16 h (oil bath temperature of 185 °C). After cooling to room temperature, the mixture was filtered and the filter was washed with CH2Cl2 (3 × 10 mL). The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 45:55, Ø 4 cm, h = 16 cm, v = 30 mL). Yellow solid, mp 220 °C, yield of 379 mg (79%). C26H26N2O7 (478.5). TLC (ethyl acetate/CH2Cl2 = 3:7): Rf = 0.38.
- Purity (HPLC): 97.4%, (tR = 19.3 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.31 (t, J = 7.1 Hz, 3H, OCH2CH3), 3.53–3.57 (m, 1H, 10a-H), 3.57 (s, 3H, 4-OCH3), 3.61 (s, 6H, 3-OCH3, 5-OCH3), 4.05 (dd, J = 9.0/3.4 Hz, 1H, 3a-H), 4.20–4.33 (m, 2H, OCH2CH3), 4.49 (d, J = 3.5 Hz, 1H, 4-H), 4.74 (d, J = 8.3 Hz, 1H, 10-H), 6.21 (s, 2H, 2-HTMP, 6-HTMP), 6.88 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 7.05 (ddd, J = 8.1/6.9/1.2 Hz, 1H, 7-H), 7.20 (dd, J = 8.0/1.1 Hz, 1H, 9-H), 7.37 (dt, J = 8.2/0.9 Hz, 1H, 6-H), 10.92 (s, 2H, 2-H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 36.9 (1C, C-4), 38.0 (1C, C-10), 42.5 (1C, C-3a), 45.5 (1C, C-10a), 55.6 (2C, 3-OCH3 5-OCH3), 59.9 (1C, 4-OCH3), 61.7 (1C, OCH2CH3), 106.0 (2C, C-2TMP, 6-CTMP), 111.3 (1C, C-9b), 111.5 (1C, C-6), 118.4 (1C, C-9), 118.7 (1C, C-8), 121.6 (1C, C-7), 125.4 (1C, C-9a), 128.5 (1C, C-4a), 136.2 (1C, C-4TMP), 136.4 (1C, C-1TMP), 136.6 (1C, C-5a), 152.1 (2C, C-3TMP, C-5TMP), 171.1 (1C, C-4-C=O), 177.7 (1C, C-1), 179.5 (1C, C-3). Exact mass (APCI): m/z = 479.1785 (calcd. 479.1813 for C26H27N2O7 [MH]+). IR (neat): ṽ [cm−1] = 2987 (w, C-Haliph.), 1775 (w, C=O), 1709 (s, C=O), 1119 (s, C-O), 752 (m, C-Harom.).

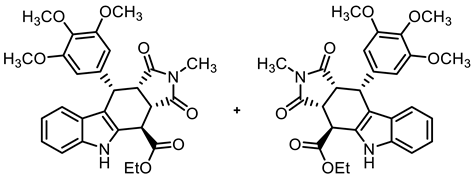
- Ethyl (3aRS,4SR,10RS,10aRS)-2-methyl-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate (9b)
![Molecules 30 00063 i008]()
- Under N2, indolylacetate 6 (3.01 g, 14.8 mmol), N-methylmaleimide (8b, 5.00 g, 45.0 mmol), and 3,4,5-trimethoxybenzaldehyde (7a, 4.42 g, 22.5 mmol) were dissolved in dry o-xylene (100 mL). Crushed CuSO4 ∙ 5 H2O (351 mg, 1.41 mmol) was added to the solution and the mixture was heated to reflux for 16 h. After cooling to -20 °C, the mixture was filtered and the obtained solid was washed with water (3 × 25 mL) and n-pentane (5 × 25 mL) to give the product without further purification. Colorless solid, mp 215 °C, yield of 6.32 g (83%). C27H28N2O7 (492.5). TLC (ethyl acetate/cyclohexane = 1:1): Rf = 0.41. Purity (HPLC): 97.3%, (tR = 20.5 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.30 (t, J = 7.1 Hz, 3H; OCH2CH3), 2.35 (s, 3H, NCH3), 3.55 (s, 3H, 4-OCH3), 3.61 (s, 6H, 3-OCH3, 5-OCH3), 3.63 (t, J = 8.2 Hz, 1H, 10a-H), 4.06 (dd, J = 8.7/3.3 Hz, 1H, 3a-H), 4.21–4.33 (m, 2H, OCH2CH3), 4.57 (d, J = 3.0 Hz, 1H, 4-H), 4.76 (d, J = 8.0 Hz, 1H, 10-H), 6.13 (s, 2H, 2-HTMP, 6-HTMP), 6.88 (ddd, J = 7.9/7.0/1.0 Hz, 1H, 8-H), 7.05 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.19 (d, J = 7.9 Hz, 1H, 9-H), 7.38 (dt, J = 8.0/0.9 Hz, 1H, 6-H), 10.96 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 23.7 (1C, NCH3), 36.6 (1C, C-4), 38.0 (1C, C-10), 41.4 (1C, C-3a), 44.7 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 61.8 (1C, OCH2CH3), 106.0 (2C, C-2TMP, C-6TMP), 110.8 (1C, C-9b), 111.5 (1C, C-6), 118.4 (1C, C-9), 118.7 (1C, C-7), 121.7 (1C, C-8), 125.4 (1C, C-9a), 128.4 (1C, C-4a), 136.1 (1C, C-1TMP), 136.3 (1C, C-4TMP), 136.7 (1C, C-5a), 152.1 (2C, C-3TMP, C-5TMP), 171.0 (1C, C-4-C=O), 176.4 (1C, C-1), 178.0 (1C, C-3). Exact mass (ESI): m/z = 493.1958 (calcd. 493.1969 for C27H29N2O7 [MH]+). IR (neat): ṽ [cm−1] = 2970 (w, C-Haliph.), 1743,1701 (s, C=O), 1180, 1115 (s, C-O).
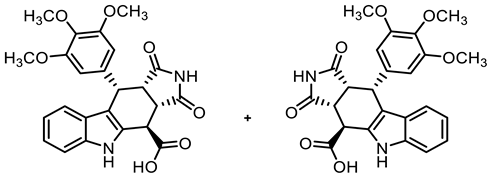
- (3aRS,4SR,10RS,10aRS)-1,3-Dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylic acid (10) [40]
![Molecules 30 00063 i009]()
- A solution of NaOH (2.60 g, 6.50 mmol) in water (50 mL) was added to a solution of ester 9a (5.06g, 10.56 mmol) in THF (50 mL). The mixture was stirred for 30 min at room temperature. Ethyl acetate (100 mL) was added and the phases were separated. The aqueous layer was washed with ethyl acetate (3 × 50 mL) to remove non-acidic impurities. HCl solution (2 M in water, 30 mL) was added to the aqueous layer and the resulting suspension was extracted with ethyl acetate (3 × 50 mL). The combined organic layers of the last extraction step were dried (Na2SO4), filtered, and concentrated in vacuo to obtain the product without further purification. Yellow solid, mp 162 °C, yield of 4.58 g (96%). C24H22N2O7 (450.4). TLC (MeOH/CH2Cl2 = 15:85 + 0.1% acetic acid): Rf = 0.33. Purity (HPLC): 94.7%, (tR = 16.5 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.54 (t, J = 8.4 Hz, 1H, 10a-H), 3.56 (s, 3H, 4-OCH3), 3.62 (s, 6H, 3-OCH3, 5-OCH3), 4.09 (dd, J = 9.0/3.6 Hz, 1H, 3a-H), 4.41 (d, J = 3.6 Hz, 1H, 4-H), 4.72 (d, J = 8.2 Hz, 1H, 10-H), 6.22 (s, 2H, 2-HTMP, 6-HTMP), 6.87 (ddd, J = 7.8/6.8/0.9 Hz, 1H, 8-H), 7.02 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.21 (d, J = 7.9 Hz, 1H, 9-H), 7.39 (d, J = 8.1 Hz, 1H, 6-H), 10.89 (s, 1H, 2-H), 10.92 (s, 1H, 5-H), 13.40 (s, 1H, COOH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 37.0 (1C, C-4), 38.1 (1C, C-10), 42.3 (1C, C-3a), 45.5 (1C, C-10a), 55.6 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 106.0 (2C, C-2TMP, C-6TMP), 111.1 (1C, C-9b), 111.6 (1C, C-6), 118.2 (1C, C-9), 118.6 (1C, C-8), 121.4 (1C, C-7), 125.4 (1C, C-9a), 128.9 (1C, C-4a), 136.1 (1C, C-4TMP), 136.5 (1C, C-1TMP), 136.6 (1C, C-5a), 152.1 (2C, C-3TMP, C-5TMP), 172.4 (1C, COOH), 177.8 (1C, C-1), 179.9 (1C, C-3). Exact mass (ESI): m/z = 451.1477 (calcd. 451.1500 for C24H23N2O7 [MH]+). IR (neat): ṽ [cm−1] = 2939, 2835 (m, C-Haliph.), 1709 (s, C=O), 1119 (m, C-O), 745 (m, C-Harom.).
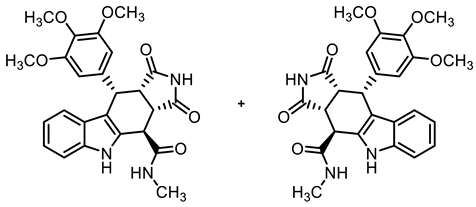
- (3aRS,4SR,10RS,10aRS)-N-Methyl-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (11)
![Molecules 30 00063 i010]()
- Carboxylic acid 10 (212 mg, 0.47 mmol), COMU® (242 mg, 0.56 mmol), and DIPEA (140 mg, 1.08 mmol) were dissolved in dry THF (15 mL) at 0 °C. Then, a solution of methylamine (17.6 mg, 0.56 mmol) in THF (2 mL) was added dropwise. The mixture was stirred for 2 h at 0 °C. After the completion of the transformation, the solution was allowed to warm to ambient temperature and ethyl acetate (30 mL) was added. The mixture was extracted with HCl solution (1 M in water, 2 × 30 mL) and saturated NaCl solution. The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by automatic flash column chromatography (cartridge: SNAP 50 g, flow rate of 50 mL/min, ethyl acetate) to yield a yellow solid. For further purification, the obtained solid was dissolved in ethyl acetate (50 mL) and extracted with NaHCO3 solution (0.1 M in water, 3 × 50 mL). The organic layer was dried (Na2SO4), filtered, and the solvent was removed in vacuo. Yellow solid, mp 215 °C, yield of 106 mg (49%). C25H25N3O6 (463.5). TLC (ethyl acetate): Rf = 0.31. Purity (HPLC): 95.6%, (tR = 16.0 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 2.81 (d, J = 4.6 Hz, 3H, NHCH3), 3.52 (dd, J = 9.6/7.9 Hz, 1H, 10a-H), 3.55 (s, 3H, 4-OCH3), 3.65 (s, 6H, 3-OCH3, 5-OCH3), 4.00 (dd, J = 9.5/5.0 Hz, 1H, 3a-H), 4.36 (d, J = 5.0 Hz, 1H, 4-H), 4.74 (d, J = 7.9 Hz, 1H, 10-H), 6.27 (s, 2H, 2-HTMP, 6-HTMP), 6.90 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 7.03 (ddd, J = 8.1/7.0/1.1 Hz, 1H, 7-H) 7.34–7.36 (m, 1H, 9-H), 7.36–7.37 (m, 1H, 6-H), 8.61 (q, J = 4.6 Hz, 1H, NHamide), 10.67 (s, 1H, 2-H), 10.88 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 26.3 (1C, NHCH3), 38.1 (1C, C-10), 38.6 (1C, C-4), 42.6 (1C, C-3a), 46.2 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.7 (2C, C-2TMP, C-6TMP), 111.4 (1C, C-9b), 111.6 (1C, C-6), 117.8 (1C, C-9), 118.6 1C, C-8), 121.2 (1C, C-7), 125.5 (1C, C-9a), 131.3 (1C, C-4a), 136.1 (1C, C-4TMP), 136.3 (1C, C-1TMP), 137.0 (1C, C-5a), 152.2 (1C, C-3TMP, C-5TMP), 170.5 (1C, COamide), 178.1 (1C, C-1), 180.4 (1C, C-3). Exact mass (ESI): m/z = 464.1818 (calcd. 451.1816 for C25H25N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2889 (m, C-Haliph.), 1697, 1678 (s, C=O), 1119 (m, C-O), 741 (m, C-Harom.).
