
The Benzoylpiperidine Fragment as a Privileged Structure in Medicinal Chemistry: A Comprehensive Review





Abstract
1. Introduction
2. Synthesis of the Benzoylpiperidine Fragment
3. Benzoylpiperidine-Based Small Molecules as Therapeutics and/or Diagnostics
3.1. Cancer
3.1.1. MAGL Inhibitors
3.1.2. Tankyrase Inhibitors
3.1.3. Complex I Inhibitors
3.2. Neuropsychiatric and Neurodegenerative Diseases
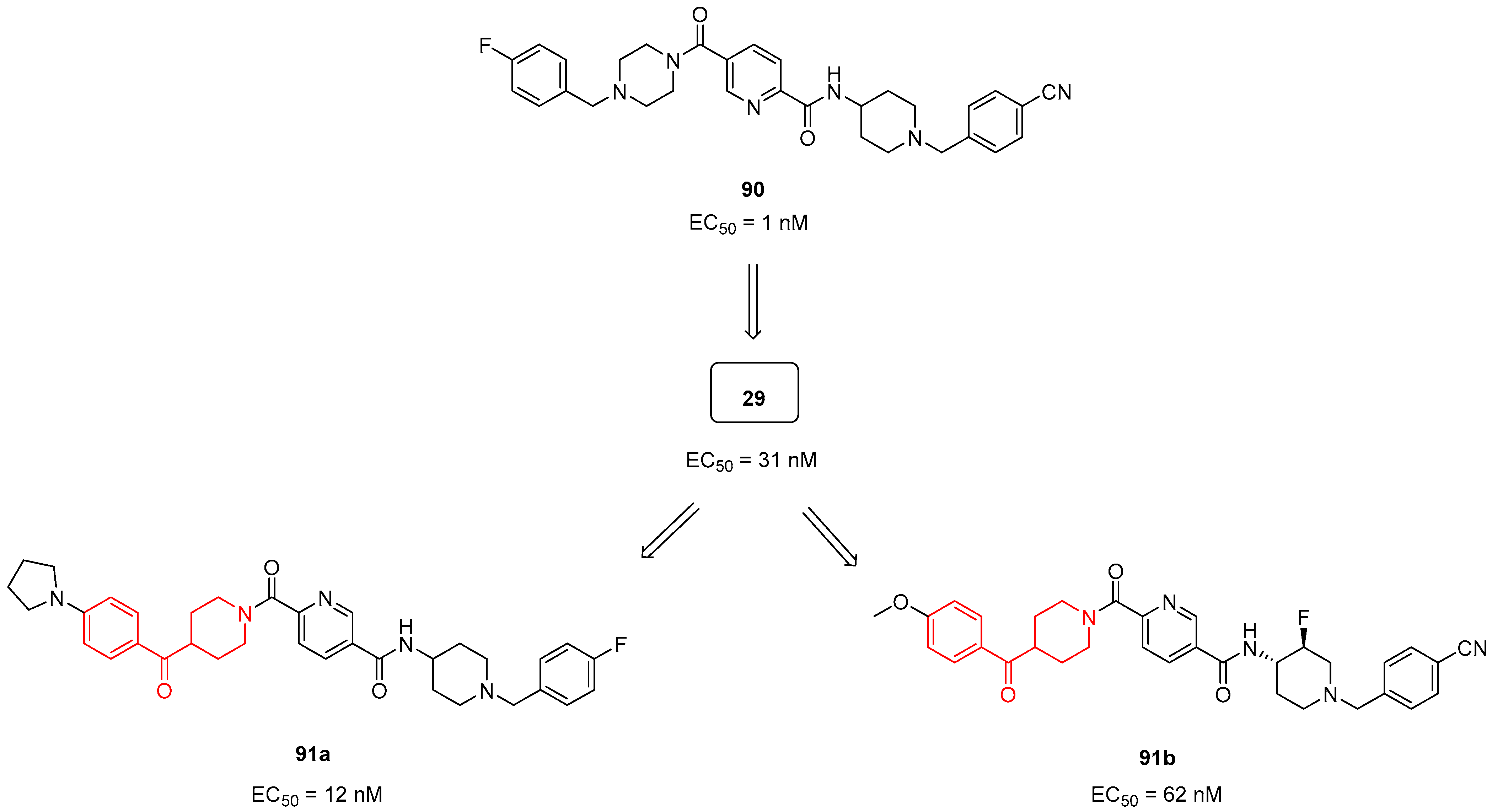
3.2.1. Serotoninergic and Dopaminergic Receptor Ligands
3.2.2. GlyT1 Inhibitors
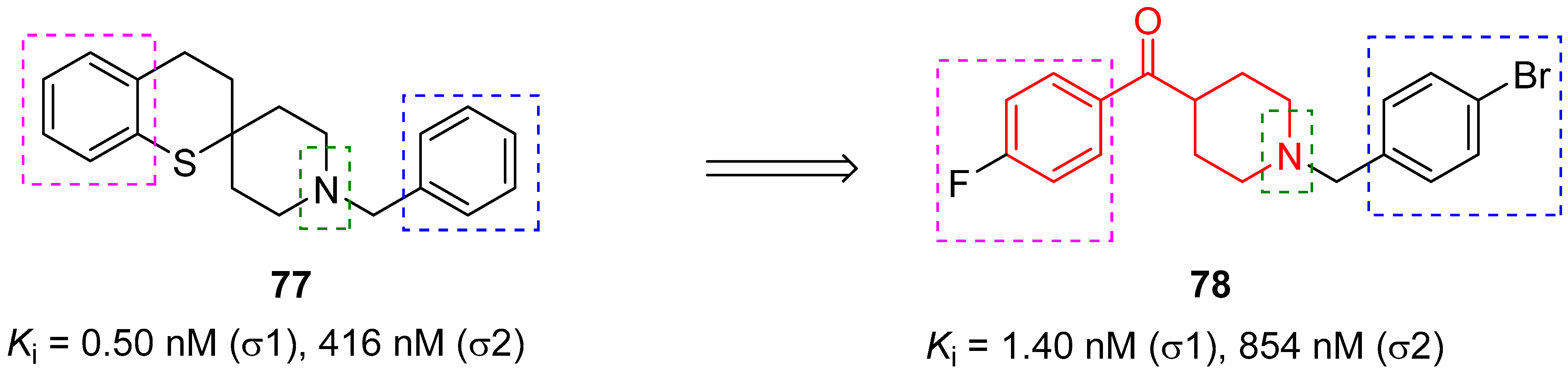
3.2.3. σ1 Receptor Inhibitors
