
Designing Antitrypanosomal and Antileishmanial BODIPY Derivatives: A Computational and In Vitro Assessment
 , , , and
, , , and 
Abstract

1. Introduction
2. Results and Discussion
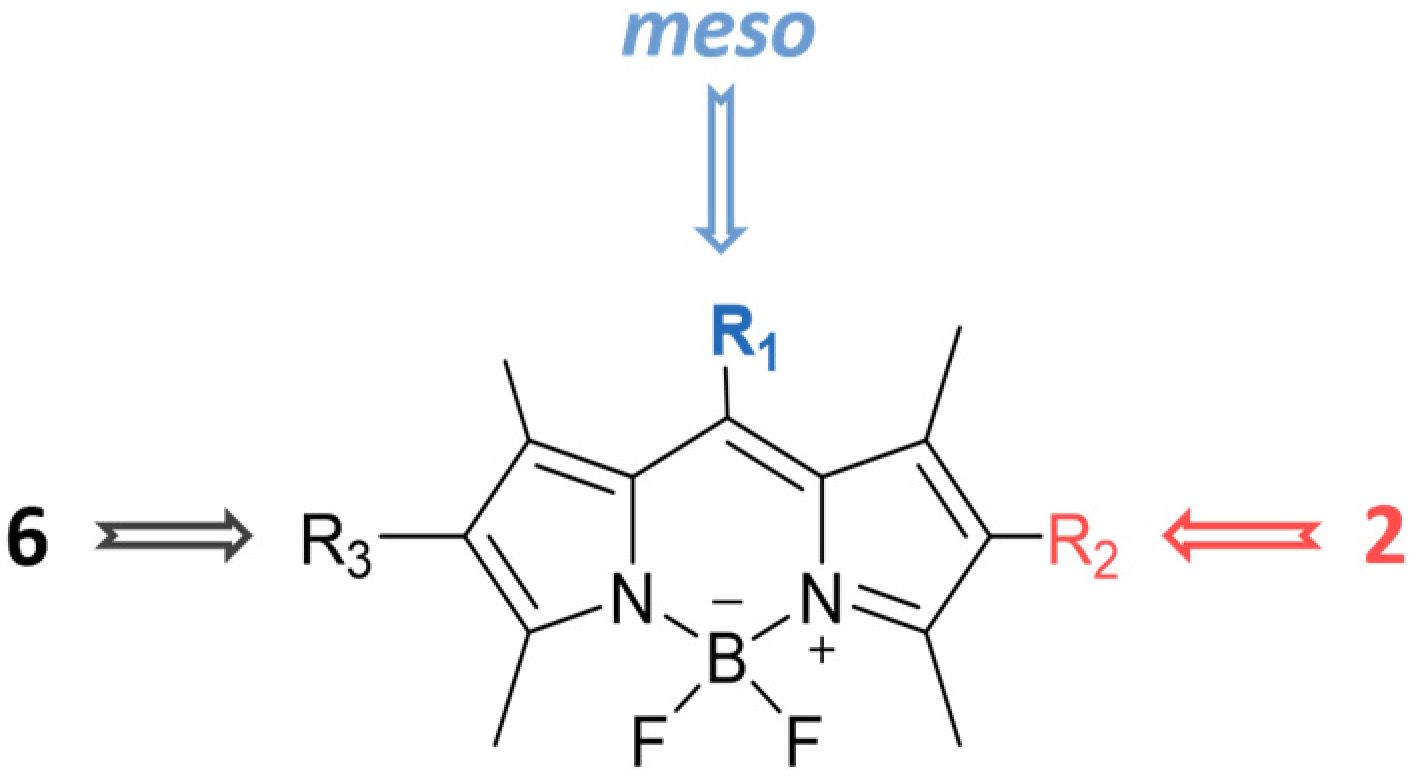
2.1. Synthesis and Characterization of BODIPY Derivatives
2.2. Antiparasitic and Cytotoxicity Activity
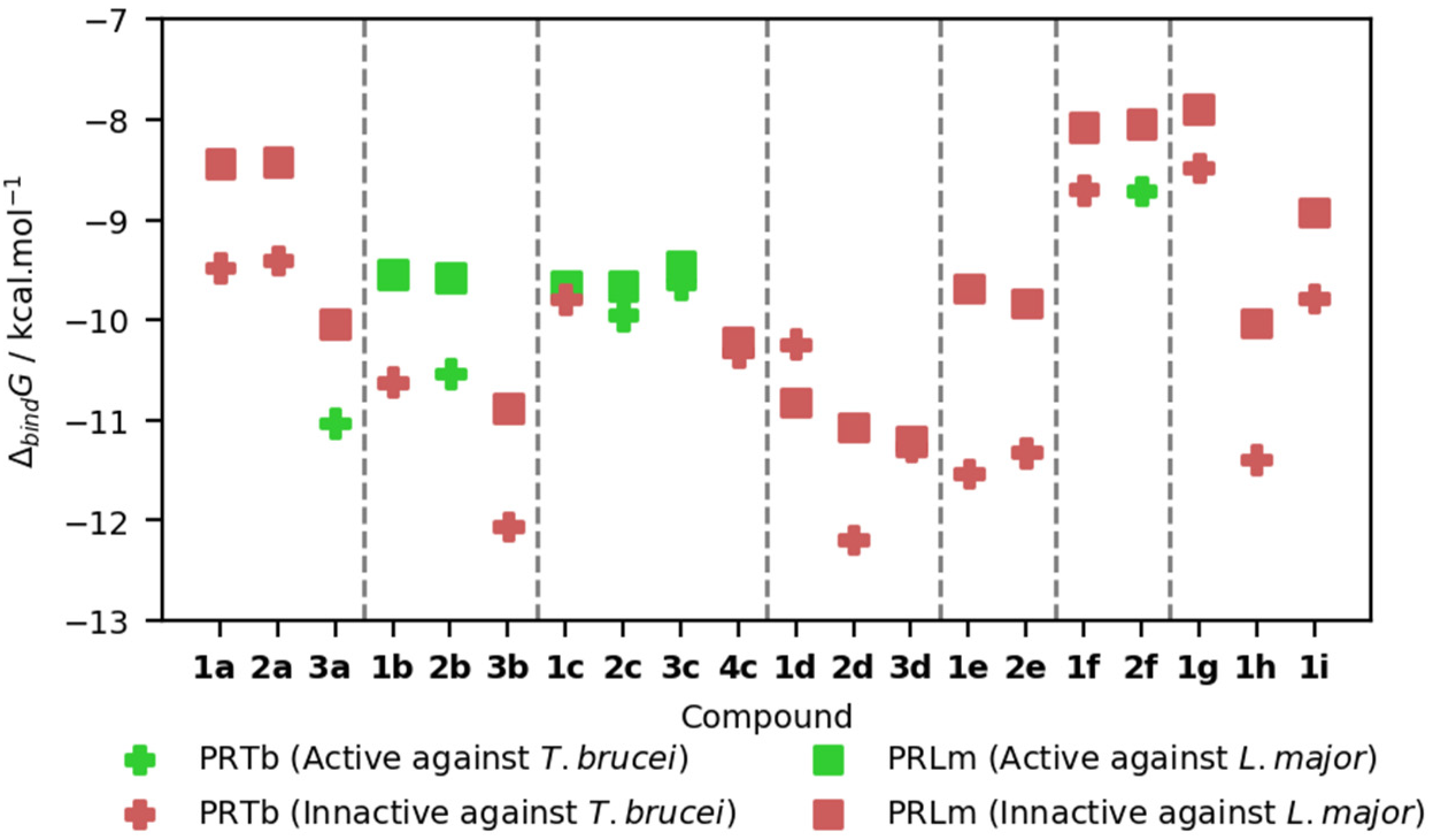
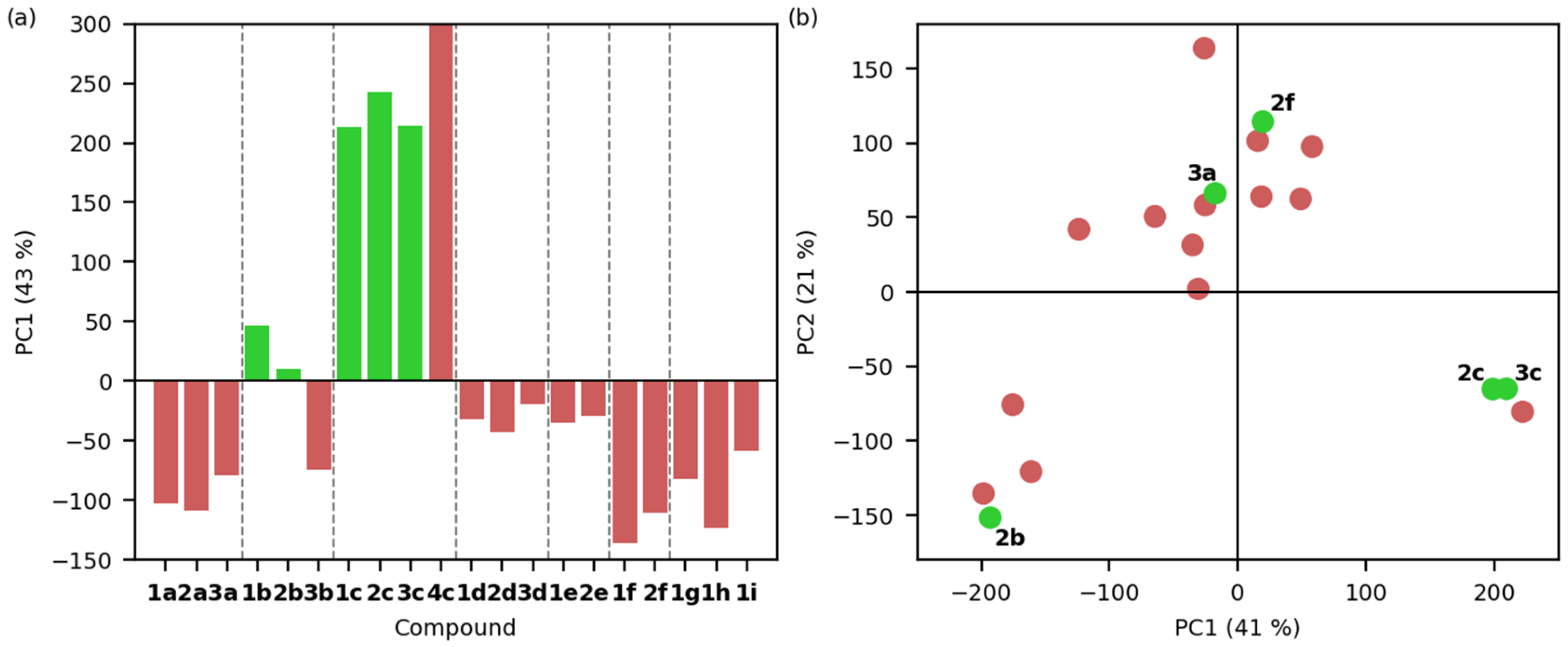
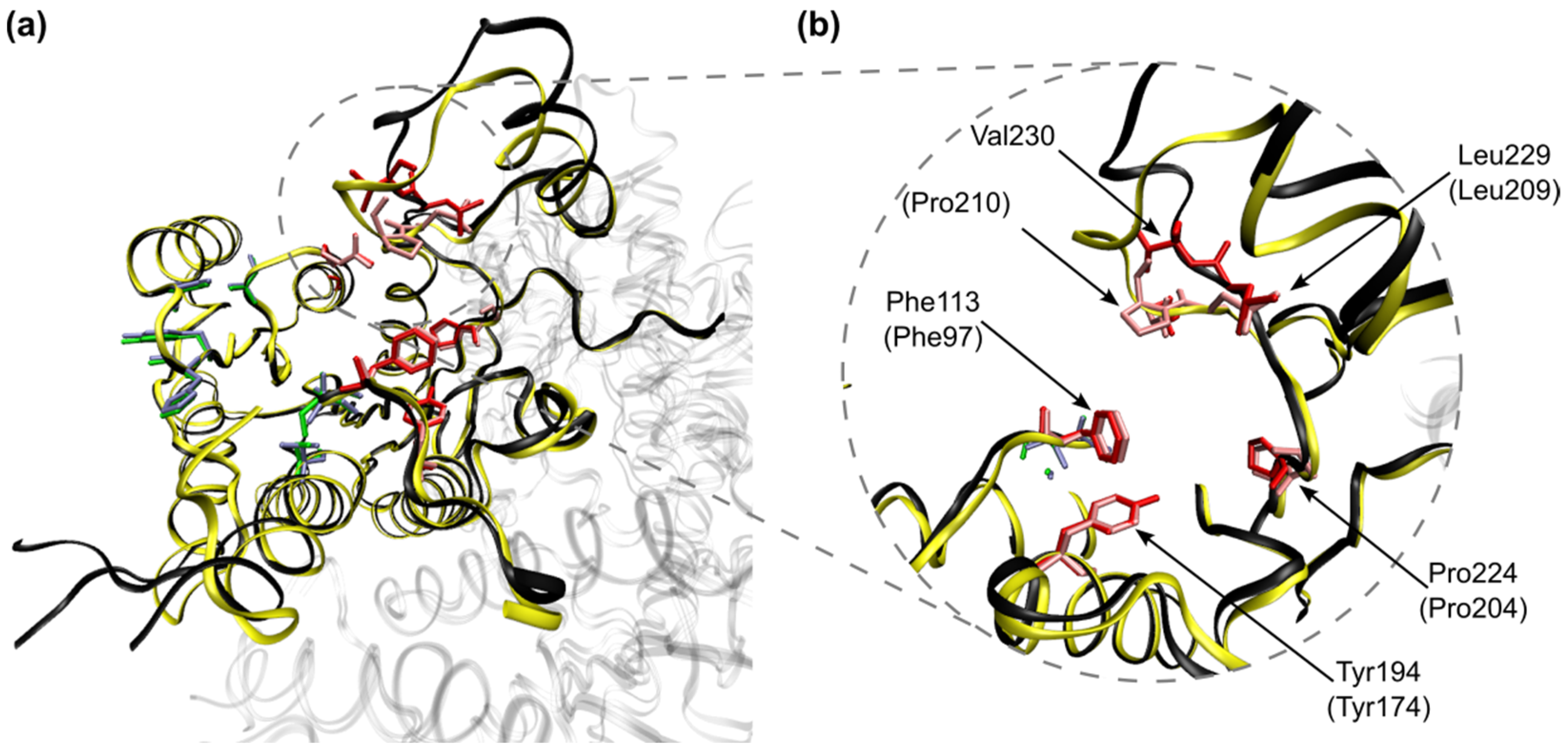
2.3. Molecular Docking Studies
3. Conclusions
4. Experimental Section
4.1. General
4.2. Biological Activity
4.2.1. Parasite and Cell Culturing
4.2.2. Antiparasitic Activity
4.2.3. Cytoxicity
4.2.4. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-Trypanosomatid Drug Discovery: Progress and Challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Peñalver, P.; Costa, S.P.G.; Morales, J.C.; Raposo, M.M.M. Polyaromatic Bis(Indolyl)Methane Derivatives with Antiproliferative and Antiparasitic Activity. Molecules 2023, 28, 7728. [Google Scholar] [CrossRef] [PubMed]
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An Overview on Target-Based Drug Design against Kinetoplastid Protozoan Infections: Human African Trypanosomiasis, Chagas Disease and Leishmaniases. Molecules 2021, 26, 4629. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. Drug Discovery for Neglected Diseases: Molecular Target-Based and Phenotypic Approaches: Miniperspectives Series on Phenotypic Screening for Antiinfective Targets. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Pozzi, C.; Tassone, G.; Mangani, S. X-Ray Crystallography Contributions to Drug Discovery Against Parasite. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 51, pp. 175–230. ISBN 978-0-12-815143-3. [Google Scholar]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate Reductase Inhibitors as Antibacterial Agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and Other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J. Antibiot. 2020, 73, 5–27. [Google Scholar] [CrossRef]
- Nzila, A. The Past, Present and Future of Antifolates in the Treatment of Plasmodium Falciparum Infection. J. Antimicrob. Chemother. 2006, 57, 1043–1054. [Google Scholar] [CrossRef]