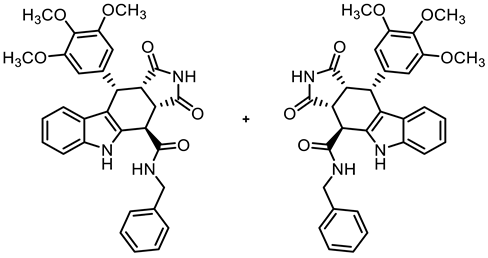
- (3aRS,4SR,10RS,10aRS)-N-Benzyl-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (12)
![Molecules 30 00063 i011]()
- Carboxylic acid 10 (135 mg, 0.30 mmol), COMU® (168 mg, 0.39 mmol), and DIPEA (90.6 mg, 0.70 mmol) were dissolved in dry THF (5 mL) at room temperature. Then, a solution of benzylamine (42.1 mg, 0.39 mmol) in THF (2 mL) was added dropwise. The mixture was stirred for 2 h at room temperature. After the completion of the transformation, ethyl acetate (50 mL) was added. The mixture was extracted successively with NaHCO3 solution (0.1 M in water, 3 × 25 mL), HCl solution (1 M in water, 3 × 25 mL), and saturated NaCl solution (3 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 6:4, Ø 2 cm, h = 18 cm, v = 10 mL). Yellow solid, mp 157 °C, yield of 136 mg (84%). C31H29N3O6 (539.6). TLC (ethyl acetate/cyclohexane = 6:4): Rf = 0.41. Purity (HPLC): 96.9%, (tR = 19.3 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.56 (s, 3H, 4-OCH3), 3.57 (dd, J = 9.5/8.0 Hz, 1H, 10a-H), 3.64 (s, 6H, 3-OCH3, 5-OCH3), 4.03 (dd, J = 9.0/4.9 Hz, 1H, 3a-H), 4.26 (dd, J = 15.2/4.2 Hz, 1H, PhCH2NH), 4.48 (d, J = 5.0 Hz, 1H, 4-H), 4.75 (dd, J = 15.3/7.3 Hz, 1H, PhCH2NH), 4.76 (d, J = 8.1 Hz, 1H, 10-H), 6.29 (s, 2H, 2-HTMP, 6-HTMP), 6.91 (ddd, J = 7.9/7.0/1.0 Hz, 1H, 8-H), 7.04 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.28 (tt, J = 7.3/1.7 Hz, 1H, 4-Hbenzyl), 7.33–7.39 (m, 4H, 6-H, 9-H, 3-Hbenzyl, 5-Hbenzyl), 7.40–7.42 (m, 2H, 2-Hbenzyl, 6-Hbenzyl), 9.09 (dd, J = 7.3/4.3 Hz, 1H, NHamide), 10.73 (s, 1H, 2-H), 10.91 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 38.1 (1C, C-10), 38.7 (1C, C-4), 42.9 (1C, C-3a), 43.1 (1C, PhCH2NH), 46.1 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.8 (2C, C-2TMP, C-6TMP), 111.3 (1C, C-9b) 111.7 (1C, C-6), 117.9 (1C, C-9), 118.7 (1C, C-8), 121.3 (1C, C-7), 125.5 (1C, C-9a), 126.9 (1C, C-4benzyl), 127.5 (2C, C-2benzyl, C-6benzyl), 128.3 (2C, C-3benzyl, C-5benzyl), 131.2 (1C, C-4a), 136.1 (1C, C-4TMP), 136.3 (1C, C-1TMP), 137.0 (1C, C-5a), 139.0 (1C, C-1benzyl), 152.2 (2C, C-3TMP, C-5TMP), 170.4 (1C, COamide), 178.1 (1C, C-1), 180.3 (1C, C-3). Exact mass (ESI): m/z = 540.2125 (calcd. 540.2129 for C31H30N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2889 (s, C-Haliph.), 1712 (s, C=O), 1153,1122 (s, C-O), 741 (m, C-Harom.).
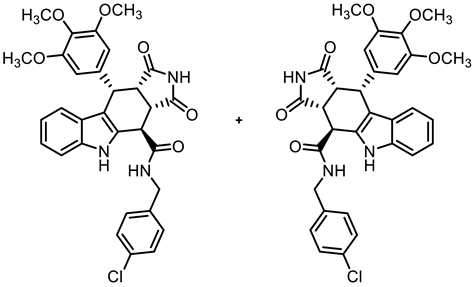
- (3aRS,4SR,10RS,10aRS)-N-(4-Chlorobenzyl)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (13)
![Molecules 30 00063 i012]()
- Carboxylic acid 10 (135 mg, 0.30 mmol), COMU® (155 mg, 0.36 mmol), and DIPEA (86.1 mg, 0.67 mmol) were dissolved in dry THF (10 mL) at 0 °C. Then, a solution of 4-chlorobenzylamine (51.2 mg, 0.36 mmol) in THF (2 mL) was added dropwise. The mixture was stirred for 30 min at 0 °C and 1 h at room temperature. After the completion of the transformation, ethyl acetate (30 mL) was added. The mixture was extracted with HCl solution (1 M in water, 5 × 30 mL) and saturated NaCl solution (2 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was adsorbed onto silica and purified by automatic flash column chromatography (cartridge: SNAP 50g, flow rate of 50 mL/min, ethyl acetate/cyclohexane = 30:70 → 0:100). Yellow solid, mp 204 °C, yield of 123 mg (71%). C31H28ClN3O6 (574.0). TLC (ethyl acetate/cyclohexane = 6:4): Rf = 0.38. Purity (HPLC): 96.9%, (tR = 20.3 min). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 3.56 (s, 3H, 4-OCH3), 3.56 (dd, J = 9.6/8.0 Hz, 1H, 10a-H), 3.64 (s, 6H, 3-OCH3, 5-OCH3), 4.00 (dd, J = 9.5/4.9 Hz, 1H, 3a-H), 4.25 (dd, J = 15.4/4.4 Hz, 1H, PhCH2NH), 4.46 (d, J = 4.9 Hz, 1H, 4-H), 4.71 (dd, J = 15.4/7.1 Hz, 1H, PhCH2NH), 4.76 (d, J = 8.0 Hz, 1H, 10-H), 6.28 (s, 2H, 2-HTMP, 6-HTMP), 6.91 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 7.04 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.34 (d, J = 7.8 Hz, 1H, 9-H), 7.37 (d, J = 8.0 Hz, 1H, 6-H), 7.40–7.45 (m, 4H, 2-Hbenzyl, 3-Hbenzyl, 5-Hbenzyl, 6-Hbenzyl), 9.11 (dd, J = 7.1/4.5 Hz, 1H, NHamide), 10.75 (s, 1H, 2-H), 10.92 (s, 1H, 5-H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 38.0 (1C, C-10), 38.7 (1C, C-4), 42.4 (1C, PhCH2NH), 42.9 (1C, C-3a), 46.1 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.8 (2C, C-2TMP, C-6TMP), 111.3 (1C, C-9b) 111.7 (1C, C-6), 118.0 (1C, C-9), 118.7 (1C, C-8), 121.3 (1C, C-7), 125.5 (1C, C-9a), 128.2 (2C, C-2benzyl, C-6benzyl), 129.3 (2C, C-3benzyl, C-5benzyl), 131.1 (1C, C-4a), 131.4 (1C, C-4benzyl), 136.1 (1C, C-4TMP), 136.3 (1C, C-1TMP), 137.0 (1C, C-5a), 138.1 (1C, C-1benzyl), 152.2 (2C, C-3TMP, C-5TMP), 170.5 (1C, COamide), 178.1 (1C, C-1), 180.3 (1C, C-3). Exact mass (ESI): m/z = 574.1730 (calcd. 574.1739 for C31H29ClN3O6 [MH]+). IR (neat): ṽ [cm−1] = 2970, 2940 (s, C-Haliph.), 1717, 1682 (s, C=O), 1173, 1126 (s, C-O), 745 (m, C-Harom.).
- (3aRS,4SR,10RS,10aRS)-N-[2-(Indol-3-yl)ethyl]-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (14)
![Molecules 30 00063 i013]()
- Carboxylic acid 10 (136 mg, 0.30 mmol), COMU® (154 mg, 0.36 mmol), and DIPEA (117 mg, 0.90 mmol) were dissolved in ethyl acetate (10 mL) at room temperature. Then, 2-(indol-3-yl)ethanamine (58.4 mg, 0.36 mmol) was added to the solution. The mixture was stirred for 4 h at room temperature. After the completion of the transformation, ethyl acetate (30 mL) was added. The mixture was extracted with HCl solution (1 M in water, 3 × 20 mL) and saturated NaCl solution (2 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by automatic flash column chromatography (cartridge: SNAP 50 g, flow rate of 50 mL/min, ethyl acetate/cyclohexane = 35:65 → 0:100) to obtain a yellow solid. For further purification, the solid was recrystallized (ethyl acetate/cyclohexane), filtered, and washed with cold ethanol (2 × 10 mL). Colorless solid, mp 182 °C, yield of 93 mg (52%). C34H32N4O6 (592.7). TLC (ethyl acetate/cyclohexane = 7:3): Rf = 0.33. Purity (HPLC): 99.4%, (tR = 19.5 min). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 3.01 (t, J = 7.6 Hz, 2H, NHCH2CH2), 3.40–3.47 (m, 1H, NHCH2CH2), 3.53 (dd, J = 9.5/8.0 Hz, 1H, 10a-H), 3.56 (s, 3H, 4-OCH3), 3.65 (s, 6H, 3-OCH3, 5-OCH3), 3.62–3.68 (m, 1H, NHCH2CH2), 3.99 (dd, J = 9.4/4.6 Hz, 1H, 3a-H), 4.42 (d, J = 4.6 Hz, 1H, 4-H), 4.74 (d, J = 8.0 Hz, 1H, 10-H), 6.27 (s, 2H, 2-HTMP, 6-HTMP) 6.89 (ddd, J = 7.9/7.0/1.0 Hz, 1H, 8-H), 7.00 (ddd, J = 7.9/7.0/1.1 Hz, 1H, 5-Hindolyl), 7.02 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.09 (ddd, J = 8.1/6.9/1.2 Hz, 1H, 6-Hindolyl), 7.24 (d, J = 2.2 Hz, 1H, 2-Hindolyl), 7.31 (dt, J = 8.2/0.8 Hz, 1H, 6-H), 7.32 (d, J = 8.0 Hz, 1H, 9-H), 7.36 (dt, J = 8.1/0.9 Hz, 1H, 7-Hindolyl), 7.62 (d, J = 7.9 Hz, 1H, 4-Hindolyl), 8.73 (t, J = 5.6 Hz, 1H, NHamide), 10.42 (s, 1H, 5-H), 10.87 (d, J = 2.4 Hz, 1H, 1-Hindolyl), 10.89 (s, 1H, 2-H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 25.0 (1C, NHCH2CH2), 38.1 (1C, C-10), 38.5 (1C, C-4), 40.3 (1C, NHCH2CH2), 42.7 (1C, C-3a), 46.1 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.9 (2C, C-2TMP, C-6TMP), 111.4 (1C, C-6), 111.4 (1C, C-7indolyl), 111.6 (1C, C-9b), 111.7 (1C, C-3indolyl), 117.9 (1C, C-9), 118.3 (1C, C-5indolyl), 118.3 (1C, C-4indolyl), 118.7 (1C, C-8), 121.0 (1C, C-6indolyl), 121.3 (1C, C-7), 122.7 (1C, C-2indolyl), 125.6 (1C, C-9a), 127.2 (1C, C-3aindolyl), 131.2 (1C, C-4a), 136.1 (1C, C-4TMP), 136.2 (1C, C-5a), 136.3 (1C, C-7aindolyl), 136.9 (1C, C-1TMP), 152.2 (2C, C-3TMP, C-5TMP), 170.1 (1C, COamide), 178.1 (1C, C-1), 180.3 (1C, C-3). Exact mass (ESI): m/z = 593.2370 (calcd. 593.2395 for C34H33N4O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2940 (s, C-Haliph.), 1701 (s, C=O), 1177, 1123 (s, C-O), 733 (s, C-Harom.).

