

Thorough Validation of Optimized Size Exclusion Chromatography-Total Organic Carbon Analysis for Natural Organic Matter in Fresh Waters
 , , , ,
, , , ,
Abstract

1. Introduction
2. Results and Discussion
2.1. Method Development
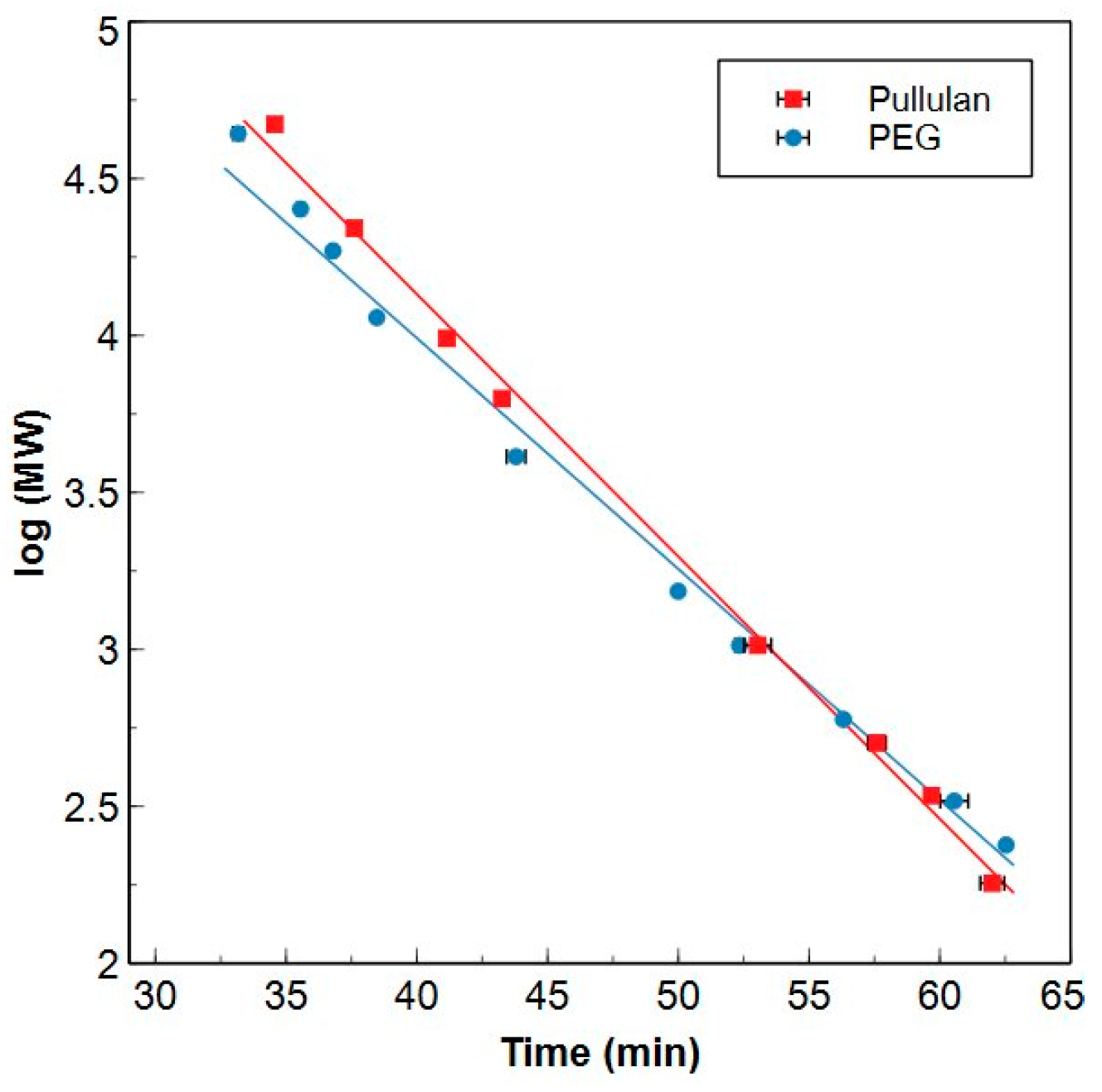
2.1.1. HPSEC-TOC Calibration
2.1.2. Definition of the Integration Ranges