- Gilbert, I.H. Inhibitors of Dihydrofolate Reductase in Leishmania and Trypanosomes. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2002, 1587, 249–257. [Google Scholar] [CrossRef]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine Reductase Mechanism Correlates Pterin Metabolism with Drug Resistance in Trypanosomatid Parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef]
- Panecka-Hofman, J.; Poehner, I. Structure and Dynamics of Pteridine Reductase 1: The Key Phenomena Relevant to Enzyme Function and Drug Design. Eur. Biophys. J. 2023, 52, 521–532. [Google Scholar] [CrossRef]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of Potent Pteridine Reductase Inhibitors to Guide Antiparasite Drug Development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Kimuda, M.P.; Laming, D.; Hoppe, H.C.; Tastan Bishop, Ö. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma Brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules 2019, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Panecka-Hofman, J.; Poehner, I.; Wade, R.C. Anti-Trypanosomatid Structure-Based Drug Design—Lessons Learned from Targeting the Folate Pathway. Expert Opin. Drug Discov. 2022, 17, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Pöhner, I.; Quotadamo, A.; Panecka-Hofman, J.; Luciani, R.; Santucci, M.; Linciano, P.; Landi, G.; Di Pisa, F.; Dello Iacono, L.; Pozzi, C.; et al. Multitarget, Selective Compound Design Yields Potent Inhibitors of a Kinetoplastid Pteridine Reductase 1. J. Med. Chem. 2022, 65, 9011–9033. [Google Scholar] [CrossRef] [PubMed]
- Treibs, A.; Kreuzer, F. Difluorboryl-Komplexe von Di- Und Tripyrrylmethenen. Justus Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Boens, N.; Verbelen, B.; Dehaen, W. Postfunctionalization of the BODIPY Core: Synthesis and Spectroscopy. Eur. J. Org. Chem. 2015, 2015, 6577–6595. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Kowada, T.; Maeda, H.; Kikuchi, K. BODIPY-Based Probes for the Fluorescence Imaging of Biomolecules in Living Cells. Chem. Soc. Rev. 2015, 44, 4953–4972. [Google Scholar] [CrossRef]