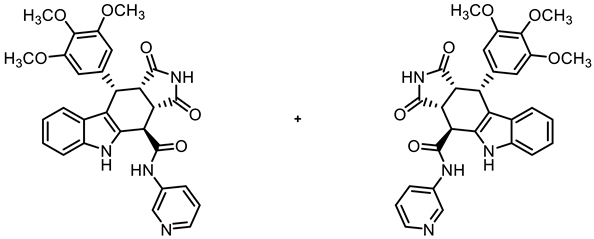
- (3aRS,4SR,10RS,10aRS)-1,3-Dioxo-N-(pyridin-3-yl)-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (15)
![Molecules 30 00063 i014]()
- Carboxylic acid 10 (135 mg, 0.30 mmol), COMU® (155 mg, 0.36 mmol), and DIPEA (85.5 mg, 0.66 mmol) were dissolved in dry THF (10 mL) at room temperature. Then, pyridin-3-amine (37.7 mg, 0.40 mmol) was added to the solution. The mixture was stirred for 12 h at room temperature. After the completion of the transformation, ethyl acetate (50 mL) was added. The mixture was extracted with HCl solution (1 M in water, 5 × 20 mL) and saturated NaCl solution (2 × 20 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by automatic flash column chromatography (cartridge: SNAP 50g, flow rate of 50 mL/min, MeOH/CH2Cl2 = 0:100 → 10:90). Colorless solid, mp 198 °C, yield of 21 mg (12%). C29H26N4O6 (526.5). TLC (MeOH/CH2Cl2 = 5:95): Rf = 0.14. Purity (HPLC): 97.6%, (tR = 15.7 min). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 3.57 (s, 3H, 4-OCH3), 3.60 (dd, J = 9.6/8.0 Hz, 1H, 10a-H), 3.68 (s, 6H, 3-OCH3, 5-OCH3), 4.14 (dd, J = 9.5/4.9 Hz, 1H, 3a-H), 4.67 (d, J = 5.0 Hz, 1H, 4-H), 4.81 (d, J = 7.9 Hz, 1H, 10-H), 6.31 (s, 2H, 2-HTMP, 6-HTMP), 6.92 (ddd, J = 7.9/7.1/1.0 Hz, 1H, 8-H), 7.04 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.34 (d, J = 8.1 Hz, 1H, 6-H), 7.40 (d, J = 7.9 Hz, 1H, 9-H), 7.45 (dd, J = 8.3/4.7 Hz, 1H, 5-Hpyridinyl), 8.22 (ddd, J = 8.3/2.6/1.5 Hz, 1H, 6-Hpyridinyl), 8.36 (dd, J = 4.7/1.5 Hz, 1H, 4-Hpyridinyl), 8.93 (d, J = 2.5 Hz, 1H, 2-Hpyridinyl), 10.95 (s, 1H, 5-H), 10.97 (s, 1H, 2-H), 11.02 (s, 1H, NHamide). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 38.0 (1C, C-10), 42.8 (1C, C-3a), 46.0 (1C, C-10a), 55.8 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.9 (2C, C-2TMP, C-6TMP), 111.3 (1C, C-9b), 112.0 (1C, C-6), 118.0 (1C, C-9), 118.8 (1C, C-8), 121.5 (1C, C-7), 123.7 (1C, C-5pyridinyl), 125.4 (1C, C-9a), 126.7 (1C, C-6pyridinyl), 130.4 (1C, C-4a), 135.9 (1C, C-3pyridinyl), 136.2 (1C, C-4TMP), 136.4 (1C, C-5a), 137.0 (1C, C-1TMP), 141.2 (1C, C-2pyridinyl), 144.5 (1C, C-4pyridinyl), 152.3 (2C, C-3TMP, C-5TMP), 169.7 (1C, COamide), 178.0 (1C, C-1), 180.3 (1C, C-3). The signal for C-4 C-atom is overlaid by the DMSO signals at 39.5 ppm. Exact mass (ESI): m/z = 527.1919 (calcd. 527.1925 for C29H27N4O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2932 (s, C-Haliph.), 1709 (s, C=O), 1173, 1123 (s, C-O), 745 (s, C-Harom.).
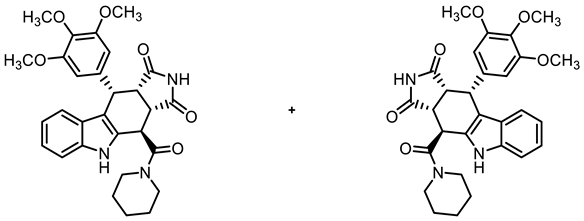
- (3aRS,4SR,10RS,10aRS)-4-[(Piperidin-1-yl)carbonyl]-10-(3,4,5-trimethoxyphenyl)-4,5,10,10a-tetrahydropyrrolo[3,4-b]carbazole-1,3(2H,3aH)-dione (16)
![Molecules 30 00063 i015]()
- Carboxylic acid 10 (136 mg, 0.30 mmol), COMU® (168 mg, 0.39 mmol), and DIPEA (89.6 mg, 0.69 mmol) were dissolved in dry THF (10 mL) at 0 °C. Then, a solution of piperidine (32.2 mg, 0.38 mmol) in THF (3 mL) was added dropwise. The mixture was stirred for 2 h at 0 °C. After the completion of the transformation, the solution was allowed to warm to ambient temperature and ethyl acetate (30 mL) was added. The mixture was extracted with NaHCO3 solution (0.1 M in water, 3 × 25 mL), HCl solution (1 M in water, 3 × 25 mL), and saturated NaCl solution (1 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 6:4 → 7:3, Ø 2 cm, h = 25 cm, v = 10 mL). Colorless solid, mp 293 °C, yield of 103 mg (66%). C29H31N3O6 (517.6). TLC (ethyl acetate/cyclohexane = 6:4): Rf = 0.29. Purity (HPLC): 96.7%, (tR = 18.4 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.43–1.74 (m, 6H, N(CH2CH2)2CH2), 3.22 (ddd, J = 12.8/9.7/3.0 Hz, 1H, N(CH2CH2)2CH2), 3.51 (dd, J = 9.3/8.0 Hz, 1H, 10a-H), 3.57 (s, 3H, 4-OCH3), 3.59–3.62 (m, 1H, N(CH2CH2)2CH2), 3.63 (s, 6H, 3-OCH3, 5-OCH3), 3.92 (dt, J = 13.7/4.2 Hz, 1H, N(CH2CH2)2CH2), 3.97 (dd, J = 9.1/4.3 Hz, 1H, 3a-H), 3.95–4.02 (m, 1H, N(CH2CH2)2CH2), 4.76 (d, J = 8.3 Hz, 1H, 10-H), 4.77 (d, J = 4.0 Hz, 1H, 4-H), 6.28 (s, 2H, 2-HTMP, 6-HTMP), 6.89 (ddd, J = 7.9/7.0/1.0 Hz, 1H, 8-H), 7.03 (ddd, J = 8.1/7.0/1.2 Hz, 1H, 7-H), 7.27 (dd, J = 8.0/1.0 Hz, 1H, 9-H), 7.33 (dd, J = 8.1/0.9 Hz, 1H, 6-H), 10.78 (s, 1H, 5-H), 10.91 (s, 1H, 2-H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 24.2 (1C, N(CH2CH2)2CH2) 25.3 (1C, N(CH2CH2)2CH2), 26.0 (1C, N(CH2CH2)2CH2), 34.3 (1C, C-4), 38.3 (1C, C-10), 43.3 (1C, N(CH2CH2)2CH2), 44.0 (1C, C-3a), 46.3 (1C, C-10a), 47.0 (1C, N(CH2CH2)2CH2), 55.5 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.9 (2C, C-2TMP, C-6TMP), 111.3 (1C, C-6), 111.5 (1C, C-9b), 118.0 (1C, C-9), 118.6 (1C, C-8), 121.2 (1C, C-7), 125.5 (1C, C-9a), 131.1 (1C, C-4a), 136.1 (1C, C-4TMP), 136.4 (1C, C-5a), 136.6 (1C, C-1TMP), 152.2 (2C, C-3TMP, C-5TMP), 168.5 (1C, COamide), 178.0 (1C, C-1), 180.2 (1C, C-3). Exact mass (ESI): m/z = 518.2280 (calcd. 518.2286 for C29H32N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2886 (s, C-Haliph.), 1709 (s, C=O), 1150, 1119 (s, C-O), 799, 745 (s, C-Harom.).
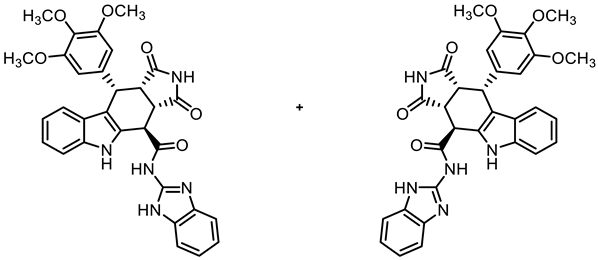
- (3aRS,4SR,10RS,10aRS)-N-(1H-Benzimidazol-2-yl)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxamide (17)
![Molecules 30 00063 i016]()
- Carboxylic acid 10 (136 mg, 0.30 mmol), COMU® (167 mg, 0.39 mmol), and DIPEA (128 mg, 0.99 mmol) were dissolved in dry THF (10 mL) at room temperature. Then, benzimidazol-2-amine (52.2 mg, 0.39 mmol) was added. The mixture was stirred for 2 h at room temperature. After the completion of the transformation, ethyl acetate (50 mL) was added. The mixture was extracted with saturated NH4Cl solution (3 × 25 mL), NaHCO3 solution (0.1 M in water, 3 × 25 mL), and saturated NaCl solution (1 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in ethyl acetate (10 mL), and n-hexane (30 mL) was added. The resulting mixture was filtered, and the precipitate was washed with cold Et2O. Colorless solid, mp 233 °C, yield of 39 mg (23%). C31H27N5O6 (565.6). TLC (ethyl acetate/cyclohexane = 6:4): Rf = 0.16. Purity (HPLC): 97.3%, (tR = 17.7 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.57 (s, 3H, 4-OCH3), 3.65 (dd, J = 9.8/7.9 Hz, 1H, 10a-H), 3.70 (s, 6H, 3-OCH3, 5-OCH3), 4.19 (dd, J = 9.7/5.8 Hz, 1H, 3a-H), 4.79 (d, J = 5.8 Hz, 1H, 4-H), 4.83 (d, J = 8.0 Hz, 1H, 10-H), 6.34 (s, 2H, 2-HTMP, 6-HTMP), 6.93 (ddd, J = 7.9/6.9/1.0 Hz, 1H, 8-H), 7.03 (ddd, J = 8.1/6.9/1.2 Hz, 1H, 7-H), 7.11–7.16 (m, 2H, 5-HBI, 6-HBI), 7.30–7.34 (m, 1H, 6-H), 7.44 (d, J = 7.9 Hz, 1H, 9-H), 7.50–7.54 (m, 2H, 4-HBI, 7-HBI), 11.03 (s, 1H, 5-H), 11.03 (s, 1H, 2-H), 12.20 (s, 2H, NHamide, 1-HBI). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 38.1 (1C, C-10), 42.9 (1C, C-3a), 46.1 (1C, C-10a), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.5 (2C, C-2TMP, C-6TMP), 111.4 (1C, C-6), 112.1 (1C, C-9b), 117.9 (1C, C-9), 118.8 (1C, C-8), 121.3 (2C, C-5BI, C-6BI), 121.4 (1C, C-7), 125.4 (1C, C-9a), 130.4 (1C, C-4a), 136.1 (1C, C-4TMP), 136.3 (1C, C-5a), 137.1 (1C, C-1TMP), 152.3 (2C, C-3TMP, C-5TMP), 172.0 (1C, COamide), 178.1 (1C, C-1), 180.3 (1C, C-3). Signals for C-3aBI, C-4BI, C-7BI, and C-7aBI C-atoms were not observed in the spectrum. The signal for C-4 C-atom was overlaid by the DMSO signal at 39.5 ppm. Exact mass (ESI): m/z = 566.2037 (calcd. 566.2034 for C31H28N5O6 [MH]+). IR (neat): ṽ [cm−1] = 2909, 2835 (w, C-Haliph.), 1701 (s, C=O), 1177, 1126 (s, C-O), 798, 745 (s, C-Harom.).