- High MW fraction
- b.
- Medium MW fraction
- c.
- Low MW fraction
2.1.3. HPSEC-TOC Sample Pretreatment
2.2. Method Validation
2.2.1. LOD and LOQ Determination
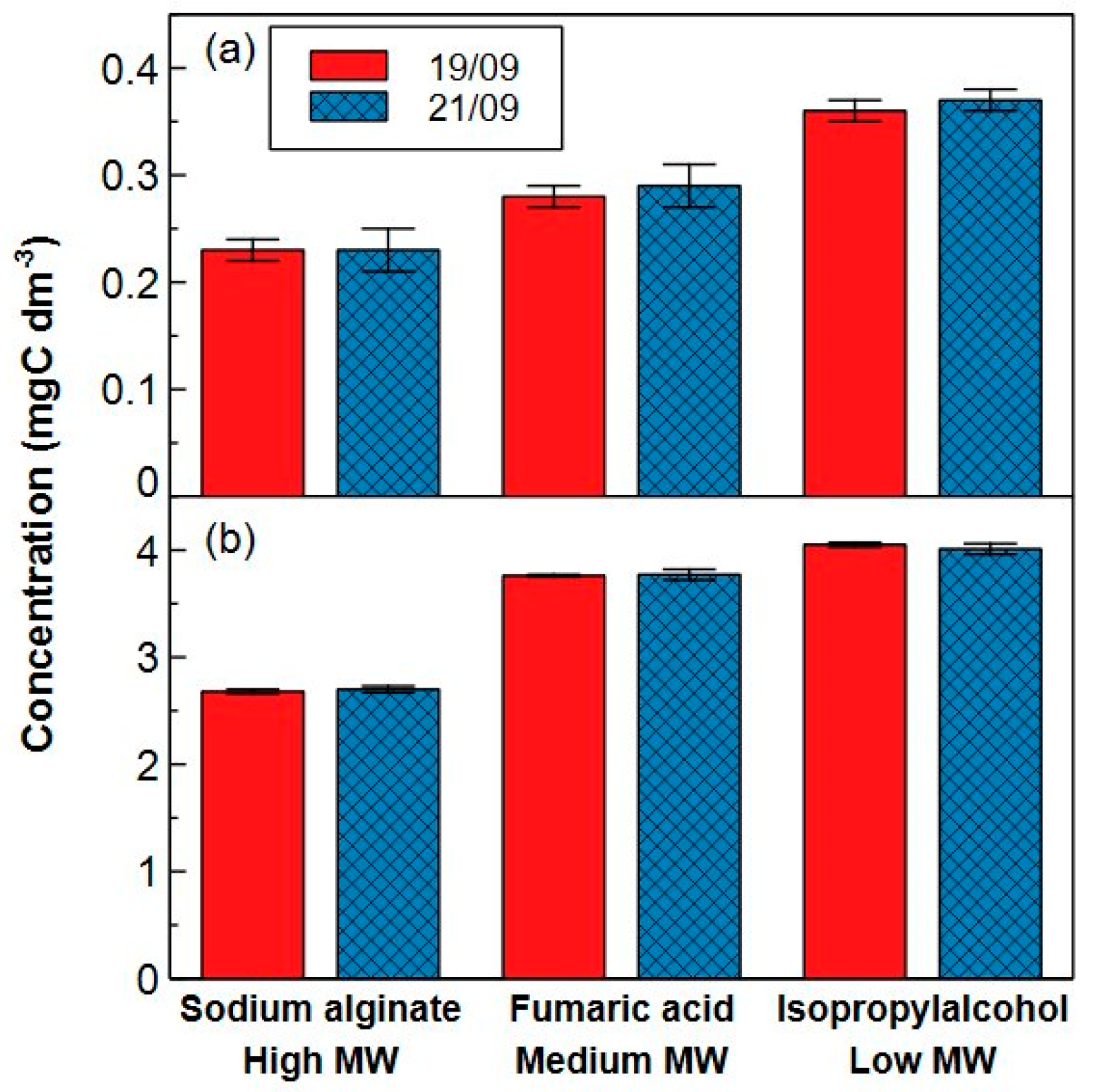
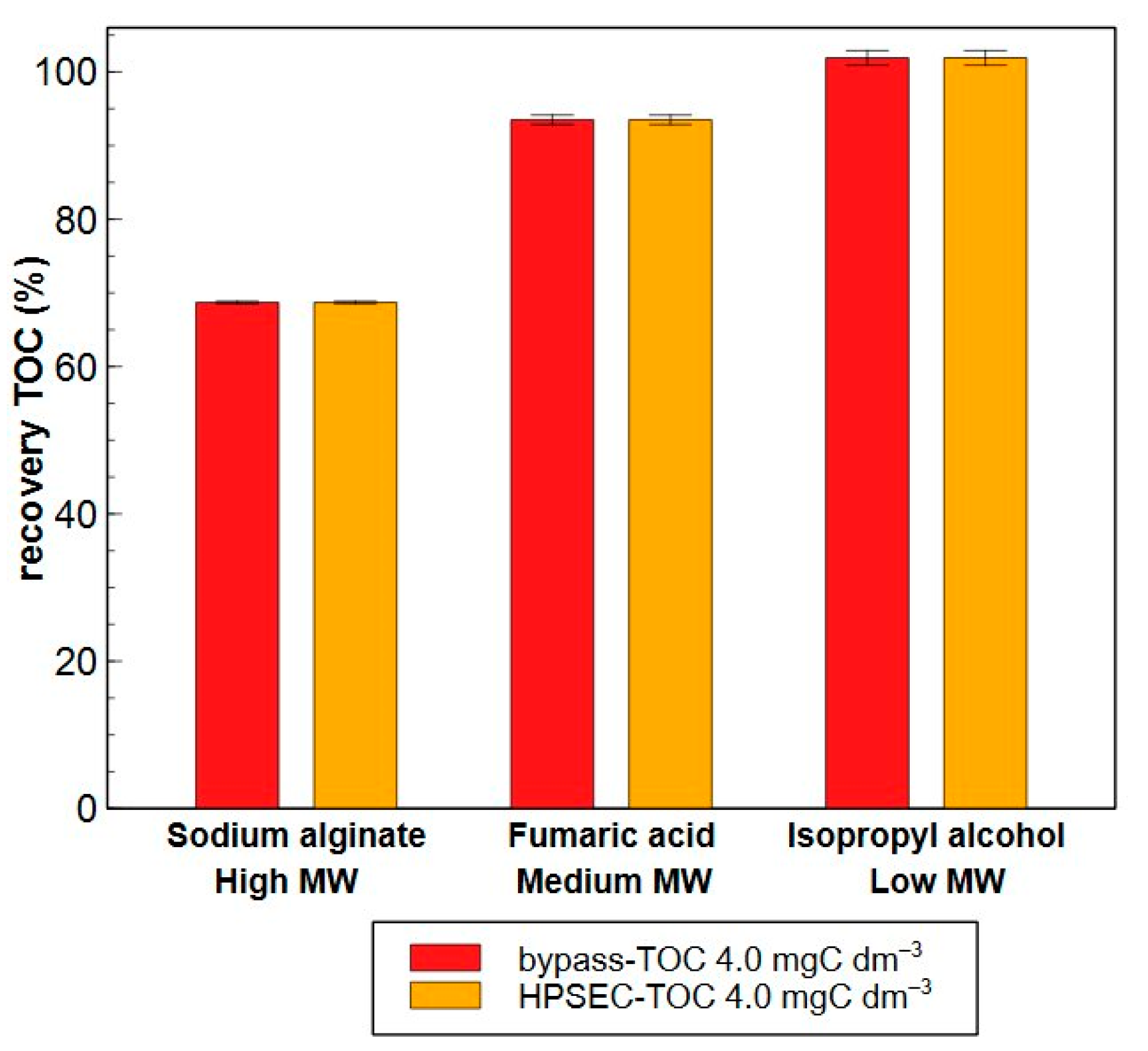
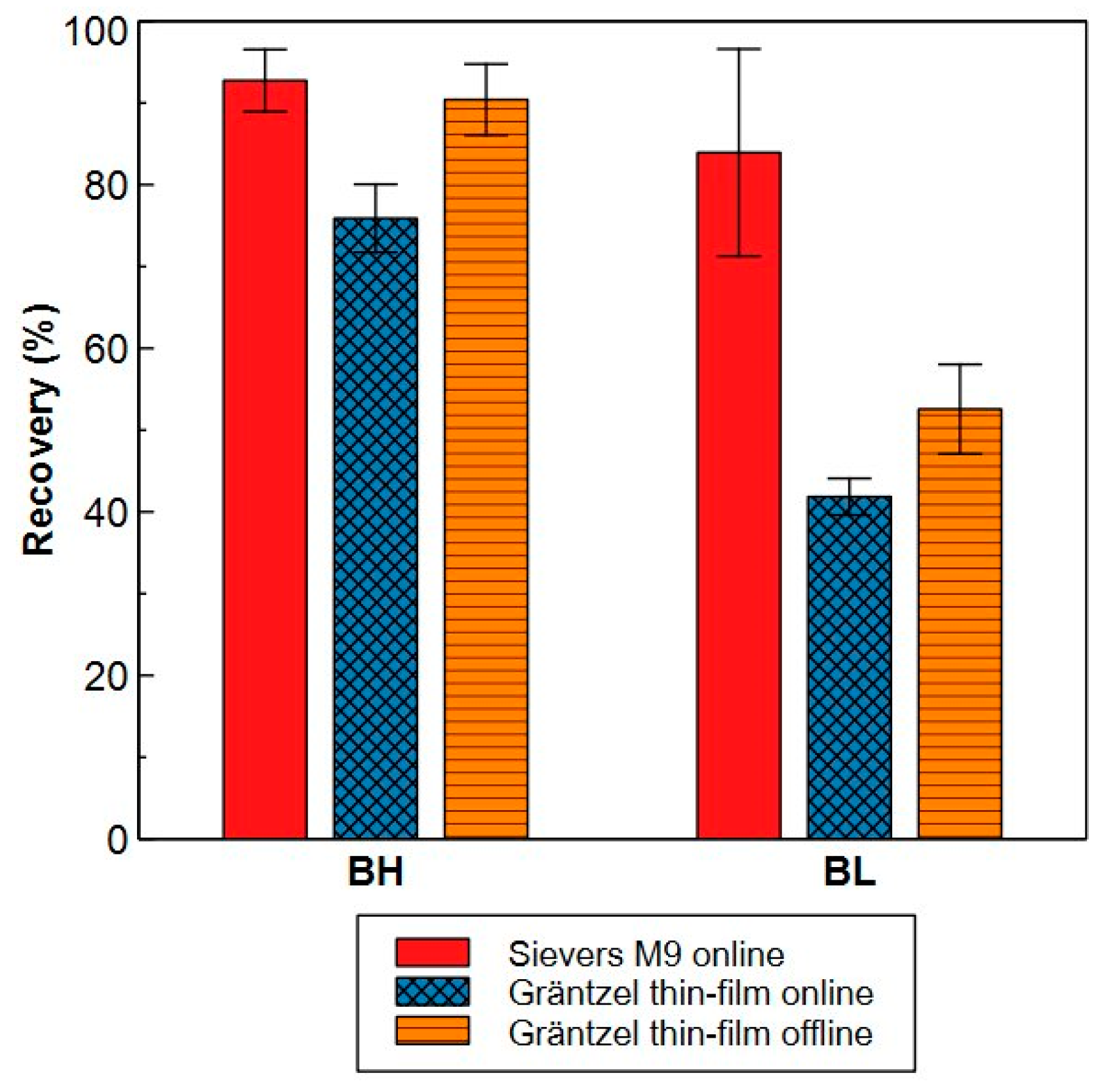
2.2.2. Precision and Trueness
- Certified reference material
- b.