- Bañuelos, J. BODIPY Dye, the Most Versatile Fluorophore Ever? Chem. Rec. 2016, 16, 335–348. [Google Scholar] [CrossRef]
- Poddar, M.; Misra, R. Recent Advances of BODIPY Based Derivatives for Optoelectronic Applications. Coord. Chem. Rev. 2020, 421, 213462. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, K. Recent Advances in the Application of BODIPY in Bioimaging and Chemosensing. J. Mater. Chem. C 2019, 7, 11361–11405. [Google Scholar] [CrossRef]
- Prieto-Montero, R.; Prieto-Castañeda, A.; Sola-Llano, R.; Agarrabeitia, A.R.; García-Fresnadillo, D.; López-Arbeloa, I.; Villanueva, A.; Ortiz, M.J.; De La Moya, S.; Martínez-Martínez, V. Exploring BODIPY Derivatives as Singlet Oxygen Photosensitizers for PDT. Photochem. Photobiol. 2020, 96, 458–477. [Google Scholar] [CrossRef]
- Carpenter, B.; Situ, X.; Scholle, F.; Bartelmess, J.; Weare, W.; Ghiladi, R. Antiviral, Antifungal and Antibacterial Activities of a BODIPY-Based Photosensitizer. Molecules 2015, 20, 10604–10621. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, Q.; Wang, L.; Hao, E.; Jiao, L. The Main Strategies for Tuning BODIPY Fluorophores into Photosensitizers. J. Porphyr. Phthalocyanines 2020, 24, 603–635. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Pina, J.; Costa, S.P.G.; Raposo, M.M.M. Synthesis and Characterization of Aryl-Substituted BODIPY Dyes Displaying Distinct Solvatochromic Singlet Oxygen Photosensitization Efficiencies. Dye. Pigment. 2021, 196, 109784. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Pinto, S.C.S.; Pina, J.; Gomes-da-Silva, L.C.; Costa, S.P.G.; Raposo, M.M.M. Investigating the Photophysical Properties and Biological Efficacy of BODIPY Derivatives as Photosensitizers in Photodynamic Therapy. Chem. Proc. 2023, 14, 71. [Google Scholar] [CrossRef]
- De La Torre, B.G.; Hornillos, V.; Luque-Ortega, J.R.; Abengózar, M.A.; Amat-Guerri, F.; Ulises Acuña, A.; Rivas, L.; Andreu, D. A BODIPY-Embedding Miltefosine Analog Linked to Cell-Penetrating Tat(48-60) Peptide Favors Intracellular Delivery and Visualization of the Antiparasitic Drug. Amino Acids 2014, 46, 1047–1058. [Google Scholar] [CrossRef]