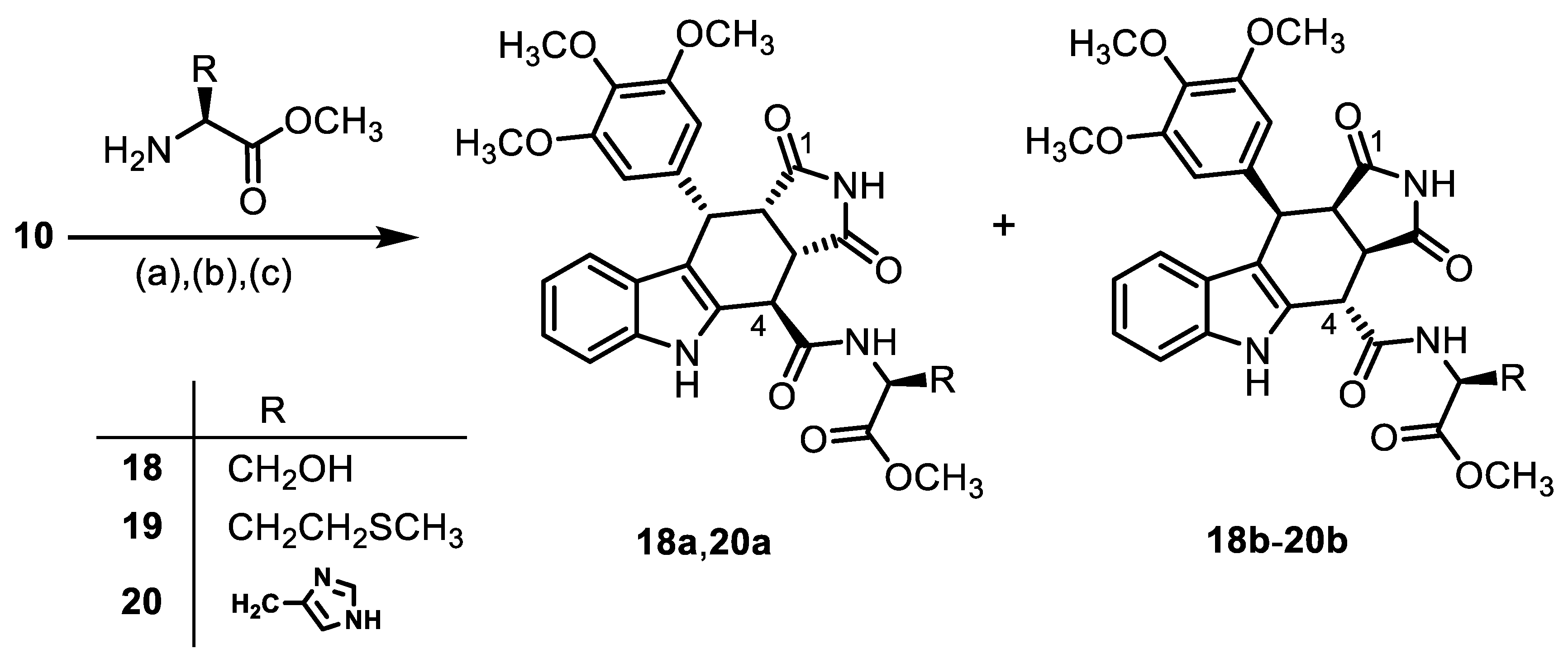
- Methyl N-{[(3aS,4R,10S,10aS)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazol-4-yl]carbonyl}-(S)-serinate ((+)-18a) and methyl N-{[(3aR,4S,10R,10aR)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazol-4-yl]carbonyl}-(S)-serinate ((-)-18b)
![Molecules 30 00063 i017]()
- Carboxylic acid 10 (136 mg, 0.30 mmol), COMU® (168 mg, 0.39 mmol), and DIPEA (128 mg, 0.99 mmol) were dissolved in dry DMF (10 mL) at 0 °C. After the addition of (S)-serine methyl ester HCl (38.9 mg, 0.25 mmol), the mixture was stirred for 2 h at 0 °C. After the completion of the transformation, the solution was allowed to warm to ambient temperature, and ethyl acetate (30 mL) was added. The mixture was washed with HCl solution (1 M in water, 3 × 25 mL) and saturated NaCl solution (1 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 8:2, Ø 2 cm, h = 15 cm, v = 10 mL) to give both diastereomers.
- (+)-18a [Rf = 0.41 (ethyl acetate/cyclohexane = 8:2)]: Yellow solid, mp 174 °C, yield of 37 mg (27%). C28H29N3O9 (551.6). Purity (HPLC): 93.7%, (tR = 16.5 min). Specific rotation: +100 (c = 1.7, THF). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.51 (dd, J = 9.6/7.8 Hz, 1H, 10a-H), 3.56 (s, 3H, 4-OCH3), 3.68 (s, 6H, 3-OCH3, 5-OCH3), 3.78 (s, 3H, CO2CH3), 3.78–3.84 (m, 1 H, CHCH2OH), 3.97 (dt, J = 11.0/5.1 Hz, 1H, CHCH2OH), 4.08 (dd, J = 9.6/5.2 Hz, 1H, 3a-H), 4.60–4.69 (m, 1H, CHCH2OH), 4.67 (d, J = 5.3 Hz, 1H, 4-H), 4.76 (d, J = 7.8 Hz, 1H, 10-H), 5.41 (t, J = 5.2 Hz, 1H, CHCH2OH), 6.33 (s, 2H, 2-HTMP, 6-HTMP), 6.95 (ddd, J = 7.9/7.0/1.0 Hz, 1H, 8-H), 7.08 (ddd, J = 8.1/7.0/1.2 Hz, 1H, 7-H), 7.34 (dt, J = 8.0/0.9 Hz, 1H, 6-H), 7.42 (dd, J = 7.9/1.0 Hz, 1H, 9-H), 9.35 (d, J = 7.9 Hz, 1H), NHamide), 10.38 (s, 1H, 5-H), 10.93 (s, 1H, 2-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 38.1 (1C, C-10), 38.2 (1C, C-4), 41.7 (1C, C-3a), 46.3 (1C, C-10a), 52.7 (1C, CO2CH3), 55.3 (1C, CHCH2OH), 55.8 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 61.0 (1C, CHCH2OH), 105.8 (2C, C-2TMP, C-5TMP), 111.0 (1C, C-6), 112.0 (1C, C-9b), 118.1 (1C, C-9), 119.0 (1C, C-8), 121.6 (1C, C-7), 125.6 (1C, C-9a), 131.3 (1C, C-4a), 135.7 (1C, C-5a), 136.2 (1C, C-4TMP), 136.9 (1C, C-1TMP), 152.3 (2C, C-3TMP, C-5TMP), 170.3 (1C, CO2CH3), 172.7 (C-4-C=O), 178.1 (1C, C-1), 180.5 (1C, C-3). Exact mass (ESI): m/z = 552.1972 (calcd. 552.1977 for C28H30N3O9 [MH]+). IR (neat): ṽ [cm−1] = 3314 (m, OH), 2931, 2847 (w, C-Haliph.), 1708, 1674 (s, C=O), 1123 (s, C-O), 745 (s, C-Harom.).
- (−)-18b [Rf = 0.35 (ethyl acetate/cyclohexane = 8:2)]: Yellow solid, mp 183 °C, yield of 37 mg (27%). C28H29N3O9 (551.6). Purity (HPLC): 93.2%, (tR = 16.0 min). Specific rotation: −115 (c = 1.3, THF). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.50 (dd, J = 9.5/7.8 Hz, 1H, 10a-H), 3.56 (s, 3H, 4-OCH3), 3.66 (s, 6H, 3-OCH3, 5-OCH3), 3.71 (s, 3H, CO2CH3), 3.82–3.89 (m, 1H, CHCH2OH), 3.94 (dt, J = 10.8/4.7 Hz, 1H, CHCH2OH), 4.03 (dd, J = 9.7/4.4 Hz, 1H, 3a-H), 4.62 (d, J = 4.6 Hz, 1H, 4-H), 4.62–4.65 (m, 1H, CHCH2OH), 4.75 (d, J = 7.8 Hz, 1H, 10-H), 5.83 (t, J = 5.3 Hz, 1H, CHCH2OH), 6.29 (s, 2H, C-2TMP, C-6TMP), 6.92 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 7.05 (ddd, J = 8.2/6.9/1.2 Hz, 1H, 7-H), 7.30 (dt, J = 8.1/0.9 Hz, 1H, 6-H), 7.36 (d, J = 7.9 Hz, 1H, 9-H), 9.15 (d, J = 8.1 Hz, 1H, NHamide), 10.58 (s, 1H, 5-H), 10.90 (s, 1H, 2-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 38.0 (1C, C-10), 38.2 (1C, C-4), 41.9 (1C, C-3a), 46.3 (1C, C-10a), 52.1 (1C, CO2CH3), 55.1 (1C, CHCH2OH), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 60.8 (1C, CHCH2OH), 105.8 (2C, C-2TMP, C-6TMP), 111.0 (1C, C-6), 111.7 (1C, C-9b), 118.0 (1C, C-9), 118.8 (1C, C-8), 121.4 (1C, C-7), 125.6 (1C, C-9a), 130.9 (1C, C-4a), 136.0 (1C, C-5a), 136.1 (1C, C-4TMP), 136.8 (1C, C-1TMP), 152.2 (2C, C-3TMP, C-5TMP), 170.5 (1C, C-4-C=O), 170.6 (CO2CH3), 178.1 (1C, C-1), 180.3 (1C, C-3). Exact mass (ESI): m/z = 552.1972 (calcd. 552.1977 for C28H30N3O9 [MH]+). IR (neat): ṽ [cm−1] = 3309 (m, OH), 2927, 2850 (w, C-Haliph.), 1708, 1674 (s, C=O), 1123 (s, C-O), 745 (s, C-Harom.).

- Methyl N-{[(3aR,4S,10R,10aR)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazol-4-yl]carbonyl}-(S)-methioninate ((-)-19b)
![Molecules 30 00063 i018]()
- Under N2, carboxylic acid 10 (136 mg, 0.30 mmol) and CDI (53.5 mg, 0.33 mmol) were dissolved in dry CH2Cl2 (10 mL) at room temperature. The mixture was stirred for 2 h. Then, (S)-methionine methyl ester HCl (120 mg, 0.60 mmol) was added. The mixture was stirred for 48 h at room temperature. After the completion of the transformation, the resulting suspension was filtered through Celite®, followed by rinsing with CH2Cl2 (40 mL). The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 6:4, Ø 2 cm, h = 20 cm, v = 10 mL). Yellow solid, mp 127 °C, yield of 39 mg (22%). C30H33N3O8S (595.7). TLC (ethyl acetate/cyclohexane = 6:4): Rf = 0.24. Purity (HPLC): 98.1%, (tR = 19.6 min). Specific rotation: −113 (c = 1.2, DMSO). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 2.02–2.07 (m, 1H, CH3SCH2CH2CH), 2.09 (s, 3H, SCH3), 2.11–2.17 (m, 1H, CH3SCH2CH2CH), 2.58–2.69 (m, 2H, CH3SCH2CH2CH), 3.55 (dd, J = 9.7/8.0 Hz, 1H, 10a-H), 3.57 (s, 3H, 4-OCH3), 3.67 (s, 6H, 3-OCH3, 5-OCH3), 3.76 (s, 3H, CO2CH3), 4.04 (dd, J = 9.6/5.3 Hz, 1H, 3a-H), 4.52 (d, J = 5.3 Hz, 1H, 4-H), 4.69 (ddd, J = 9.0/7.6/4.7 Hz, 1H, CH3SCH2CH2CH), 4.78 (d, J = 8.0 Hz, 1H, 10-H), 6.32 (s, 2H, 2-HTMP,6-HTMP), 6.95 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 7.07 (ddd, J = 8.2/7.0/1.2 Hz, 1H, 7-H), 7.36 (dt, J = 8.1/0.9 Hz, 1H, 6-H), 7.42 (d, J = 8.0 Hz, 1H, 9-H), 9.18 (d, J = 7.7 Hz, 1H, NHamide), 10.34 (s, 1H, 5-H), 10.97 (s, 1H, 2-H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 14.6 (1C, SCH3), 29.5 (1C, CH3SCH2CH2CH), 30.7 (1C, CH3SCH2CH2CH), 38.0 (1C, C-10), 38.4 (1C, C-4), 42.1 (1C, C-3a), 46.0 (1C, C-10a), 51.6 (1C, CH3SCH2CH2CH), 52.7 (1C, CO2CH3), 55.8 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.8 (2C, C-2TMP, C-6TMP), 111.1 (1C, C-6), 112.1 (1C, C-9b), 118.1 (1C, C-9), 119.0 (1C, C-8), 121.6 (1C, C-7), 125.6 (1C, C-9a), 130.8 (1C, C-4a), 135.9 (1C, C-5a), 136.2 (1C, C-4TMP), 137.0 (1C, C-1TMP), 152.3 (2C, C-3TMP, C-5TMP), 170.3 (1C, COamide), 173.5 (1C, CO2CH3), 178.1 (1C, C-1), 180.3 (1C, C-3). Exact mass (ESI): m/z = 596.2062 (calcd. 566.2061 for C30H34N3O8S [MH]+). IR (neat): ṽ [cm−1] = 2978, 2889 (w, C-Haliph.), 1774, (w, C=O), 1713 (s, C=O), 1643 (w, C=O), 1173, 1123 (s, C-O), 745 (s, C-Harom.).