- Model compounds
- c.
- Real water samples
2.2.3. Measurement Uncertainty
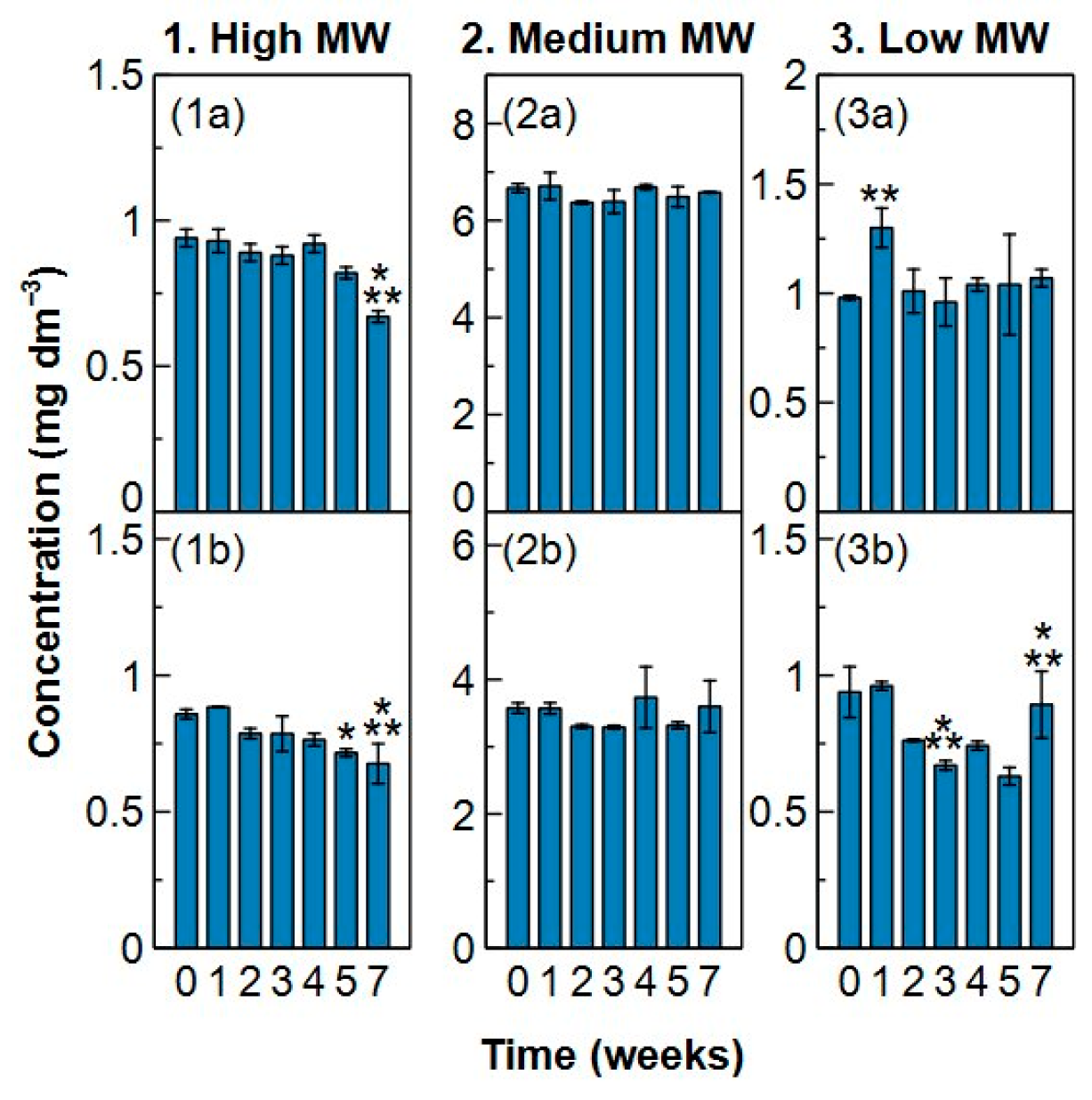
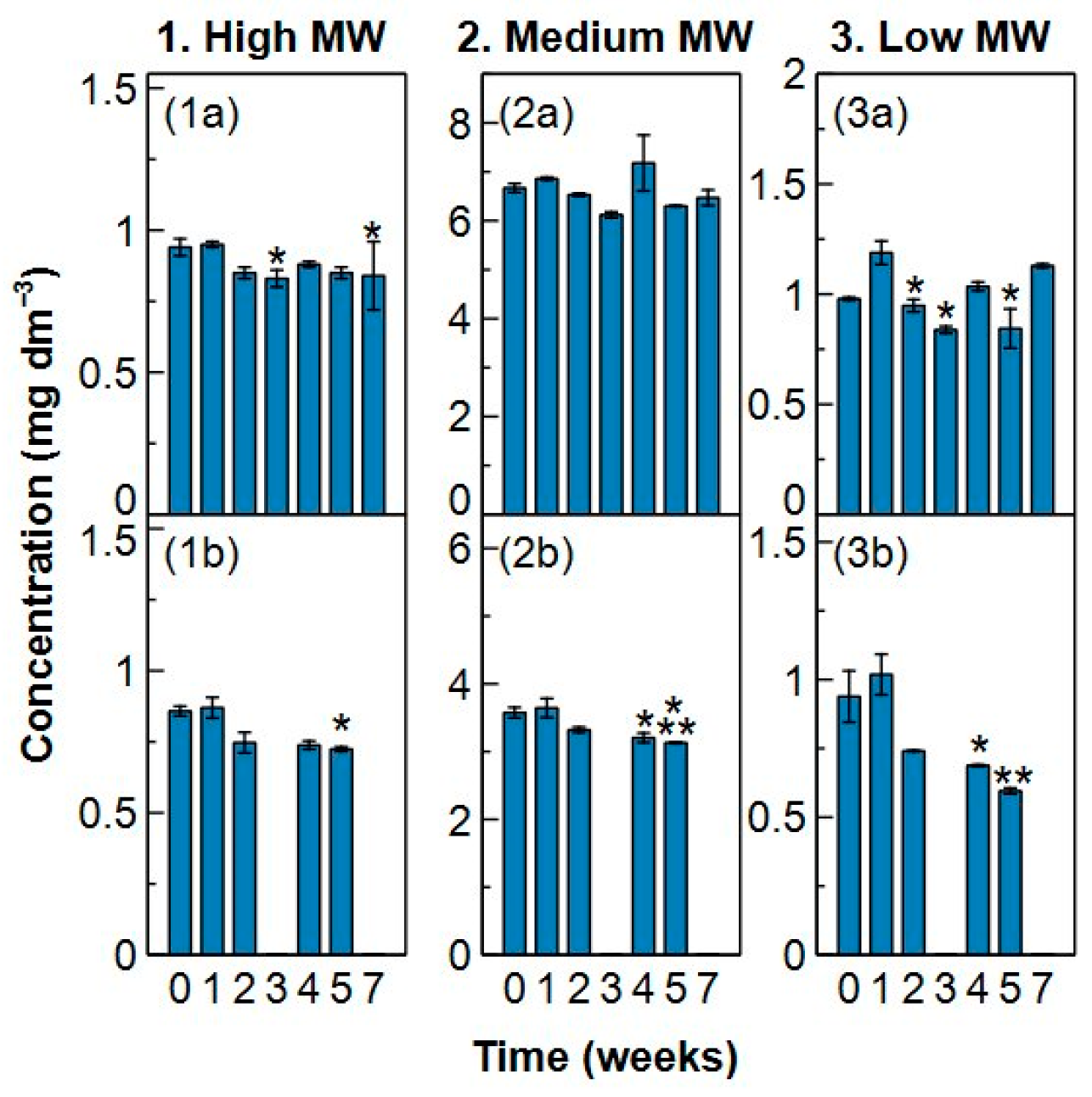
2.3. Sample Stability
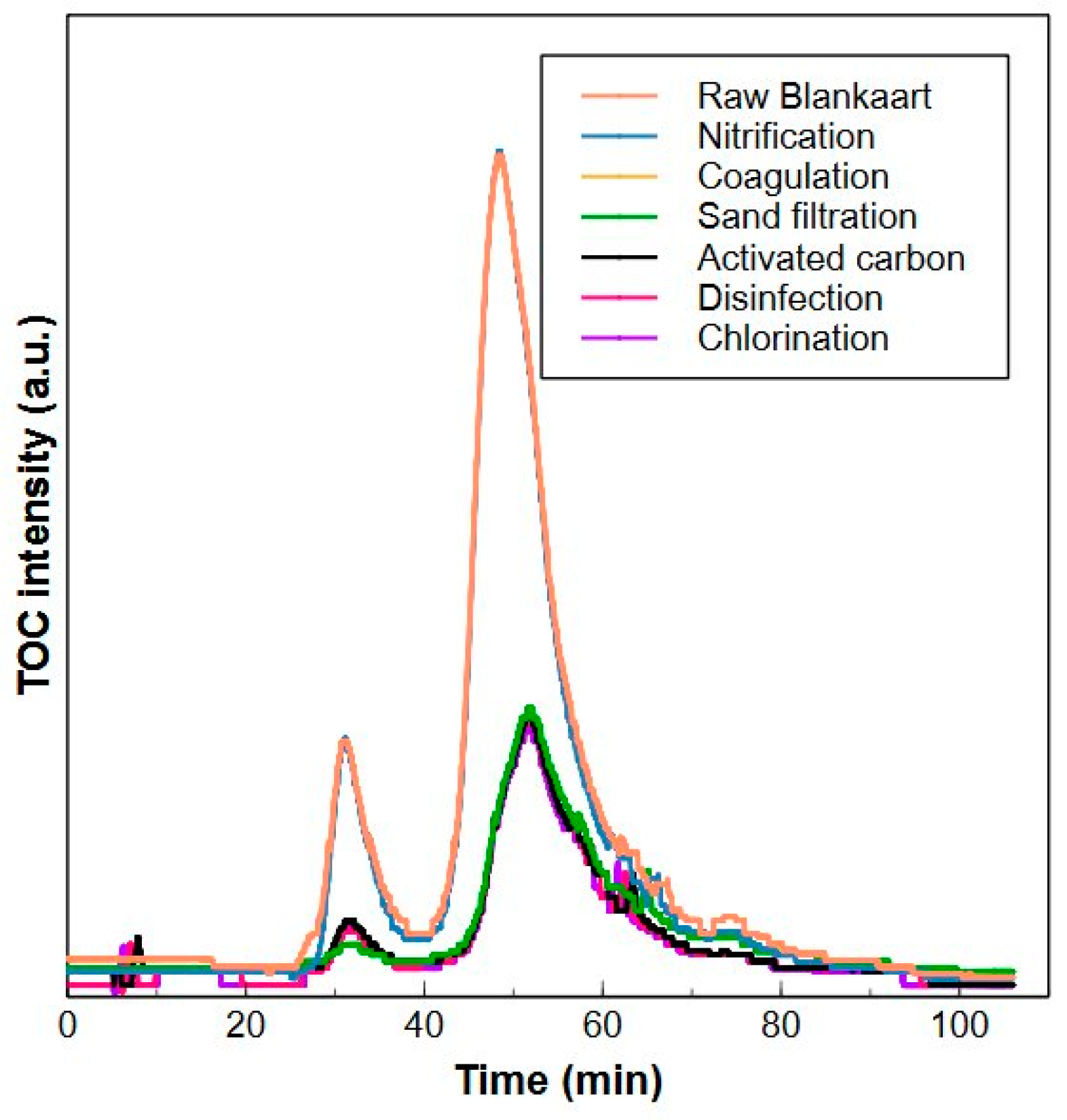
2.4. Application of the HPSEC-TOC Method in a Drinking Water Treatment Plant
3. Materials and Methods
3.1. Chemicals
3.2. Water Sources
3.3. Instruments
3.3.1. TOC Detectors
3.3.2. HPSEC-TOC System
3.4. Sample Preparation
- The sample (aliquot of minimum 8 cm3 for practical reasons) was transferred into a TOC vial.
- The pH (InoLab pH Level 1) of the sample was adjusted to pH 6 using 1 M H3PO4.
- The sample was purged with N2-gas at 70 cm3 min−1 for at least 30 min.
- A 20-times concentrated mobile phase solution was gradually added with a micropipette to the sample to assimilate the conductivity of the sample to the one of the mobile phase (5.3 mS).
- A 0.45 µm filter (Chromafil PET-45/15, Macherey-Nagel) was pre-filtered with 2 mL sample to remove possible impurities of the filter whereafter another 2 mL was filtered and transferred into an HPLC vial.