- Rodríguez, G.; Nargoli, J.; López, A.; Moyna, G.; Álvarez, G.; Fernández, M.; Osorio-Martínez, C.A.; González, M.; Cerecetto, H. Synthesis and in Vivo Proof of Concept of a BODIPY-Based Fluorescent Probe as a Tracer for Biodistribution Studies of a New Anti-Chagas Agent. RSC Adv. 2017, 7, 7983–7989. [Google Scholar] [CrossRef]
- Hornillos, V.; Carrillo, E.; Rivas, L.; Amat-Guerri, F.; Acuña, A.U. Synthesis of BODIPY-Labeled Alkylphosphocholines with Leishmanicidal Activity, as Fluorescent Analogues of Miltefosine. Bioorg. Med. Chem. Lett. 2008, 18, 6336–6339. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Nogueira, M.B.; Costa, S.P.G.; Raposo, M.M.M. BODIPY Derivatives: Synthesis and Evaluation of Their Optical Properties. Proceedings 2019, 9, 10. [Google Scholar] [CrossRef]
- Pinto, S.C.S.; Gonçalves, R.C.R.; Costa, S.P.G.; Raposo, M.M.M. Synthesis and Characterization of a Meso-Anthracene-BODIPY Derivative for Colorimetric Recognition of Cu2+ and Fe3+. Chem. Proc. 2021, 3, 79. [Google Scholar] [CrossRef]
- Pinto, S.C.S.; Gonçalves, R.C.R.; Costa, S.P.G.; Raposo, M.M.M. Synthesis, Characterization and Evaluation of a Carbazolyl-BODIPY as a Fluorimetric Chemosensor for F−. Chem. Proc. 2022, 8, 20. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Boland, M.L.; Costa, S.P.G.; Raposo, M.M.M. A BODIPY Derivative for Selective Fluorescent Chemosensing of Iron (III). Eng. Proc. 2022, 27, 7. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Belmonte-Reche, E.; Pina, J.; Costa Da Silva, M.; Pinto, S.C.S.; Gallo, J.; Costa, S.P.G.; Raposo, M.M.M. Bioimaging of Lysosomes with a BODIPY pH-Dependent Fluorescent Probe. Molecules 2022, 27, 8065. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.C.R.; Pinto, S.C.S.; Costa, S.P.G.; Raposo, M.M.M. Synthesis, Characterization and Evaluation of a Novel BODIPY Derivative as a Colorimetric Chemosensor for Fe3+ Recognition. Proceedings 2019, 41, 40. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Pinto, S.C.S.; Costa, S.P.G.; Raposo, M.M.M. A Meso-Triphenylamine-BODIPY Derivative for the Optical Chemosensing of Metal Ions. Chem. Proc. 2021, 3, 65. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Boland, M.L.; Costa, S.P.G.; Raposo, M.M.M. Anion Dual Mode Fluoro-Chromogenic Chemosensor Based on a BODIPY Core. Eng. Proc. 2022, 27, 6. [Google Scholar] [CrossRef]