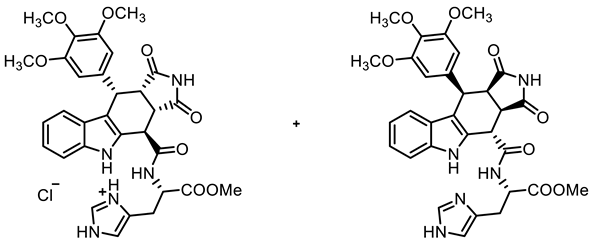
- Methyl N-{[(3aS,4R,10S,10aS)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazol-4-yl]carbonyl}-(S)-histidinate∙HCl ((+(-20a HCl) and methyl N-{[(3aR,4S,10R,10aR)-1,3-dioxo-10-(3,4,5-trimethoxyphenyl)-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazol-4-yl]carbonyl}-(S)-histidinate ((-)-20b)
![Molecules 30 00063 i019]()
- Carboxylic acid 10 (450 mg, 1.00 mmol), COMU® (471 mg, 1.10 mmol), and (S)-histidine methyl ester HCl (267 mg, 1.10 mmol) were dissolved in dry THF (20 mL) at room temperature. Then, DIPEA (426 mg, 3.30 mmol) was added dropwise. The mixture was stirred for 16 h at room temperature. After the completion of the transformation, ethyl acetate (100 mL) was added. The mixture was extracted with NaHCO3 solution (0.1 M in water, 4 × 50 mL) and saturated NaCl solution (1 × 50 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by automatic flash column chromatography (cartridge: SNAP 100 g, flow rate of 50 mL/min, MeOH/CH2Cl2 = 2:98 → 10:90) to obtain the diastereomers 20a and 20b.
- Then, 20a was dissolved in CH2Cl2 (10 mL) and HCl solution (2 M in Et2O, 5 mL) was added. The resulting suspension was filtered and the precipitate was washed with cold CH2Cl2 (2 × 10 mL) to give 20a∙HCl.
- (+)-20a∙HCl [Rf = 0.43 (MeOH/CH2Cl2 = 10:90, free base)]: yellow solid, mp 253 °C, yield of 125 mg (20%). C31H32ClN5O8 (638.1). Purity (HPLC): 95.0%, (tR = 15.3 min). Specific rotation: +121 (c = 1.7, DMSO). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.27 (dd, J = 15.2/6.4 Hz, 1H, NHCHCH2), 3.34 (dd, J = 15.1/8.3 Hz, 1H, NHCHCH2), 3.53 (t, J = 8.9 Hz, 1H, 10a-H), 3.57 (s, 3H, 4-OCH3), 3.63 (s, 6H, 3-OCH3, 5-OCH3), 3.65 (s, 3H, CO2CH3), 3.85 (dd, J = 9.0/3.7 Hz, 1H, 3a-H), 4.52 (d, J = 3.8 Hz, 1H, 4-H), 4.67–4.75 (m, 1H, NHCHCH2), 4.72 (d, J = 8.2 Hz, 1H, 10-H), 6.25 (s, 2H, 2-HTMP, 6-HTMP), 6.88 (ddd, J = 8.0/7.1/1.0 Hz, 1H, 8-H), 7.04 (ddd, J = 8.1/7.0/1.2 Hz, 1H, 7-H), 7.20 (d, J = 7.9 Hz, 1H, 9-H), 7.39 (dt, J = 8.1/0.9 Hz, 1H, 6-H), 7.51 (s, 1H, 5-Himidaz.), 8.99 (s, 1H, 2-Himidaz.), 9.22 (d, J = 7.4 Hz, 1H, NHamide), 10.52 (s, 1H, 5-H), 10.91 (s, 1H, 2-H). A signal for the NHimidaz. proton is not observed in the spectrum. 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 26.0 (1C, NHCHCH2), 37.7 (1C, C-4), 38.1 (1C, C-10), 42.8 (1C, C-3a), 45.9 (1C, C-10-a), 52.2 (1C, NHCHCH2), 52.3 (1C, CO2CH3), 55.7 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 106.1 (2C, C-2TMP, C-6TMP), 111.4 (1C, C-6), 111.4 (1C, C-9b), 117.2 (1C, C-5imidaz.) 118.2 (1C, C-9), 118.7 (1C, C-8), 121.5 (1C, C-7), 125.6 (1C, C-9a), 130.4 (1C, C-4a), 133.9 (1C, C-2imidaz.), 136.2 (1C, C-4TMP), 136.3 (1C, C-5a), 136.6 (1C, C-1TMP), 152.1 (2C, C-3TMP, C-5TMP), 170.8 (1C, COamide), 171.0 (1C, CO2CH3), 178.0 (1C, C-1), 179.8 (1C, C-3). A signal for the C-4imidaz. C-atom was not observed in the spectrum. Exact mass (ESI): m/z = 602.2251 (calcd. 602.2245 for C31H32N5O8 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2886 (s, C-Haliph.), 1713 (s, C=O), 1686 (m, C=O), 1154, 1119 (s, C-O), 745 (s, C-Harom.).
- (−)-20b [Rf = 0.32 (MeOH/CH2Cl2 = 10:90)]: Yellow solid, mp 233 °C, yield of 172 mg (29%). C31H31N5O8 (601.6). Purity (HPLC): 94.3%, (tR = 15.9 min). Specific rotation: −169 (c 1.5, DMSO). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.10 (dd, J = 14.8/9.3 Hz, 1H, NHCHCH2), 3.16 (dd, J = 14.9/4.2 Hz, 1H, NHCHCH2), 3.50 (dd, J = 9.5/7.8 Hz, 1H, 10a-H), 3.56 (s, 3H, 4-OCH3), 3.66 (s, 9H, 3-OCH3, 5-OCH3, CO2CH3), 4.03 (dd, J = 9.6/4.9 Hz, 1H, 3a-H), 4.29 (s broad, 1H, NHCHCH2), 4.53 (d, J = 5.0 Hz, 1H, 4-H), 4.75 (d, J = 7.8 Hz, 1H, 10-H), 6.31 (s, 2H, 2-HTMP, 6-HTMP), 6.93 (td, J = 7.5/7.1/0.9 Hz, 1H, 8-H), 7.03 (s, 1H, 5-Himidaz.), 7.06 (ddd, J = 8.2/7.0/1.1 Hz, 1H, 7-H), 7.37–7.43 (m, 2H, 6-H, 9-H), 7.80 (s, 1H, 2-Himidaz.), 9.36 (s, 1H, NHamide), 10.90 (s, 1H, 2-H), 11.95 (s, 1H, 5-H), 12.07 (s, 1H, τ-NHimidaz.). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 27.8 (1C, NHCHCH2), 38.1 (1C, C-10), 38.3 (1C, C-4), 42.0 (1C, C-3a), 46.2 (1C, C-10-a), 52.1 (1C, CO2CH3), 52.3 (1C, NHCHCH2), 55.8 (2C, 3-OCH3, 5-OCH3), 59.9 (1C, 4-OCH3), 105.8 (2C, C-2TMP, C-6TMP), 111.2 (1C, C-6), 111.8 (1C, C-9b), 113.7 (1C, C-5imidaz.), 117.9 (1C, C-9), 118.7 (1C, C-8), 121.5 (1C, C-7), 125.5 (1C, C-9a), 130.4 (1C, C-4a), 135.1 (1C, C-2imidaz.), 136.1 (1C, C-4TMP), 136.1 (1C, C-5a), 137.0 (1C, C-1TMP), 152.3 (2C, C-3TMP, C-5TMP), 170.1 (1C, COamide), 172.1 (1C, CO2CH3), 178.1 (1C, C-1), 180.2 (1C, C-3). A signal for the C-4imidaz. C-atom was not observed in the spectrum. Exact mass (ESI): m/z = 602.2254 (calcd. 602.2245 for C31H32N5O8 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2886 (s, C-Haliph.), 1705, 1674 (s, C=O), 1153, 1125 (s, C-O), 745 (s, C-Harom.).