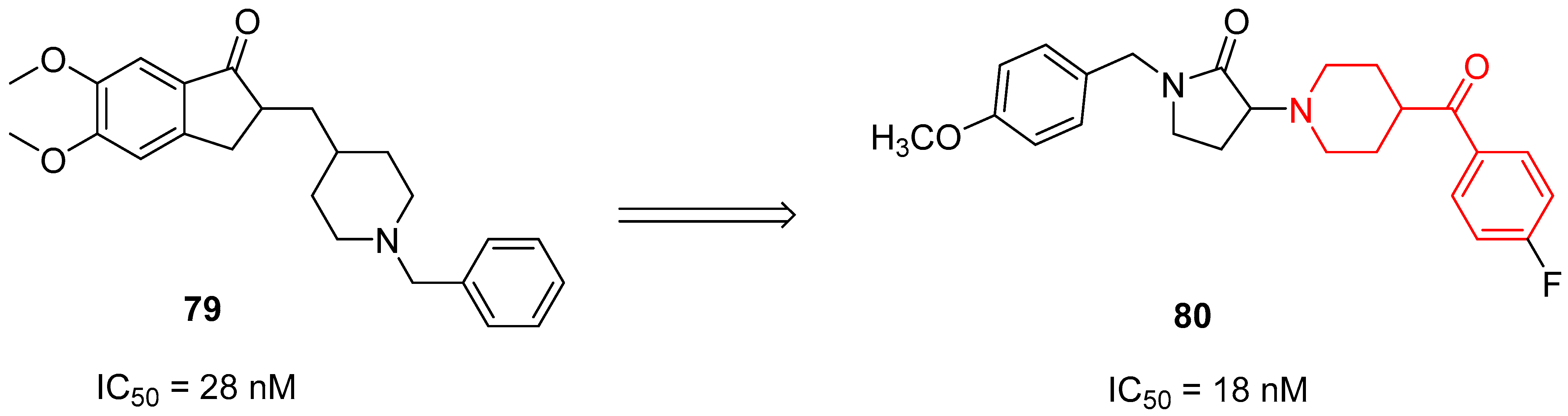
3.2.4. AChE Inhibitors
3.2.5. Other Neuroprotective Agents
3.3. Tuberculosis and Parasites
3.3.1. Antitubercular Agents
3.3.2. Antiparasitic Agents
3.4. Metabolic Syndrome, Diabetes, and Lipid-Related Diseases
3.4.1. SCD-1 Inhibitors