- Pinto, S.C.S.; Gonçalves, R.C.R.; Costa, S.P.G.; Raposo, M.M.M. Colorimetric Chemosensor for Cu2+ and Fe3+ Based on a Meso-Triphenylamine-BODIPY Derivative. Sensors 2023, 23, 6995. [Google Scholar] [CrossRef]
- Cunha Dias De Rezende, L.; Menezes Vaidergorn, M.; Biazzotto Moraes, J.C.; Da Silva Emery, F. Synthesis, Photophysical Properties and Solvatochromism of Meso-Substituted Tetramethyl BODIPY Dyes. J. Fluoresc. 2014, 24, 257–266. [Google Scholar] [CrossRef]
- Shi, W.-J.; Yan, X.-H.; Yang, J.; Wei, Y.-F.; Huo, Y.-T.; Su, C.-L.; Yan, J.; Han, D.; Niu, L. Development of Meso -Five-Membered Heterocycle BODIPY-Based AIE Fluorescent Probes for Dual-Organelle Viscosity Imaging. Anal. Chem. 2023, 95, 9646–9653. [Google Scholar] [CrossRef] [PubMed]
- Kukoyi, A.; He, H.; Wheeler, K. Quinoline-Functionalized BODIPY Dyes: Structural and Photophysical Properties. J. Photochem. Photobiol. Chem. 2022, 425, 113686. [Google Scholar] [CrossRef]
- Gonçalves, R.C.R.; Nogueira, M.B.; Costa, S.P.G.; Raposo, M.M.M. Functionalized BODIPY Derivatives as Potential Fluorescent Labels. Proceedings 2019, 9, 36. [Google Scholar] [CrossRef]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-Based Design of Pteridine Reductase Inhibitors Targeting African Sleeping Sickness and the Leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Voroshylova, I.V.; Teixeira, F.; Costa, R.; Pereira, C.M.; Cordeiro, M.N.D.S. Interactions in the Ionic Liquid [EMIM][FAP]: A Coupled Experimental and Computational Analysis. Phys. Chem. Chem. Phys. 2016, 18, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Crosby, G.A.; Demas, J.N. Measurement of Photoluminescence Quantum Yields. Review. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar] [CrossRef]
- Wirtz, E.; Leal, S.; Ochatt, C.; Cross, G.M. A Tightly Regulated Inducible Expression System for Conditional Gene Knock-Outs and Dominant-Negative Genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [Google Scholar] [CrossRef]