3.5. Method Development
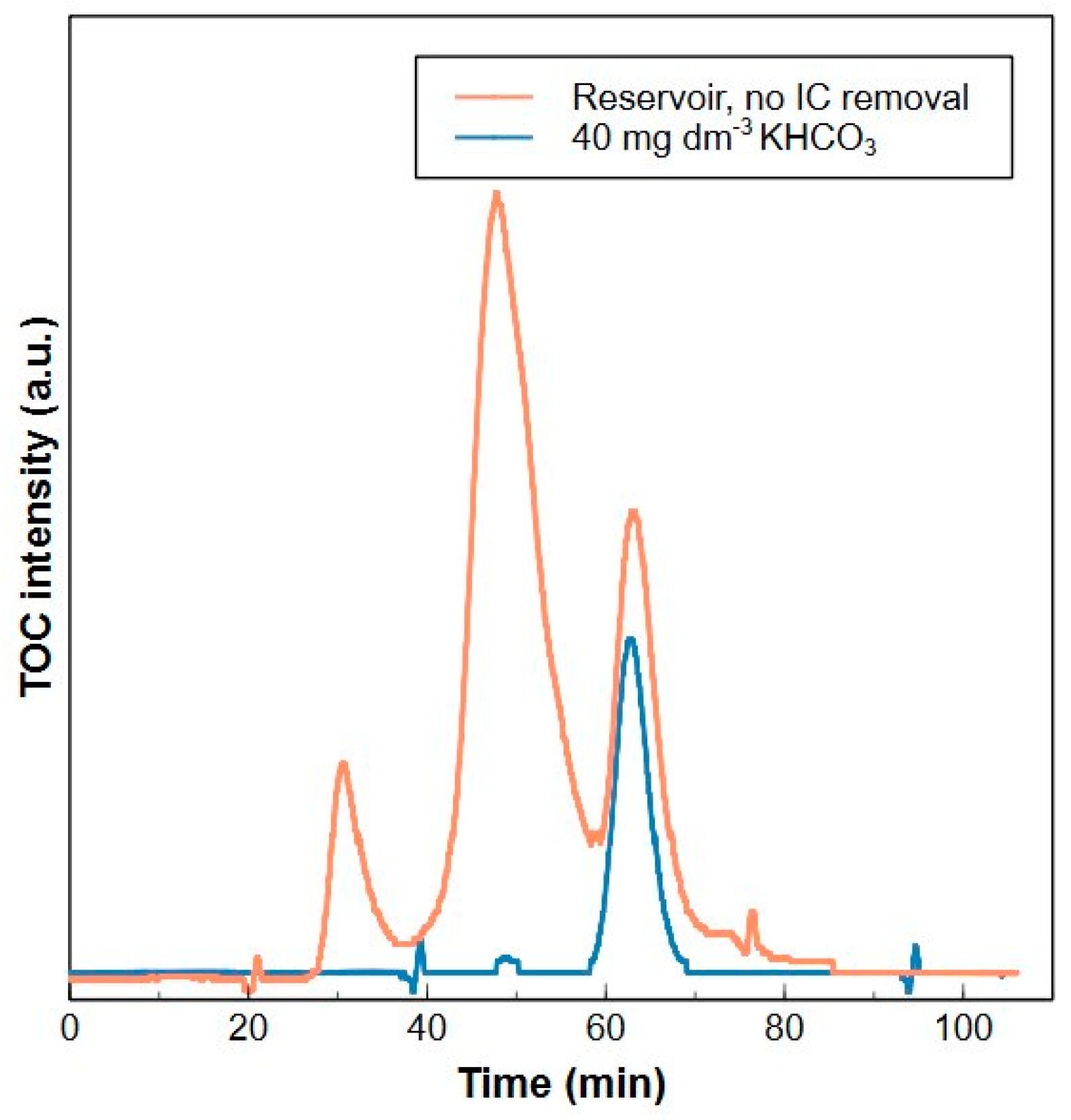
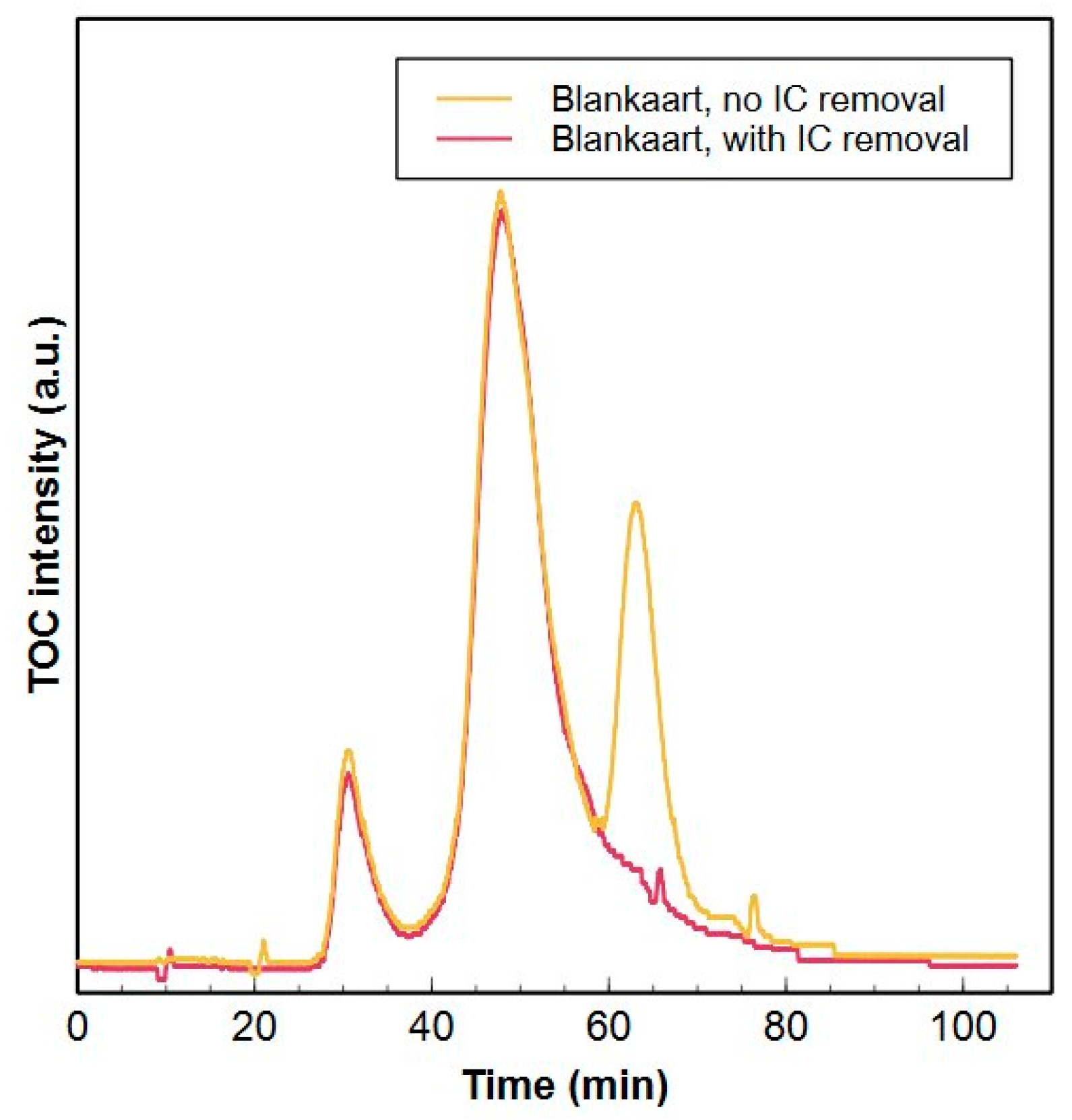
3.5.1. Inorganic Carbon Removal
3.5.2. Concentration and Molecular Weight Calibration of the HPSEC-TOC System
3.6. Method Validation
3.6.1. Limit of Detection and Limit of Quantification
3.6.2. Precision and Trueness
- Certified reference material
- b.
- Model compounds
- c.
- Real water samples
3.6.3. Method Measurement Uncertainty
3.7. Sample Stability
4. Conclusions
- Both PEG and pullulan standards have been found suitable for the calibration of the system and as quality control for the separation performance of the SEC column.
- Removal of IC by acidification of the sample to pH 6 (H3PO4) and subsequent purging prior to analysis avoids IC interference during the HPSEC-TOC method and does not modify the organic matter composition.
- The LOD of the system is 19.0 µgC dm−3. The RSDs and recoveries for model compounds are respectively between 0.26–5.4% and 60–100%. For real water samples, the recovery was in general about 80%.
- The relative measurement uncertainty Urw on routine analysis of real water samples is between 3.22–5.17%, while the measurement uncertainty on the bias Ubias, determined using a surface water sample spiked with isopropyl alcohol is 8.73%.
- Analysis of a sample should be done after a maximum preservation of two weeks in the fridge to maintain the initial composition and characteristics of the water sample. Preservation in the freezer should be avoided.