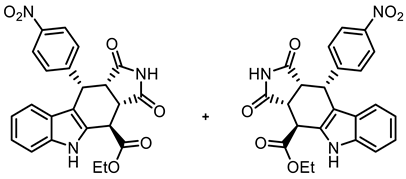
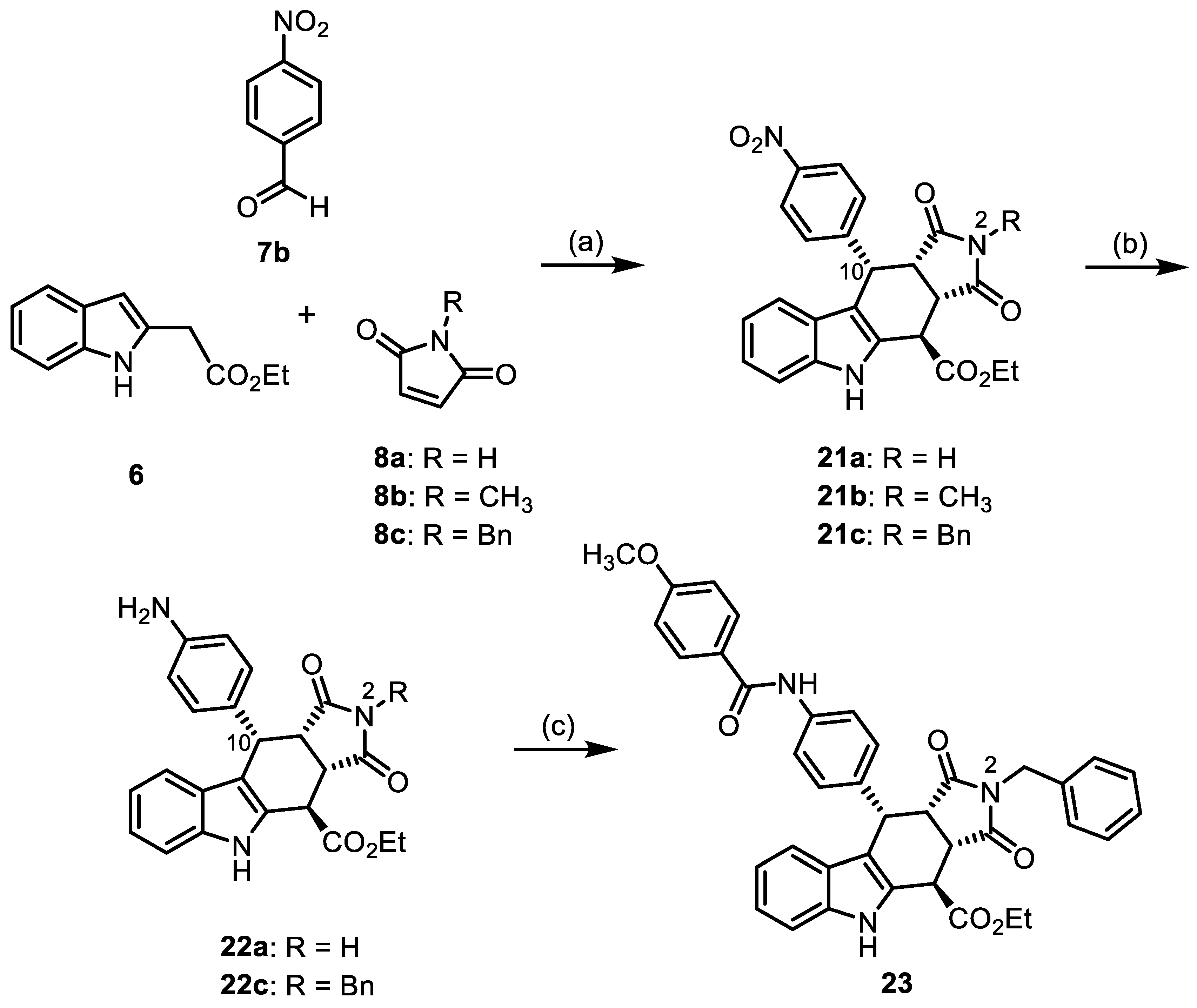
- Ethyl (3aRS,4SR,10RS,10aRS)-10-(4-nitrophenyl)-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate (21a)
![Molecules 30 00063 i020]()
- Under N2, indolylacetate 6 (205 mg, 1.01 mmol), maleimide (8a, 292 mg, 3.00 mmol), and 4-nitrobenzaldehyde (7b, 228 mg, 1.51 mmol) were dissolved in dry o-xylene (15 mL) in a pressure-resistant Schlenk tube. Crushed CuSO4 ∙ 5 H2O (26.4 mg, 0.11 mmol) was added to the solution and the mixture was heated to reflux for 40 h (oil bath temperature 180 °C). After cooling to room temperature, the mixture was diluted with CH2Cl2 until a clear solution formed. After filtration, the filter was washed with CH2Cl2 (3 × 10 mL). The filtrate was concentrated in vacuo. The residue was again dissolved in CH2Cl2 (2 mL) and adsorbed onto silica gel, followed by purification by automatic flash column chromatography (cartridge: SNAP 100g, flow rate of 50 mL/min, ethyl acetate/cyclohexane = 10:90 → 0:100). For further purification, the resulting solid was left stirring in Et2O (5 mL) for 30 min. The suspension was filtrated, washed with cold Et2O (3 × 10 mL), and dried under vacuum. Yellow solid, mp 262 °C, yield of 107 mg (25%). C23H19N3O6 (433.4). TLC (ethyl acetate/cyclohexane = 1:1): Rf = 0.26. Purity (HPLC): 94.9%, (tR = 20.1 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.31 (t, J = 7.1 Hz, 3H, COCH2CH3), 3.72 (t, J = 9.0 Hz, 1H, 10a-H), 4.09 (dd, J = 9.0/3.8 Hz, 1H, 3a-H), 4.22–4.33 (m, 2H, COCH2CH3), 4.56 (d, J = 3.7 Hz, 1H, 4-H), 4.99 (d, J = 8.9 Hz, 1H, 10-H), 6.83 (ddd, J = 7.9/6.8/1.0 Hz, 1H, 8-H), 6.98 (d, J = 7.8 Hz, 9H), 7.04 (ddd, J = 8.1/6.9/1.1 Hz, 1H, 7H), 7.25–7.30 (m, 2H, 2-HNO2Ph, 6-HNO2Ph), 7.35–7.39 (dt, J = 8.1/0.8 Hz, 1H, 6-H), 8.03–8.07 (m, 2H, 3-HNO2Ph, 5-HNO2Ph), 10.97 (s, 1H, 2-H), 11.10 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, COCH2CH3), 37.1 (1C, C-4), 37,4 (1C, C-10), 42.6 (1C, C-3a), 45.0 (1C, C-10a), 61.7 (1C, COCH2CH), 109.9 (1C, C-9b), 111.6 (1C, C-6), 118.1 (1C, C-9), 118.9 (1C, C-8), 121.7 (1C, C-7), 123.0 (2C, C-3NO2Ph, C-3NO2Ph), 125.2 (1C, C-9a), 129.2 (1C, C-4a), 130.2 (2C, C-2NO2Ph, C-6NO2Ph), 136.6 (1C, C-5a), 146.3 (1C, C-4NO2Ph), 149.1 (1C, C-1NO2Ph), 170.9 (1C, COCH2CH3), 177.5 (1C, C-1), 178.2 (1C, C-3). Exact mass (ESI): m/z = 434.1342 (calcd. 434.1347 for C23H20N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2893 (w, C-Haliph.), 1720 (s, C=O), 1508, 1342 (s, N-O) 1141 (s, C-O), 745 (m, C-Harom.).
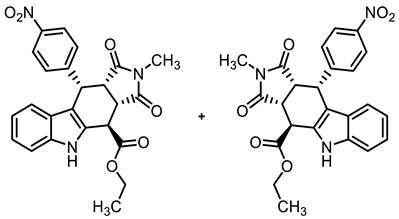
- Ethyl (3aRS,4SR,10RS,10aRS)-2-methyl-10-(4-nitrophenyl)-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate (21b)
![Molecules 30 00063 i021]()
- Under N2, indolylacetate 6 (101 mg, 0.50 mmol), N-methylmaleimide (8b, 281 mg, 2.53 mmol), and 4-nitrobenzaldehyde (7b, 132 mg, 0.87 mmol) were dissolved in dry o-xylene (15 mL) in a pressure-resistant Schlenk tube. Crushed CuSO4 ∙ 5 H2O (12.4 mg, 0.05 mmol) was added to the solution and the mixture was heated to reflux for 40 h (oil bath temperature of 180 °C). After cooling to room temperature, the mixture was filtered, and the filter was washed with CH2Cl2 (3 × 10 mL). The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 4:7, Ø 4 cm, h = 12 cm, v = 20 mL). Yellow solid, mp 237 °C, yield of 82 mg (40%). C24H21N3O6 (447.4). TLC (ethyl acetate/cyclohexane = 4:6): Rf = 0.21. Purity (HPLC): 95.3%, (tR = 21.3 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.36 (s, 3H, NHCH3), 3.80 (t, J = 8.8 Hz, 1H, 10a-H), 4.10 (dd, J = 8.6/3.4 Hz, 1H, 3a-H), 4.16–4.36 (m, 2H, OCH2CH3), 4.62 (d, J = 3.4 Hz, 1H, 4-H), 5.01 (d, J = 8.9 Hz, 1H, 10-H), 6.82 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 6.93 (d, J = 8.2 Hz, 1H, 9-H), 7.03 (ddd, J = 8.2/7.0/1.3 Hz, 1H, 7-H), 7.17–7.25 (m, 2H, 2-HNO2Ph 6-HNO2Ph), 7.37 (dt, J = 8.1/0.9 Hz, 1H, 6-H), 7.98–8.10 (m, 3-HNO2Ph, 5-HNO2Ph), 11.14 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 23.8 (1C, NHCH3), 37.0 (1C, C-4), 37.6 (1C, C-10), 41.6 (1C, C-3a), 44.0 (1C C-10a), 61.7 (1C, OCH2CH3), 109.6 (1C, C-9b), 111.5 (1C, C-6), 118.2 (1C, C-9), 118.9 (1C, C-8), 121.7 (1C, C-7), 122.9 (2C, C-3NO2Ph, C-5NO2Ph) 125.1 (1C, C-9a), 129.1 (1C, C-4a), 130.0 (2C, C-2NO2Ph, C-6NO2Ph), 136.6 (1C, C-5a), 146.3 (1C, C-4NO2Ph), 148.8 (1C, C-1NO2Ph), 170.8 (1C, COester) 176.2 (1C, C-1), 177.4 (1C, C-3). Exact mass (ESI): m/z = 446.1506 (calcd. 448.1503 for C24H22N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978 (w, C-Haliph.), 1782, (w, C=O), 1717 (s, C=O), 1196,1150 (s, C-O), 745 (m, C-Harom.).
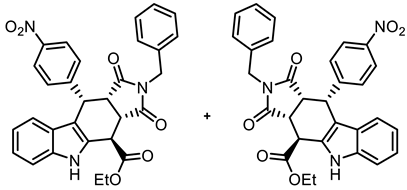
- Ethyl (3aRS,4SR,10RS,10aRS)-2-benzyl-10-(4-nitrophenyl)-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate (21c)
![Molecules 30 00063 i022]()
- Under N2, indolylacetate 6 (104 mg, 0.52 mmol), N-benzylmaleimide (8c, 282 mg, 1.50 mmol), and 4-nitrobenzaldehyde (7b, 113 mg, 0.75 mmol) were dissolved in dry o-xylene (15 mL) in a pressure-resistant Schlenk tube. Crushed CuSO4 ∙ 5 H2O (12.9 mg, 0.05 mmol) was added to the solution and the mixture was heated to reflux for 24 h (oil bath temperature of 180 °C). The mixture was allowed to cool down to ambient temperature and CH2Cl2 (20 mL) was added until a clear solution formed. After filtration, the filter was washed with CH2Cl2 (3 × 10 mL). The filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate/CH2Cl2 = 2:98, Ø 3 cm, h = 22 cm, v = 20 mL). Yellow solid, mp 199 °C, yield of 109 mg (40%). C30H25N3O6 (523.5). TLC (ethyl acetate/CH2Cl2 = 2:98): Rf = 0.33. Purity (HPLC): 98.7%, (tR = 23.2 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) 1.33 (t, J = 7.1 Hz, 3H, OCH2CH3), 3.86 (t, J = 9.0 Hz, 1H, 10a-H), 4.15 (d, J = 14.8 Hz, 1H, NCH2Ph), 4.20 (d, J = 14.8 Hz, 1H, NCH2Ph), 4.24 (dd, J = 9.3/4.6 Hz, 1H, 3a-H), 4.26–4.40 (m, 2H, OCH2CH3), 4.62 (d, J = 4.5 Hz, 1H, 4-H), 5.04 (d, J = 8.7 Hz, 1H, 10-H), 6.83 (ddd, J = 8.1/7.1/1.4 Hz, 1H, 8-H), 6.88–6.95 (m, 2H, C-2benzyl, C-6benzyl), 7.02–7.08 (m, 5H, 7-H, 9-H, 3-Hbenzyl, 4-Hbenzyl, 5-Hbenzyl), 7.10–7.13 (m, 2H, 2-HNO2Ph, 6-HNO2Ph), 7.35–7.40 (m, 1H, 6-H), 7.75–7.79 (m, 2H, 3-HNO2Ph, 5-HNO2Ph), 11.07 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 37.3 (1C, C-10), 37.6 (1C, C-4), 41.5 (1C, NCH2Ph), 41.6 (1C, C-3a), 43.8 (1C, C-10a), 61.9 (1C, OCH2CH3), 110.3 (1C, C-9b), 111.6 (1C, C-6), 118.0 (1C, C-9), 118.9 (1C, C-8), 121.7 (1C, C-7), 123.0 (2C, C-3NO2Ph, C-5NO2Ph), 125.1 (1C, C-9a), 127.1 (1C, C-4benzyl), 127.9 (2C, C-3benzyl, C-5benzyl), 128.0 (2C, C-2benzyl, C-6benzyl), 129.1 (1C, C-4a), 129.8 (2C, C-2NO2Ph, C-6NO2Ph), 135.3 (1C C-1benzyl), 136.5 (1C, C-5a), 146.1 (1C, C-4NO2Ph), 148.2 (1C, C-1NO2Ph), 170.9 (1C, C-4-C=O), 175.9 (1C, C-1), 177.7 (1C, C-3). Exact mass (ESI): m/z = 524.1794 (calcd. 524.1816 for C30H26N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2928 (w, C-Haliph.), 1732, 1701 (s, C=O), 1342 (s, N-O) 1141 (s, C-Oester), 745, 689 (m, C-Harom.).