- Cabello-Donayre, M.; Malagarie-Cazenave, S.; Campos-Salinas, J.; Gálvez, F.J.; Rodríguez-Martínez, A.; Pineda-Molina, E.; Orrego, L.M.; Martínez-García, M.; Sánchez-Cañete, M.P.; Estévez, A.M.; et al. Trypanosomatid Parasites Rescue Heme from Endocytosed Hemoglobin through Lysosomal HRG Transporters: Trypanosomatid HRG Proteins Rescue Heme from Hb. Mol. Microbiol. 2016, 101, 895–908. [Google Scholar] [CrossRef]
- Belmonte-Reche, E.; Martínez-García, M.; Guédin, A.; Zuffo, M.; Arévalo-Ruiz, M.; Doria, F.; Campos-Salinas, J.; Maynadier, M.; López-Rubio, J.J.; Freccero, M.; et al. G-Quadruplex Identification in the Genome of Protozoan Parasites Points to Naphthalene Diimide Ligands as New Antiparasitic Agents. J. Med. Chem. 2018, 61, 1231–1240. [Google Scholar] [CrossRef]
- Larson, E.M.; Doughman, D.J.; Gregerson, D.S.; Obritsch, W.F. A New, Simple, Nonradioactive, Nontoxic in Vitro Assay to Monitor Corneal Endothelial Cell Viability. Investig. Ophtalmol. Vis. Sci. 1997, 38, 1929–1933. [Google Scholar]
- Pérez-Victoria, J.M.; Bavchvarov, B.I.; Torrecillas, I.R.; Martínez-García, M.; López-Martín, C.; Campillo, M.; Castanys, S.; Gamarro, F. Sitamaquine Overcomes ABC-Mediated Resistance to Miltefosine and Antimony in Leishmania. Antimicrob. Agents Chemother. 2011, 55, 3838–3844. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post- d Group 16–18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Teixeira, F.; Mosquera, R.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Charge Distribution in Mn(Salen) Complexes. Int. J. Quantum Chem. 2014, 114, 525–533. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- wwPDB consortium. Protein Data Bank: The Single Global Archive for 3D Macromolecular Structure Data. Nucleic Acids Res. 2019, 47, D520–D528. [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y. Protein Database Searches Using Compositionally Adjusted Substitution Matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Harris, R.; Olson, A.J.; Goodsell, D.S. Automated Prediction of Ligand-binding Sites in Proteins. Proteins Struct. Funct. Bioinform. 2008, 70, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | Yield (%) | λabs (nm) | λfluo (nm) | ΦF |