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Leenheer, J.A.; Croue, J.P. Characterizing aquatic dissolved organic matter. Environ. Sci. Technol. 2003, 37, 18A–26A. [Google Scholar] [CrossRef]
- Matilainen, A.; Gjessing, E.T.; Lahtinen, T.; Hed, L.; Bhatnagar, A.; Sillanpaa, M. An overview of the methods used in the characterisation of natural organic matter (NOM) in relation to drinking water treatment. Chemosphere 2011, 83, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Riyadh, A.; Peleato, N.M. Natural Organic Matter Character in Drinking Water Distribution Systems: A Review of Impacts on Water Quality and Characterization Techniques. Water 2024, 16, 446. [Google Scholar] [CrossRef]
- Anderson, L.E.; DeMont, I.; Dunnington, D.D.; Bjorndahl, P.; Redden, D.J.; Brophy, M.J.; Gagnon, G.A. A review of long-term change in surface water natural organic matter concentration in the northern hemisphere and the implications for drinking water treatment. Sci. Total Environ. 2023, 858, 159699. [Google Scholar] [CrossRef] [PubMed]
- Dejaeger, K.; Criquet, J.; Vanoppen, M.; Vignal, C.; Billon, G.; Cornelissen, E.R. Identification of disinfection by-product precursors by natural organic matter fractionation: A review. Environ. Chem. Lett. 2022, 20, 3861–3882. [Google Scholar] [CrossRef]
- Delpla, I.; Jung, A.V.; Baures, E.; Clement, M.; Thomas, O. Impacts of climate change on surface water quality in relation to drinking water production. Environ. Int. 2009, 35, 1225–1233. [Google Scholar] [CrossRef]
- Brezinski, K.; Gorczyca, B. An overview of the uses of high performance size exclusion chromatography (HPSEC) in the characterization of natural organic matter (NOM) in potable water, and ion-exchange applications. Chemosphere 2019, 217, 122–139. [Google Scholar] [CrossRef]
- Zark, M.; Dittmar, T. Universal molecular structures in natural dissolved organic matter. Nat. Commun. 2018, 9, 3178. [Google Scholar] [CrossRef]
- Ignatev, A.; Tuhkanen, T. Step-by-step analysis of drinking water treatment trains using size-exclusion chromatography to fingerprint and track protein-like and humic/fulvic-like fractions of dissolved organic matter. Environ. Sci.-Water Res. Technol. 2019, 5, 1568–1581. [Google Scholar] [CrossRef]
- Allpike, B.P.; Heitz, A.; Joll, C.A.; Kagi, R.I.; Abbt-Braun, G.; Frimmel, F.H.; Brinkmann, T.; Her, N.; Amy, G. Size exclusion chromatography to characterize DOC removal in drinking water treatment. Environ. Sci. Technol. 2005, 39, 2334–2342. [Google Scholar] [CrossRef]
- Wilske, C.; Herzsprung, P.; Lechtenfeld, O.J.; Kamjunke, N.; Einax, J.W.; von Tümpling, W. New Insights into the Seasonal Variation of DOM Quality of a Humic-Rich Drinking-Water Reservoir-Coupling 2D-Fluorescence and FTICR MS Measurements. Water 2021, 13, 1703. [Google Scholar] [CrossRef]
- Pan, Y.; Li, H.; Zhang, X.R.; Li, A.M. Characterization of natural organic matter in drinking water: Sample preparation and analytical approaches. Trends Environ. Anal. Chem. 2016, 12, 23–30. [Google Scholar] [CrossRef]
- Matilainen, A.; Sillanpaa, M. Removal of natural organic matter from drinking water by advanced oxidation processes. Chemosphere 2010, 80, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.Q.; Gao, D.; Yan, Z.R.; Gao, C.; Wang, L.L. Optical properties and spatial distribution of chromophoric dissolved organic matter (CDOM) in Poyang Lake, China. J. Great Lakes Res. 2017, 43, 700–709. [Google Scholar] [CrossRef]
- Cai, M.H.; Wu, Y.P.; Ji, W.X.; Han, Y.Z.; Li, Y.; Wu, J.C.; Shuang, C.-D.; Korshin, G.V.; Li, A.-M.; Li, W.-T. Characterizing property and treatability of dissolved effluent organic matter using size exclusion chromatography with an array of absorbance, fluorescence, organic nitrogen and organic carbon detectors. Chemosphere 2020, 243, 125321. [Google Scholar] [CrossRef] [PubMed]
- Shimotori, K.; Satou, T.; Imai, A.; Kawasaki, N.; Komatsu, K.; Kohzu, A.; Tomioka, N.; Shinohara, R.; Miura, S. Quantification and characterization of coastal dissolved organic matter by high-performance size exclusion chromatography with ultraviolet absorption, fluorescence, and total organic carbon analyses. Limnol. Oceanogr. Methods 2016, 14, 637–648. [Google Scholar] [CrossRef]
- Xu, H.C.; Guo, L.D. Molecular size-dependent abundance and composition of dissolved organic matter in river, lake and sea waters. Water Res. 2017, 117, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Raeke, J.; Lechtenfeld, O.J.; Wagner, M.; Herzsprung, P.; Reemtsma, T. Selectivity of solid phase extraction of fresh water dissolved organic matter and its effect on ultrahigh resolution mass spectra. Environ. Sci.-Process. Impacts 2016, 18, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Stalter, D.; Peters, L.I.; O’Malley, E.; Tang, J.Y.M.; Revalor, M.; Farré, M.J.; Watson, K.; von Gunten, U.; Escher, B.I. Sample Enrichment for Bioanalytical Assessment of Disinfected Drinking Water: Concentrating the Polar, the Volatiles, and the Unknowns. Environ. Sci. Technol. 2016, 50, 6495–6505. [Google Scholar] [CrossRef]
- Her, N.; Amy, G.; Foss, D.; Cho, J.; Yoon, Y.; Kosenka, P. Optimization of method for detecting and characterizing NOM by HPLC-size exclusion chromatography with UV and on-line DOC detection. Environ. Sci. Technol. 2002, 36, 1069–1076. [Google Scholar] [CrossRef]
- Huber, S.A.; Balz, A.; Abert, M.; Pronk, W. Characterisation of aquatic humic and non-humic matter with size-exclusion chromatography—Organic carbon detection—Organic nitrogen detection (LC-OCD-OND). Water Res. 2011, 45, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Mapp, K. Size Exclusion Chromatography—Toyopearl Resins for SEC; Tosoh Bioscience LLC: King of Prussia, PA, USA, 2023. [Google Scholar]
- Ruhl, A.S.; Jekel, M. Elution behaviour of low molecular weight compounds in size exclusion chromatography. J. Water Supply Res. Technol.-AQUA 2012, 61, 32–40. [Google Scholar] [CrossRef]
- Hidayah, E.N.; Chou, Y.C.; Yeh, H.H. Using HPSEC to identify NOM fraction removal and the correlation with disinfection by-product precursors. Water Sci. Technol.-Water Supply 2016, 16, 305–313. [Google Scholar] [CrossRef]
- Mojela, H.; Gericke, G.; Madhav, H.; Malinga, S.P. Seasonal variations of natural organic matter (NOM) in surface water supplied to two coal-fired power stations. Environ. Sci. Pollut. Res. 2023, 30, 15454–15463. [Google Scholar] [CrossRef] [PubMed]
- MacKeown, H.; Gyamfi, J.A.; Delaporte, M.; Schoutteten, K.; Verdickt, L.; Ouddane, B.; Criquet, J. Removal of disinfection by-product precursors by ion exchange resins. J. Environ. Chem. Eng. 2021, 9, 104602. [Google Scholar] [CrossRef]
- Carra, I.; Lozano, J.F.; Johannesen, S.; Godart-Brown, M.; Goslan, E.H.; Jarvis, P.; Judd, S. Sorptive removal of disinfection by-product precursors from UK lowland surface waters: Impact of molecular weight and bromide. Sci. Total Environ. 2021, 754, 142152. [Google Scholar] [CrossRef] [PubMed]
- Kaarela, O.; Koppanen, M.; Kesti, T.; Kettunen, R.; Palmroth, M.; Rintala, J. Natural organic matter removal in a full-scale drinking water treatment plant using ClO2 oxidation: Performance of two virgin granular activated carbons. J. Water Process Eng. 2021, 41, 102001. [Google Scholar] [CrossRef]
- Lavanya, G.S.M.; Eswarudu, M.M.; Eswaraiah, M.C.; Harisudha, K.; Spandana, B.N. Analytical Method Validation: An Updated Review. Int. J. Pharm. Sci. Res. 2013, 4, 1280–1286. [Google Scholar]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method validation and Related Topics, 2nd ed.; 2014; ISBN 978-91-87461-59-0. Available online: http://www.eurachem.org (accessed on 25 April 2024).