- Ethyl (3aRS,4SR,10RS,10aRS)-10-(4-aminophenyl)-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate∙HCl (22a.HCl)
![Molecules 30 00063 i023]()
- Aromatic nitro compound 21a (119 mg, 0.28 mmol) was dissolved in THF (20 mL), and saturated NH4Cl solution (20 mL) was added. Under vigorous stirring, a large excess of Zn dust (1.01 g, 15.4 mmol) was poured into the mixture, which was then stirred for 2 h at room temperature. After the completion of the transformation, saturated NaHCO3 solution (30 mL) was added, and the suspension was stirred for an additional 30 min at room temperature. The reaction mixture was filtered through Celite®, followed by rinsing with ethyl acetate (150 mL). The filtrate was poured into a separating funnel and the layers were separated. The aqueous layer was then extracted with ethyl acetate (2 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate/cyclohexane = 8:2, Ø 2 cm, h = 12 cm, v = 10 mL). For conversion into the HCl salt, the resulting solid (74 mg) was dissolved in CH2Cl2 (5 mL). HCl solution (1 M in Et2O, 3 mL) was added, and the suspension was concentrated in vacuo. Orange solid, mp 244 °C, yield of 75 mg (62%). C23H22ClN3O4 (439.9). TLC (ethyl acetate/cyclohexane = 8:2): Rf = 0.21 (free base). Purity (HPLC): 95.9%, (tR = 15.1 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.31 (t, J = 7.1 Hz, 3H, COCH2CH3), 3.62 (t, J = 8.9 Hz, 1H, 10a-H), 4.06 (dd, J = 9.1/3.9 Hz, 1H, 3a-H), 4.21–4.35 (m, 2H, COCH2CH3), 4.51 (d, J = 3.9 Hz, 1H, 4-H), 4.82 (d, J = 8.7 Hz, 1H, 10-H), 6.82 (ddd, J = 7.9/6.9/1.0 Hz, 1H, 8-H), 6.99 (d, J = 7.9 Hz, 1H, 9-H), 7.01–7.07 (m, 3H, 7-H, 3-HNH2Ph, 5-HNH2Ph), 7.10 (d, J = 8.1 Hz, 2H, 2-HNH2Ph, 6-HNH2Ph), 7.32–7.40 (m, 1H, 6-H), 9.79 (s, 3H, NH3+), 10.95 (s, 1H, 2-H), 11.01 (s, 1H, 5-H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, COCH2CH3), 37.1 (1C, C-4), 37.2 (1C, C-10), 42.6 (1C, C-3a), 45.2 (1C, C-10a), 61.7 (1C, COCH2CH), 110.9 (1C, C-9b), 111.5 (1C, C-6), 118.1 (1C, C-9), 118.7 (1C, C-8), 121.6 (1C, C-7), 121.8 (2C, C-2NH2Ph, C-6NH2Ph), 125.2 (1C, C-9a), 129.0 (1C, C-4a), 130.0 (2C, C-3NH2Ph, C-5NH2Ph), 136.5 (1C, C-5a), 171.0 (1C, COCH2CH3), 177.6 (1C, C-1), 179.1 (1C, C-3). Signals for C-1NH2Ph and C-4NH2Ph C-atoms were not observed in the spectrum. Exact mass (ESI): m/z = 404.1606 (calcd. 404.1605 for C23H22N3O4 [MH]+). IR (neat): ṽ [cm−1] = 2978 (w, N-Hamine salt), 2983, 2862 (w, C-Haliph.), 1782 (w, C=O), 1701 (s, C=O), 1180 (s, C-O), 1153 (m, C-O), 741 (m, C-Harom.).
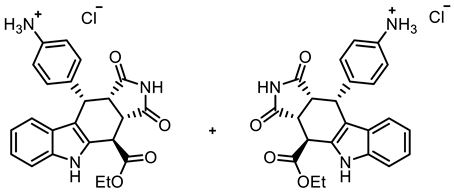
- Ethyl (3aRS,4SR,10RS,10aRS)-10-(4-aminophenyl)-2-benzyl-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate∙HCl (22c.HCl)
![Molecules 30 00063 i024]()
- Aromatic nitro compound 21c (263 mg, 0.50 mmol) was dissolved in THF (30 mL) and saturated NH4Cl solution (30 mL) was added. Under vigorous stirring, a large excess of Zn dust (5.00 g, 76.5 mmol) was poured into the mixture, which was then stirred for 2 h at room temperature. After the completion of the transformation, saturated NaHCO3 solution (30 mL) was added, and the suspension was stirred for an additional 30 min at room temperature. The reaction mixture was filtered through Celite®, followed by rinsing with ethyl acetate (80 mL). The filtrate was poured into a separating funnel and the layers were separated. The aqueous layer was then extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. For conversion into the HCl salt, the resulting solid was dissolved in CH2Cl2 (5 mL). HCl solution (1 M in Et2O, 3 mL) was added, and the suspension was concentrated in vacuo. Orange solid, mp 206 °C, yield of 126 mg (48%). C30H28ClN3O4 (530.0). TLC (ethyl acetate/cyclohexane = 7:3): Rf = 0.38 (free base). Purity (HPLC): 95.9%, (tR = 18.7 min). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 1.30 (t, J = 7.1 Hz, 3H, COCH2CH3), 3.81 (t, J = 8.8 Hz, 1H, 10a-H), 3.98 (d, J = 15.1 Hz, 1H, NCH2Ph), 4.12 (d, J = 15.1 Hz, 1H, NCH2Ph), 4.20 (dd, J = 8.9/4.0 Hz, 1H, 3a-H), 4.22–4.34 (m, 2H, COCH2CH3), 4.57 (d, J = 4.0 Hz, 1H, 4-H), 4.88 (d, J = 8.5 Hz, 1H, 10-H), 6.82 (ddd, J = 8.0/7.0/1.0 Hz, 1H, 8-H), 6.93–7.09 (m, 8H, 7-H, 9-H, 2-HNH2Ph, 3-HNH2Ph, 5-HNH2Ph, 6-HNH2Ph, 3-Hbenzyl, 5-Hbenzyl), 7.18–7.23 (m, 3H, 2-Hbenzyl, 4-Hbenzyl, 6-Hbenzyl), 7.38 (dt, J = 8.1/1.1/0.9 Hz, 1H, 6-H), 9.77 (s, 3H, NH3+), 11.08 (s, 1H, 5-H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 14.1 (1C, COCH2CH3), 37.51 (1C, C-4), 37.53 (1C, C-10), 41.1 (1C, NCH2Ph), 41.8 (1C, C-3a), 44.4 (1C, C-10a), 61.8 (1C, COCH2CH), 110.7 (1C, C-9b), 111.6 (1C, C-6, 118.2 (1C, C-9), 118.8 (1C, C-8), 121.6 (1C, C-7), 122.1 (2C, C-2NH2Ph, C-6NH2Ph), 125.2 (1C, C-9a), 127.2 (2C, C-3benzyl, C-5benzyl), 127.3 (1C, C-4benzyl), 128.3 (2C, C-2benzyl, C-6benzyl), 129.1 (1C, C-4a), 130.0 (2C, C-3NH2Ph, C-5NH2Ph), 135.5 (1C, C-1benzyl), 136.5 (1C, C-5a), 170.8 (1C, COCH2CH3), 176.2 (1C, C-1), 177.6 (1C, C-3). Signals for C-1NH2Ph and C-4NH2Ph C-atoms are not observed in the spectrum. Exact mass (APCI): m/z = 494.2064 (calcd. 494.2074 for C30H28N3O4 [MH]+). IR (neat): ṽ [cm−1] = 2978 (w, N-Hamine salt), 2978, 2808 (w, C-Haliph.), 1697 (s, C=O), 1184, 1146 (m, C-O), 745 (m, C-Harom.).

- Ethyl (3aRS,4SR,10RS,10aRS)-2-benzyl-10-[4-(4-methoxybenzamido)phenyl]-1,3-dioxo-1,2,3,3a,4,5,10,10a-octahydropyrrolo[3,4-b]carbazole-4-carboxylate (23)
![Molecules 30 00063 i025]()
- Ammonium chloride 22c∙HCl (77.5 mg, 0.14 mmol) was suspended in dry CH2Cl2 (20 mL) followed by the addition of DIPEA (80 µL, 0.47 mmol) to obtain the free base. 4-methoxybenzoyl chloride (43.1 mg, 0.28 mmol) was added and the mixture was stirred for 2 h at room temperature. After the completion of the transformation, the solution was washed with HCl solution (0.1 M, 2 × 10 mL) and saturated NaCl solution (1x 10 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (THF/CH2Cl2 = 3:97, Ø 3 cm, h = 14 cm, v = 10 mL). Yellow solid, mp 249 °C, yield of 76 mg (82%). C38H33N3O6 (627.7). TLC (THF/CH2Cl2 = 3:97): Rf = 0.19. Purity (HPLC): 94.5%, (tR = 22.8 min). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.32 (t, J = 7.1 Hz, 3H, COCH2CH3), 3.78 (t, J = 8.9 Hz, 1H, 10a-H), 3.83 (s, 3H, OCH3), 4.04 (d, J = 15.2 Hz, 1H, NCH2Ph), 4.19 (dd, J = 9.2/4.3 Hz, 1H, 3a-H), 4.20 (d, J = 15.1 Hz, 1H, NCH2Ph), 4.24–4.37 (m, 2H, COCH2CH3), 4.57 (d, J = 4.3 Hz, 1H, 4-H), 4.83 (d, J = 8.4 Hz, 1H, 10-H), 6.83–6.88 (m, 3H, 8-H, 2-HNH2Ph, 6-HNH2Ph), 6.89–6.93 (m, 2H, 2-Hbenzyl, 6-Hbenzyl), 7.01–7.09 (m, 4H, 7-H, 9-H, 3-HMeObz, 5-HMeObz), 7.09–7.18 (m, 3H, 3-Hbenzyl, 4-Hbenzyl, 5-Hbenzyl), 7.38 (dt, J = 8.3/0.9 Hz, 1H, 6-H), 7.48–7.54 (m, 2H, 3-HNH2Ph, 5-HNH2Ph), 7.90–7.96 (m, 2H, 2-HMeObz, 6-HMeObz), 9.98 (s, 1H, NHamide), 11.00 (s, 1H, 5-H). 13C NMR: (151 MHz, DMSO-d6) δ (ppm) = 14.0 (1C, COCH2CH3), 37.5 (2 × 1C, C-4, C-10), 41.2 (1C, NCH2Ph), 41.8 (1C, C-3a), 44.7 (1C, C-10a), 55.4 (1C, OCH3), 61.7 (1C, COCH2CH), 111.3 (1C, C-9b), 111.5 (1C, C-6), 113.6 (2C, C-3MeObz, C-5MeObz), 118.2 (1C, C-9), 118.8 (1C, C-8), 119.5 (2C, C-3NH2Ph, C-5NH2Ph), 121.5 (1C, C-7), 125.3 (1C, C-9a), 127.0 (1C, C-4benzyl), 127.0 (1C, C-1MeObz), 127.2 (2C, C-2benzyl, C-6benzyl) 128.1 (2C, C-3benzyl, C-5benzyl), 128.8 (2C, C-2NH2Ph, C-6NH2Ph), 128.9 (1C, C-4a), 129.5 (2C, C-2 MeObz, C-6 MeObz), 134.9 (1C, C-1NH2Ph), 135.5 (1C, C-1benzyl), 136.5 (1C, C-5a), 138.1 (1C, C-4NH2Ph), 161.8 (1C, C-4 MeObz), 164.7 (1C, COamide), 170.9 (1C, COCH2CH3), 176.2 (1C, C-1), 177.8 (1C, C-3). Exact mass (APCI): m/z = 628.2400 (calcd. 628.2442 for C38H34N3O6 [MH]+). IR (neat): ṽ [cm−1] = 2978, 2889 (m, C-Haliph.), 1775 (w, C=O), 1694 (s, C=O), 1643, 1605 (m, C=Caryl) 1184 (s, C-O), 1153 (m, C-O).