|---|---|---|---|---|---|---|---|
| 1a |  | H | H | 21 | 497 | 513 | 0.68 |
| 2a | H | CHO | 91 | 492 | 508 | 0.84 | |
| 3a | H | Benzimidazole | 77 | 507 | 517 | 0.10 | |
| 1b | 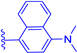 | H | H | 22 | 502 | 515 | 0.03 |
| 2b | H | CHO | 59 | 497 | 509 | 0.01 | |
| 3b | H | Benzimidazole | 31 | 512 | 514 | 0.03 | |
| 1c | 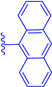 | H | H | 74 | 502 | 515 | 0.03 |
| 2c | H | CHO | 47 | 497 | 509 | 0.01 | |
| 3c | CHO | CHO | 10 | 500 | 524 | 0.009 | |
| 4c | I | I | 57 | 540 | 559 | 0.003 | |
| 1d | 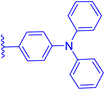 | H | H | 20 | 497 | 519 | 0.005 |
| 2d | H | CHO | 25 | 491 | 515 | 0.01 | |
| 3d | 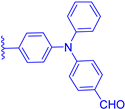 | H | CHO | 26 | 493 | 546 | 0.011 |
| 1e | 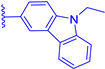 | H | H | 17 | 496 | 507 | 0.019 |
| 2e | H | CHO | 57 | 492 | 506 | 0.024 | |
| 1f |  | H | H | 8 | 508 | 519 | 0.045 |
| 2f | H | CHO | 49 | 502 | 517 | 0.30 | |
| 1g |  | H | H | 7 | 512 | 524 | 0.009 |
| 1h | 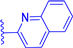 | H | H | 37 | 500 | 512 | 0.026 |
| 1i | 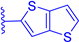 | H | H | 3 | 511 | 522 | 0.036 |
| Compounds | IC50 μM | Selectivity Index (SI) | |||
|---|---|---|---|---|---|
| T. brucei | L. major | MRC 5 | T. brucei | L. major | |
| 1a | 38.20 ± 7.29 | 19.65 ± 3.94 | 67.16 ± 4.39 | 1.76 | 3.42 |
| 2a | 74.68 ± 6.93 | >100 | 91.33 ± 0.66 | 1.22 | <0.91 |
| 3a | 5.5 ± 0.21 | >100 | 62.61 ± 3.27 | 11.38 | <0.63 |
| 1b | 18.24 ± 3.63 | 4.84 ± 1.56 | 76.84 ± 5.33 | 4.21 | 15.93 |
| 2b | 2.84 ± 0.04 | 15.75 ± 1.31 | 69.12 ± 4.90 | 24.34 | 4.39 |
| 3b | 27.87 ± 5.42 | 84.39 ± 0.32 | >100 | >3.59 | >1.18 |
| 1c | 77.11 ± 2.98 | 5.41 ± 0.43 | 82.14 ± 0.03 | 1.07 | 17.90 |
| 2c | 6.17 ± 2.35 | 10.33 ± 1.59 | 66.89 ± 7.65 | 10.84 | 6.47 |
| 3c | 14.15 ± 0.31 | 3.13 ± 0.13 | 21.65 ± 3.46 | 1.53 | 6.91 |
| 4c | 28.05 ± 1.77 | >50 | 48.22 ± 1.13 | 1.72 | <0.96 |