- EN ISO 5667-3:2018; Water Quality—Sampling—Part 3: Preservation and Handling of Water Samples. ISO: Geneva, Switzerland, 2018.
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]
- Dulaquais, G.; Breitenstein, J.; Waeles, M.; Marsac, R.; Riso, R. Measuring dissolved organic matter in estuarine and marine waters: Size-exclusion chromatography with various detection methods. Environ. Chem. 2018, 15, 436–449. [Google Scholar] [CrossRef]
- Park, Y.J.; Choi, Y.B.; Oh, S.B.; Moon, J.; Truong, T.Q.; Huynh, P.K.; Kim, S.M. Development and application of a high-performance liquid chromatography diode-array detection (HPLC-DAD) method for the simultaneous quantification of phenolic compounds in the aerial part of Glehnia littoralis. Appl. Biol. Chem. 2024, 67, 34. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, J.F.; Ma, J.; Qiang, Z.M. Fluorescence spectroscopic characterization of DOM fractions isolated from a filtered river water after ozonation and catalytic ozonation. Chemosphere 2008, 71, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Thurman, E. Organic Geochemistry of Natural Waters; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Warton, B.; Heitz, A.; Allpike, B.; Kagi, R. Size-exclusion chromatography with organic carbon detection using a mass spectrometer. J. Chromatogr. A 2008, 1207, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lee, D.; Snyder, S.A. Deconvolution of Size Exclusion Chromatograms: New Insights into the Molecular Weight Distribution of Dissolved Organic Matter in Ozone and Biological Activated Carbon. Acs Es&T Water 2021, 1, 125–133. [Google Scholar] [CrossRef]
- Humbert, H.; Gallard, H.; Suty, H.; Croue, J.P. Performance of selected anion exchange resins for the treatment of a high DOC content surface water. Water Res. 2005, 39, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.R.; Kilduff, J.E. Factors affecting selectivity during dissolved organic matter removal by anion-exchange resins. Water Res. 2007, 41, 4211–4221. [Google Scholar] [CrossRef]
- Sleighter, R.L.; Hatcher, P.G. Molecular characterization of dissolved organic matter (DOM) along a river to ocean transect of the lower Chesapeake Bay by ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Mar. Chem. 2008, 110, 140–152. [Google Scholar] [CrossRef]
- Choosing the Right Calibration for the Agilent Bio SEC-3 Column; Agilent Technologies: Santa Clara, CA, USA, 2017.
- Matilainen, A.; Vepsalainen, M.; Sillanpaa, M. Natural organic matter removal by coagulation during drinking water treatment: A review. Adv. Colloid Interface Sci. 2010, 159, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sievers M9/M9e TOC Analyzers—Operation and Maintenance Manual; GE Power & Water: Schenectady, NY, USA, 2014.
- Karanfil, T.; Schlautman, M.A.; Erdogan, I. Survey of DOC and UV measurement practices with implications for SUVA determination. J. Am. Water Works Assoc. 2002, 94, 68–80. [Google Scholar] [CrossRef]
- Dasaard, C.B.; Bayless, D.J.; Stuart, B.J. Saturated pH and Total Inorganic Carbon from CO2 Solubility Related to Algal Growth. Int. Adv. Res. J. Sci. Eng. Technol. 2016, 3, 146–150. [Google Scholar] [CrossRef]
- Allpike, B.P.; Heitz, A.; Joll, C.A.; Kagi, R.I. A new organic carbon detector for size exclusion chromatography. J. Chromatogr. A 2007, 1157, 472–476. [Google Scholar] [CrossRef]
- Zhang, W.J.; Li, L.; Wang, D.H.; Wang, R.; Yu, S.L.; Gao, N.Y. Characterizing dissolved organic matter in aquatic environments by size exclusion chromatography coupled with multiple detectors. Anal. Chim. Acta 2022, 1191, 339358. [Google Scholar] [CrossRef]
- Li, X.; Rao, N.R.H.; Linge, K.L.; Joll, C.A.; Khan, S.; Henderson, R.K. An evaluation of measurement techniques for algal-derived organic nitrogen. Water Res. 2019, 165, 114998. [Google Scholar] [CrossRef]
- Lankes, U.; Mueller, M.B.; Weber, M.; Frimmel, F.H. Reconsidering the quantitative analysis of organic carbon concentrations in size exclusion chromatography. Water Res. 2009, 43, 915–924. [Google Scholar] [CrossRef]
- Horwitz, W.; Albert, R. The Horwitz ratio (HorRat): A useful index of method performance with respect to precision. J. AOAC Int. 2006, 89, 1095–1109. [Google Scholar] [CrossRef]
- Hammes, F.; Meylan, S.; Salhi, E.; Koster, O.; Egli, T.; Von Gunten, U. Formation of assimilable organic carbon (AOC) and specific natural organic matter (NOM) fractions during ozonation of phytoplankton. Water Res. 2007, 41, 1447–1454. [Google Scholar] [CrossRef]
- Hem, L.J.; Efraimsen, H. Assimilable organic carbon in molecular weight fractions of natural organic matter. Water Res. 2001, 35, 1106–1110. [Google Scholar] [CrossRef]
- Vanderkooij, D.; Visser, A.; Hijnen, W.A.M. Determining the concentration of easily assimilable organic-carbon in drinking-water. J. Am. Water Work Assoc. 1982, 74, 540–545. [Google Scholar] [CrossRef]
- Payandi-Rolland, D.; Shirokova, L.S.; Labonne, F.; Benezeth, P.; Pokrovsky, O.S. Impact of freeze-thaw cycles on organic carbon and metals in waters of permafrost peatlands. Chemosphere 2021, 279, 130510. [Google Scholar] [CrossRef]
- Peacock, M.; Freeman, C.; Gauci, V.; Lebron, I.; Evans, C.D. Investigations of freezing and cold storage for the analysis of peatland dissolved organic carbon (DOC) and absorbance properties. Environ. Sci.-Process. Impacts 2015, 17, 1290–1301. [Google Scholar] [CrossRef]
- Chen, J.; Xue, S.; Lin, Y.Z.; Wang, C.; Wang, Q.; Han, Q. Effect of freezing-thawing on dissolved organic matter in water. Desalination Water Treat. 2016, 57, 17230–17240. [Google Scholar] [CrossRef]
- Lloyd, C.E.M.; Johnes, P.J.; Pemberton, J.A.; Yates, C.A.; Jones, D.; Evershed, R.P. Sampling, storage and laboratory approaches for dissolved organic matter characterisation in fresh waters: Moving from nutrient fraction to molecular-scale characterisation. Sci. Total Environ. 2022, 827, 154105. [Google Scholar] [CrossRef]
- Cook, S.; Peacock, M.; Evans, C.D.; Page, S.E.; Whelan, M.; Gauci, V.; Khoon, K.L. Cold storage as a method for the long-term preservation of tropical dissolved organic carbon (DOC). Mires Peat 2016, 18, 1–8. [Google Scholar] [CrossRef]