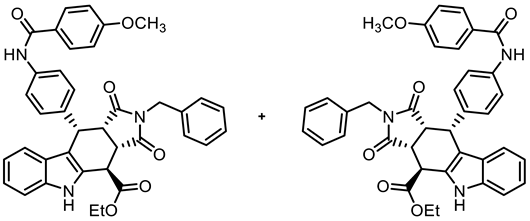
4.4. Microscale Thermophoresis Assay
4.5. Capillary Electrophoresis Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Garand, C.; Belda, H.; Gagnon-Arsenault, I.; Treeck, M.; Elowe, S.; Landry, C.R. The substrate quality of CK2 target sites has a determinant role on their function and evolution. Cell Syst. 2024, 15, 544–562. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Montenarh, M.; Götz, C. Protein kinase CK2 and ion channels (Review). Biomed. Rep. 2020, 13, 55. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzyme Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 1999, 20, 391–408. [Google Scholar] [CrossRef]
- Montenarh, M. Protein kinase CK2 in DNA damage and repair. Transl. Cancer Res. 2016, 5, 49–63. [Google Scholar]
- Götz, C.; Montenarh, M. Protein kinase CK2 in development and differentiation. Biomed. Rep. 2017, 6, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.G. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [PubMed]
- Martel, V.; Filhol, O.; Nueda, A.; Cochet, C. Dynamic Localization/Association of Protein Kinase CK2 Subunits in Living Cells. Ann. N. Y. Acad. Sci. 2002, 973, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Bidwai, A.P.; Reed, J.C.; Glover, C.V. Phosphorylation of calmodulin by the catalytic subunit of casein kinase II is inhibited by the regulatory subunit. Arch. Biochem. Biophys. 1993, 300, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Martiel, J.-L.; Cochet, C. Protein kinase CK2: A new view of an old molecular complex. EMBO Rep. 2004, 5, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Okur, V.; Cho, M.T.; Henderson, L.; Retterer, K.; Schneider, M.; Sattler, S.; Niyazov, D.; Azage, M.; Smith, S.; Picker, J.; et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum. Genet. 2016, 135, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Hubert, L.; Viot, G.; Rio, M.; Billuart, P.; Besmond, C.; Bienvenu, T. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum. Mutat. 2017, 38, 932–941. [Google Scholar] [CrossRef]
- Iimoto, D.S.; Masliah, E.; DeTeresa, R.; Terry, R.D.; Saitoh, T. Aberrant casein kinase II in Alzheimer’s disease. Brain Res. 1990, 507, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.Y.; Kim, D.W.; Arima, K.; Mouradian, M.M.; Kim, S.U.; Lee, G. Localization of CKII beta subunits in Lewy bodies of Parkinson’s disease. J. Neurol. Sci. 2008, 266, 9–12. [Google Scholar] [CrossRef]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—Potential clinical relevance. Cell Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Kren, B.T.; Afzal, M.; Scaria, G.A.; Klein, M.A.; Ahmed, K. Protein kinase CK2—Diverse roles in cancer cell biology and therapeutic promise. Mol. Cell. Biochem. 2023, 478, 899–926. [Google Scholar] [CrossRef]
- Wang, G.; Unger, G.; Ahmad, K.A.; Slaton, J.W.; Ahmed, K. Downregulation of CK2 induces apoptosis in cancer cells--a potential approach to cancer therapy. Mol. Cell. Biochem. 2005, 274, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: A patent review. Expert Opin. Ther. Pat. 2012, 22, 1081–1097. [Google Scholar] [CrossRef]
- Cozza, G.; Bortolato, A.; Moro, S. How druggable is protein kinase CK2? Med. Res. Rev. 2010, 30, 419–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. [Google Scholar] [CrossRef] [PubMed]
- Grygier, P.; Pustelny, K.; Nowak, J.; Golik, P.; Popowicz, G.M.; Plettenburg, O.; Dubin, G.; Menezes, F.; Czarna, A. Silmitasertib (CX-4945), a Clinically Used CK2-Kinase Inhibitor with Additional Effects on GSK3beta and DYRK1A Kinases: A Structural Perspective. J. Med. Chem. 2023, 66, 4009–4024. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Farina, H.G.; Gil, J.; Rodriguez, A.; Benavent, F.; Castellanos, L.; Gomez, R.E.; Acevedo, B.E.; Alonso, D.F.; Perea, S.E. Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol. Cancer Ther. 2009, 8, 1189–1196. [Google Scholar] [CrossRef]
- Borad, M.J.; Bai, L.-Y.; Richards, D.; Mody, K.; Hubbard, J.; Rha, S.Y.; Soong, J.; McCormick, D.; Tse, E.; O’Brien, D.; et al. Silmitasertib plus gemcitabine and cisplatin first-line therapy in locally advanced/metastatic cholangiocarcinoma: A Phase 1b/2 study. Hepatology 2023, 77, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, A.; Gratz, A.; Bollacke, A.; Weyrich, M.; Kuckländer, U.; Wünsch, B.; Götz, C.; Niefind, K.; Jose, J. A pi-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors. Pharmaceuticals 2018, 11, 23. [Google Scholar] [CrossRef]
- Rumler, H.; Schmithals, C.; Werner, C.; Bollacke, A.; Aichele, D.; Götz, C.; Niefind, K.; Wünsch, B.; Jose, J. Discovery of 7,9-Dibromodihydrodibenzofuran as Potent CK2 inhibitor: Synthesis, Biological Evaluation and Structural Studies on E-/Z isomers. ACS Pharmacol. Translat. Sci. 2024, 7, 3846–3866. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Non-ATP Competitive Protein Kinase Inhibitors. Curr. Med. Chem. 2010, 17, 2804–2821. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Bestgen, B.; Brear, P.; Prudent, R.; Laudet, B.; Moucadel, V.; Ettaoussi, M.; Sautel, C.F.; Krimm, I.; Engel, M.; et al. Discovery of holoenzyme-disrupting chemicals as substrate-selective CK2 inhibitors. Sci. Rep. 2019, 9, 15893. [Google Scholar] [CrossRef]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 2007, 408, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Iegre, J.; Brear, P.; Baker, D.J.; Tan, Y.S.; Atkinson, E.L.; Sore, H.F.; O’ Donovan, D.H.; Verma, C.S.; Hyvönen, M.; Spring, D.R. Efficient development of stable and highly functionalised peptides targeting the CK2α/CK2β protein-protein interaction. Chem. Sci. 2019, 10, 5056–5063. [Google Scholar] [CrossRef] [PubMed]
- Lindenblatt, D.; Horn, M.; Götz, C.; Niefind, K.; Neundorf, I.; Pietsch, M. Design of CK2β-Mimicking Peptides as Tools to Study the CK2α/CK2β Interaction in Cancer Cells. ChemMedChem 2019, 14, 833–841. [Google Scholar] [CrossRef]
- Brear, P.; North, A.; Iegre, J.; Hadje Georgiou, K.; Lubin, A.; Carro, L.; Green, W.; Sore, H.F.; Hyvönen, M.; Spring, D.R. Novel non-ATP competitive small molecules targeting the CK2 α/β interface. Bioorg. Med. Chem. 2018, 26, 3016–3020. [Google Scholar] [CrossRef] [PubMed]
- Laudet, B.; Moucadel, V.; Prudent, R.; Filhol, O.; Wong, Y.-S.; Royer, D.; Cochet, C. Identification of chemical inhibitors of protein-kinase CK2 subunit interaction. Mol. Cell. Biochem. 2008, 316, 63–69. [Google Scholar] [CrossRef]
- Kröger, L.; Daniliuc, C.G.; Ensan, D.; Borgert, S.; Nienberg, C.; Lauwers, M.; Steinkrüger, M.; Jose, J.; Pietsch, M.; Wünsch, B. Synthesis and SAR of tetracyclic inhibitors of protein kinase CK2 derived from furocarbazole W16. ChemMedChem 2020, 15, 871–881. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Royer, D.; Wong, Y.-S.; Plé, S.; Chiaroni, A.; Diker, K.; Lévy, J. Diastereodivergence and appendage diversity in the multicomponent synthesis of aryl-pyrrolo-tetrahydrocarbazoles. Tetrahedron 2008, 64, 9607–9618. [Google Scholar] [CrossRef]
- Kröger, L.; Wünsch, B. Stereochemistry of the Levy reaction. Eur. J. Org. Chem. 2018, 45, 6297–6303. [Google Scholar] [CrossRef]
- Sperry, J.B.; Minteer, C.J.; Tao, J.Y.; Johnson, R.; Duzguner, R.; Hawksworth, M.; Oke, S.; Richardson, P.F.; Barnhart, R.; Bill, D.R.; et al. Thermal Stability Assessment of Peptide Coupling Reagents Commonly Used in Pharmaceutical Manufacturing. Org. Process Res. Dev. 2018, 22, 1262–1275. [Google Scholar] [CrossRef]
- Pietsch, M.; Viht, K.; Schnitzler, A.; Ekambaram, R.; Steinkrüger, M.; Enkvist, E.; Nienberg, C.; Nickelsen, A.; Lauwers, M.; Jose, J.; et al. Unexpected CK2β-antagonistic functionality of bisubstrate inhibitors targeting protein kinase CK2. Bioorg. Chem. 2020, 96, 103608. [Google Scholar] [CrossRef]
- Gratz, A.; Götz, C.; Jose, J. A CE-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis 2010, 31, 634–640. [Google Scholar] [CrossRef]
- Raaf, J.; Brunstein, E.; Issinger, O.G.; Niefind, K. The interaction of CK2α and CK2β, the subunits of protein kinase CK2, requires CK2β in a preformed conformation and is enthalpically driven. Protein Sci. 2008, 17, 2180–2186. [Google Scholar] [CrossRef]
- Grankowski, N.; Boldyreff, B.; Issinger, D.G. Isolation and characterization of recombinant human casein kinase II subunits α and β from bacteria. Eur. J. Biochem. 1991, 198, 25–30. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compd. | R | R′ | Conc. Test Compd. (µM) | CK2α/CK2β Interaction KD′ [nM] a | Inhibition of CK2α/CK2β Interaction Ki [µM] |
| (±)-9a [40] | OEt | H | 100 | 59 ± 25 | 32 d |
| (±)-9b | OEt | CH3 | 20 | 14 ± 6 b | n.s. |
| (±)-10 [40] | OH | H | 100 | 758 ± 338 | 1.9 d |
| (±)-11 | NHCH3 | H | 50 | 132 ± 43 | 6.0 |
| (±)-12 | NHCH2C6H5 | H | 50 | 62 ± 19 | 14 |
| (±)-13 | NHCH2-4-ClC6H4 | H | 50 | 17 ± 3 c | n.s. |
| (±)-14 | NH(CH2)2-indol-3-yl | H | 50 | 158 ± 17 | 3.8 |
| (±)-15 | 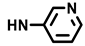 | H | 50 | 89 ± 13 | 7.4 |
| (±)-16 | 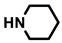 | H | 50 | 72 ± 12 | 9.7 |
| (±)-17 | 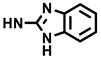 | H | 50 | 93 ± 13 | 7.1 |
| (+)-18a | CH2OH | H | 50 | 72 ± 2 | 9.1 |
| (–)-19b | CH2CH2SCH3 (S-RSRR-config.) | H | 50 | 94 ± 24 | 7.7 |
| (+)-20a | 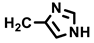 | H | 50 | 101 ± 4 | 6.1 |
| (–)-20b | 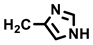 (S-RSRR-config.) | H | 20 | 29 ± 7 b | n.s. |
| (±)-21a | NO2 | H | 100 | 20 ± 10 c | n.s. |
| (±)-22a | NH2 | H | 100 | 31 ± 11 c | n.s. |
| (±)-22c | NH2 | Bn | 100 | 25 ± 15 c | n.s. |
| (±)-23 | NHC(=O)aryl | Bn | 100 | 24 ± 9 c | n.s. |
| (+)-4 (W16) [40] | 100 | 61 ± 17 | 31 d | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kröger, L.; Borgert, S.; Lauwers, M.; Steinkrüger, M.; Jose, J.; Pietsch, M.; Wünsch, B. Structure–Activity Relationship Studies of Tetracyclic Pyrrolocarbazoles Inhibiting Heterotetrameric Protein Kinase CK2. Molecules 2025, 30, 63. https://doi.org/10.3390/molecules30010063
Kröger L, Borgert S, Lauwers M, Steinkrüger M, Jose J, Pietsch M, Wünsch B. Structure–Activity Relationship Studies of Tetracyclic Pyrrolocarbazoles Inhibiting Heterotetrameric Protein Kinase CK2. Molecules. 2025; 30(1):63. https://doi.org/10.3390/molecules30010063
Chicago/Turabian StyleKröger, Lukas, Sebastian Borgert, Miriam Lauwers, Michaela Steinkrüger, Joachim Jose, Markus Pietsch, and Bernhard Wünsch. 2025. "Structure–Activity Relationship Studies of Tetracyclic Pyrrolocarbazoles Inhibiting Heterotetrameric Protein Kinase CK2" Molecules 30, no. 1: 63. https://doi.org/10.3390/molecules30010063
APA StyleKröger, L., Borgert, S., Lauwers, M., Steinkrüger, M., Jose, J., Pietsch, M., & Wünsch, B. (2025). Structure–Activity Relationship Studies of Tetracyclic Pyrrolocarbazoles Inhibiting Heterotetrameric Protein Kinase CK2. Molecules, 30(1), 63. https://doi.org/10.3390/molecules30010063