| 1d | 48.84 ± 3.29 | >100 | 91.29 ± 6.31 | 1.87 | <0.91 |
| 2d | 22.74 ± 1.88 | >100 | 81.06 ± 3.90 | 3.56 | <0.81 |
| 3d | 38.26 ± 0.93 | >100 | 87.13 ± 3.28 | 2.28 | <0.87 |
| 1e | 18.65 ± 2.67 | >100 | 88.89 ± 3.82 | 4.77 | <0.89 |
| 2e | 15.23 ± 0.86 | 20.71 ± 1.79 | 89.65 ± 9.08 | 5.89 | 4.33 |
| 1f | 36.93 ± 7.43 | 64.55 ± 8.43 | 57.96 ± 4.76 | 1.57 | 0.90 |
| 2f | 12.37 ± 4.02 | 19.72 ± 3.21 | 80.89 ± 1.06 | 6.54 | 4.10 |
| 1g | 55.67 ± 1.65 | >100 | 82.82 ± 1.20 | 1.49 | <0.83 |
| 1h | 77.36 ± 3.46 | >100 | 95.58 ± 3.45 | 1.24 | <0.96 |
| 1i | 61.47 ± 10.94 | 92.21 ± 10.02 | 96.82 ± 4.50 | 1.58 | 1.05 |
| Positive Loadings | Negative Loadings | ||
|---|---|---|---|
| Amino Acid | Value | Amino Acid | Value |
| Ser40 | +0.43 | Phe113 | −0.49 |
| Ala15 | +0.40 | Arg17 | −0.23 |
| Ser111 | +0.23 | Tyr194 | −0.23 |
| His38 | +0.23 | Gly225 | −0.15 |
| Tyr37 | +0.17 | Pro224 | −0.14 |
| Ser112 | +0.16 | Val230 | −0.11 |
| Ser146 | +0.14 | Leu229 | −0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, R.C.R.; Teixeira, F.; Peñalver, P.; Costa, S.P.G.; Morales, J.C.; Raposo, M.M.M. Designing Antitrypanosomal and Antileishmanial BODIPY Derivatives: A Computational and In Vitro Assessment. Molecules 2024, 29, 2072. https://doi.org/10.3390/molecules29092072
Gonçalves RCR, Teixeira F, Peñalver P, Costa SPG, Morales JC, Raposo MMM. Designing Antitrypanosomal and Antileishmanial BODIPY Derivatives: A Computational and In Vitro Assessment. Molecules. 2024; 29(9):2072. https://doi.org/10.3390/molecules29092072
Chicago/Turabian StyleGonçalves, Raquel C. R., Filipe Teixeira, Pablo Peñalver, Susana P. G. Costa, Juan C. Morales, and M. Manuela M. Raposo. 2024. "Designing Antitrypanosomal and Antileishmanial BODIPY Derivatives: A Computational and In Vitro Assessment" Molecules 29, no. 9: 2072. https://doi.org/10.3390/molecules29092072
APA StyleGonçalves, R. C. R., Teixeira, F., Peñalver, P., Costa, S. P. G., Morales, J. C., & Raposo, M. M. M. (2024). Designing Antitrypanosomal and Antileishmanial BODIPY Derivatives: A Computational and In Vitro Assessment. Molecules, 29(9), 2072. https://doi.org/10.3390/molecules29092072