- Baghoth, S.A.; Sharma, S.K.; Amy, G.L. Tracking natural organic matter (NOM) in a drinking water treatment plant using fluorescence excitation-emission matrices and PARAFAC. Water Res. 2011, 45, 797–809. [Google Scholar] [CrossRef]
- Krzeminski, P.; Vogelsang, C.; Meyn, T.; Köhler, S.J.; Poutanen, H.; de Wit, H.A.; Uhl, W. Natural organic matter fractions and their removal in full-scale drinking water treatment under cold climate conditions in Nordic capitals. J. Environ. Manag. 2019, 241, 427–438. [Google Scholar] [CrossRef]
- Gibert, O.; Lefèvre, B.; Fernández, M.; Bernat, X.; Paraira, M.; Pons, M. Fractionation and removal of dissolved organic carbon in a full-scale granular activated carbon filter used for drinking water production. Water Res. 2013, 47, 2821–2829. [Google Scholar] [CrossRef]
- Bi, Z.H.; Li, T.; Xing, X.C.; Qi, P.; Li, Z.S.; Hu, C.; Xu, X.; Sun, Z.; Xu, G.; Chen, C.; et al. Contribution of extracellular polymeric substances and microbial community on the safety of drinking water quality: By mean of Cu/activated carbon biofiltration. Chemosphere 2022, 286, 131686. [Google Scholar] [CrossRef]
- Shen, H.; Tang, X.C.; Wu, N.X.; Chen, H.B. Leakage of soluble microbial products from biological activated carbon filtration in drinking water treatment plants and its influence on health risks. Chemosphere 2018, 202, 626–636. [Google Scholar] [CrossRef]
- Godec, R.; O’Neill, K.J.; Hutte, R. Method and Apparatus for The Measurement of Dissolved Carbon. U.S. Patent No. 5,820,823, 1999. [Google Scholar]
- Ming, C.A. High Sensitivity Total Organic Carbon Analysis; Shimadzu Puplication no AD-0007-TOC; Customer Support Centre, Shimadzu (Asia Pacific) Pte Ltd.: Singapore, 2003. [Google Scholar]
- Godec, R.; Kosenka, P.; Hutte, R. Method and Apparatus for the Determination of Dissolved Carbon in Water. U.S. Patent US5132094A, 1992. [Google Scholar]
- Vial, J.; Jardy, A. Experimental comparison of the different approaches to estimate LOD and LOQ of an HPLC method. Anal. Chem. 1999, 71, 2672–2677. [Google Scholar] [CrossRef]
- FASFC. Microbiology—Estimationg Measurement Uncertainty; FASFC: Brussels, Belgium, 2009; Available online: https://favv-afsca.be/ (accessed on 25 April 2024).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recovery (%) | Sodium-Alginate | Fumaric Acid | Isopropyl Alcohol | |||
|---|---|---|---|---|---|---|
| 19/09 | 21/09 | 19/09 | 21/09 | 19/09 | 21/09 | |
| 0.4 mgC dm−3 | 59 ± 2 | 58 ± 4 | 70 ± 3 | 72 ± 4 | 90 ± 1 | 92 ± 1 |
| 4.0 mgC dm−3 | 67 ± 0.4 | 68 ± 0.8 | 94 ± 0.2 | 94 ± 1 | 100 ± 10 | 100 ± 1 |
| Sample | Average Concentration (mgC dm−3) | d (%) | CV (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| High MW | Medium MW | Low MW | High MW | Medium MW | Low MW | High MW | Medium MW | Low MW | |
| A Blankaart 1 | 0.616 | 5.170 | 1.220 | 2.44 | 0.43 | 5.49 | 17 | 12 | 16 |
| B Coupure microfiltrated | 0.597 | 2.605 | 1.573 | 3.69 | 3.72 | 2.10 | 17 | 14 | 15 |
| C Blankaart microfiltrated | 0.554 | 4.407 | 1.109 | 6.50 | 0.41 | 9.56 | 17 | 13 | 16 |
| D tap water | <LOQ a | 0.772 | 0.109 | n.a.b | 3.24 | 11.01 | n.a. b | 17 | 22 |
| E Essen groundwater | <LOQ a | 1.365 | 0.386 | n.a. b | 0.22 | 10.12 | n.a b | 15 | 18 |
| F Spannenburg groundwater | <LOQ a | 6.543 | 0.723 | n.a. b | 1.86 | 4.28 | n.a b | 12 | 17 |
| G Mol groundwater | <LOQ a | 0.524 | 0.365 | n.a. b | 1.72 | 0.55 | n.a. b | 18 | 19 |
| H Merksplas groundwater | <LOQ a | 1.879 | 0.326 | n.a. b | 1.97 | 4.29 | n.a. b | 15 | 19 |
| I Oud-Turnhout groundwater | <LOQ a | 2.122 | 0.443 | n.a. b | 1.23 | 8.58 | n.a. b | 14 | 18 |
| J Blankaart nanofiltrated | 0.314 | 0.090 | 2.056 | 0.64 | 6.67 | 0.63 | 19 | 23 | 14 |
| pH (−) | Conduct. (µS cm−1) | UV245 (m−1) | TOC (mgC dm−3) | IC (mgC dm−3) | |
|---|---|---|---|---|---|
| Blankaart | 8.27 ± 0.03 | 800 ± 100 | 20 ± 3 | 7.7 ± 0.5 | 50 ± 10 |
| Coupure | 7.6 ± 0.7 | 800 ± 100 | 14 ± 4 | 8.5 ± 0.8 | 54 ± 5 |
| Mol b | 8.1 | 219 | 1.7 | 0.8 | 21.77 |
| Merksplas b | 7.7 | 430 | 7.2 | 2.7 | 50.33 |
| Oud-Turnhout b | 7.7 | 314 | 7.1 | 2.9 | 37.01 |
| Essen b | 8 | 344 | 5.9 | 2/0 | 40.74 |
| Spannenburg | 6.82 ± 0.08 | 660 ± 30 | n.a. a | n.a. a | 120 ± 10 |
| Farys | 7.8 ± 0.2 | 500 ± 100 | n.a. a | 2 ± 1 | 40 ± 10 |
| Oxidation | Detection | Used Mode a | |
|---|---|---|---|
| Sievers® M9 | photochemical + ammonium persulfate | Conductometric | Online/offline |
| Sievers® 900 | photochemical + ammonium persulfate | Conductometric | offline |
| Shimadzu TOC VCPN/VCSH | High-temperature catalytic combustion | Non dispersive infrared | offline |
| Gräntzel thin-film reactor | Photochemical | Infrared | Online/offline |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laforce, E.; Dejaeger, K.; Vanoppen, M.; Cornelissen, E.; De Clercq, J.; Vermeir, P. Thorough Validation of Optimized Size Exclusion Chromatography-Total Organic Carbon Analysis for Natural Organic Matter in Fresh Waters. Molecules 2024, 29, 2075. https://doi.org/10.3390/molecules29092075
Laforce E, Dejaeger K, Vanoppen M, Cornelissen E, De Clercq J, Vermeir P. Thorough Validation of Optimized Size Exclusion Chromatography-Total Organic Carbon Analysis for Natural Organic Matter in Fresh Waters. Molecules. 2024; 29(9):2075. https://doi.org/10.3390/molecules29092075
Chicago/Turabian StyleLaforce, Elien, Karlien Dejaeger, Marjolein Vanoppen, Emile Cornelissen, Jeriffa De Clercq, and Pieter Vermeir. 2024. "Thorough Validation of Optimized Size Exclusion Chromatography-Total Organic Carbon Analysis for Natural Organic Matter in Fresh Waters" Molecules 29, no. 9: 2075. https://doi.org/10.3390/molecules29092075
APA StyleLaforce, E., Dejaeger, K., Vanoppen, M., Cornelissen, E., De Clercq, J., & Vermeir, P. (2024). Thorough Validation of Optimized Size Exclusion Chromatography-Total Organic Carbon Analysis for Natural Organic Matter in Fresh Waters. Molecules, 29(9), 2075. https://doi.org/10.3390/molecules29092075