3.4.2. AMPK Activators (Complex I Inhibitors)
3.5. Cardiovascular Diseases
3.5.1. hERG K+ Ligands
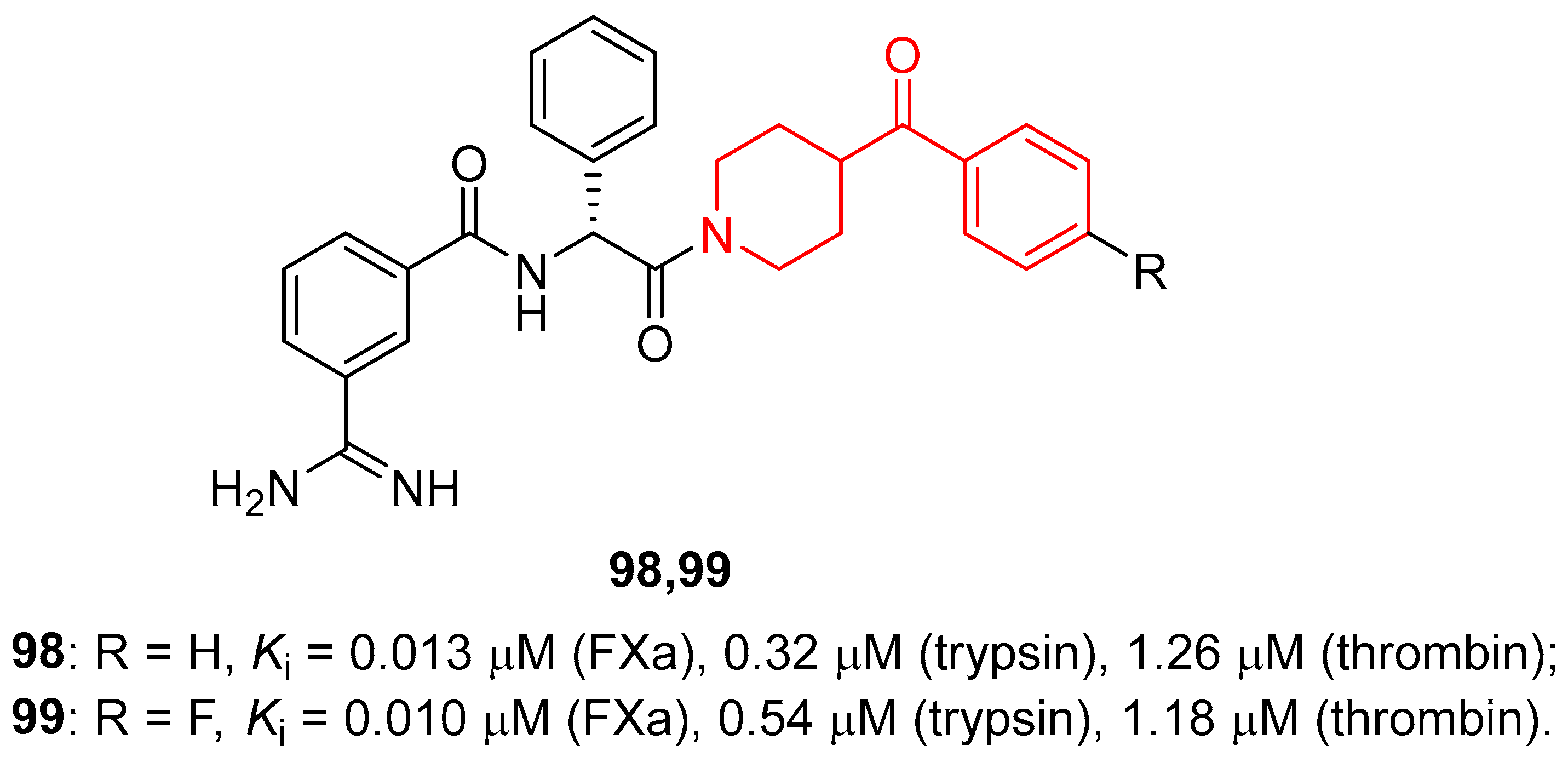
3.5.2. Factor Xa Inhibitors
3.6. Other Disorders
Beta-Adrenoceptor Ligands
3.7. Diagnostic Agents
3.7.1. VAChT Ligands for PET Brain Imaging
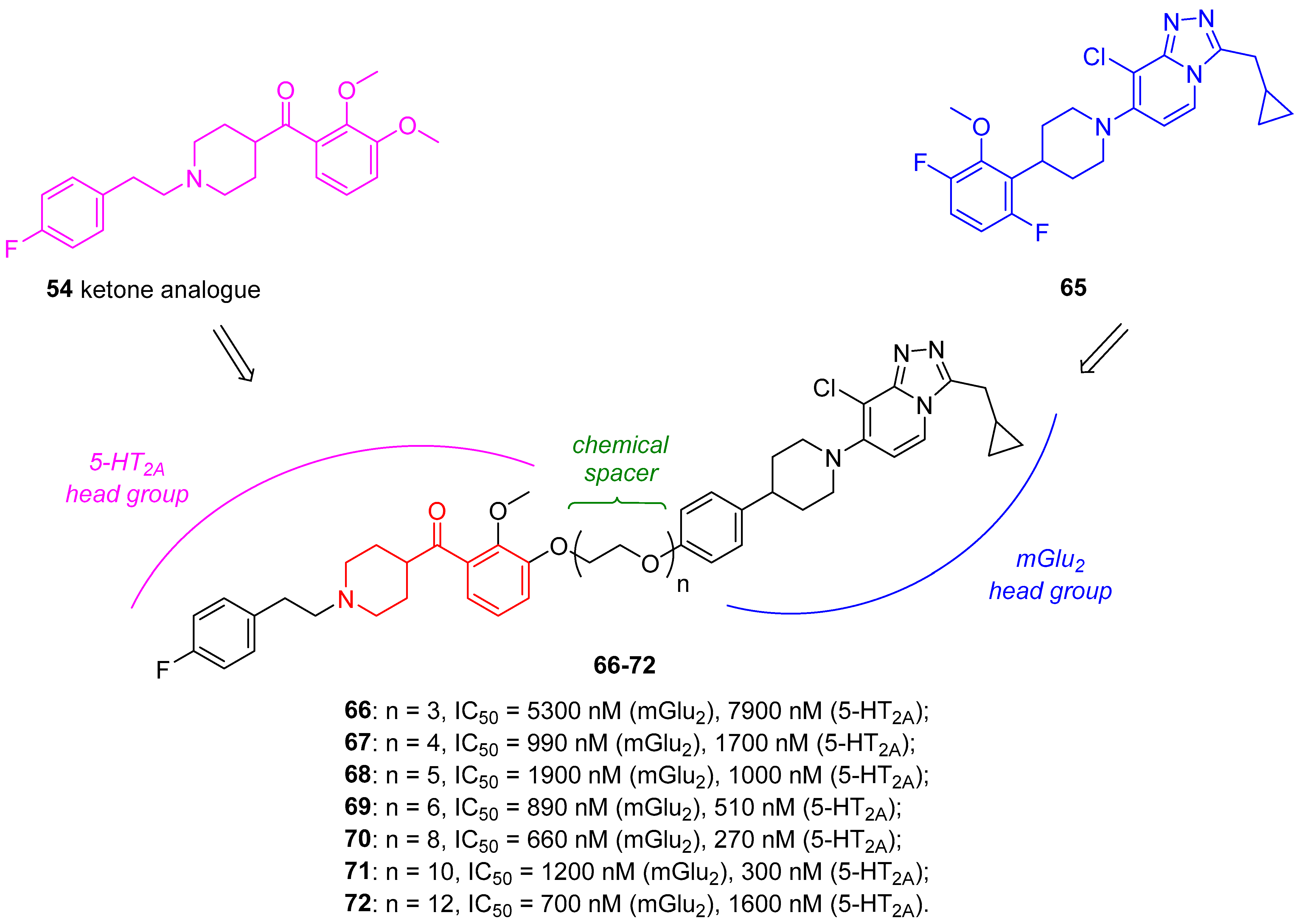
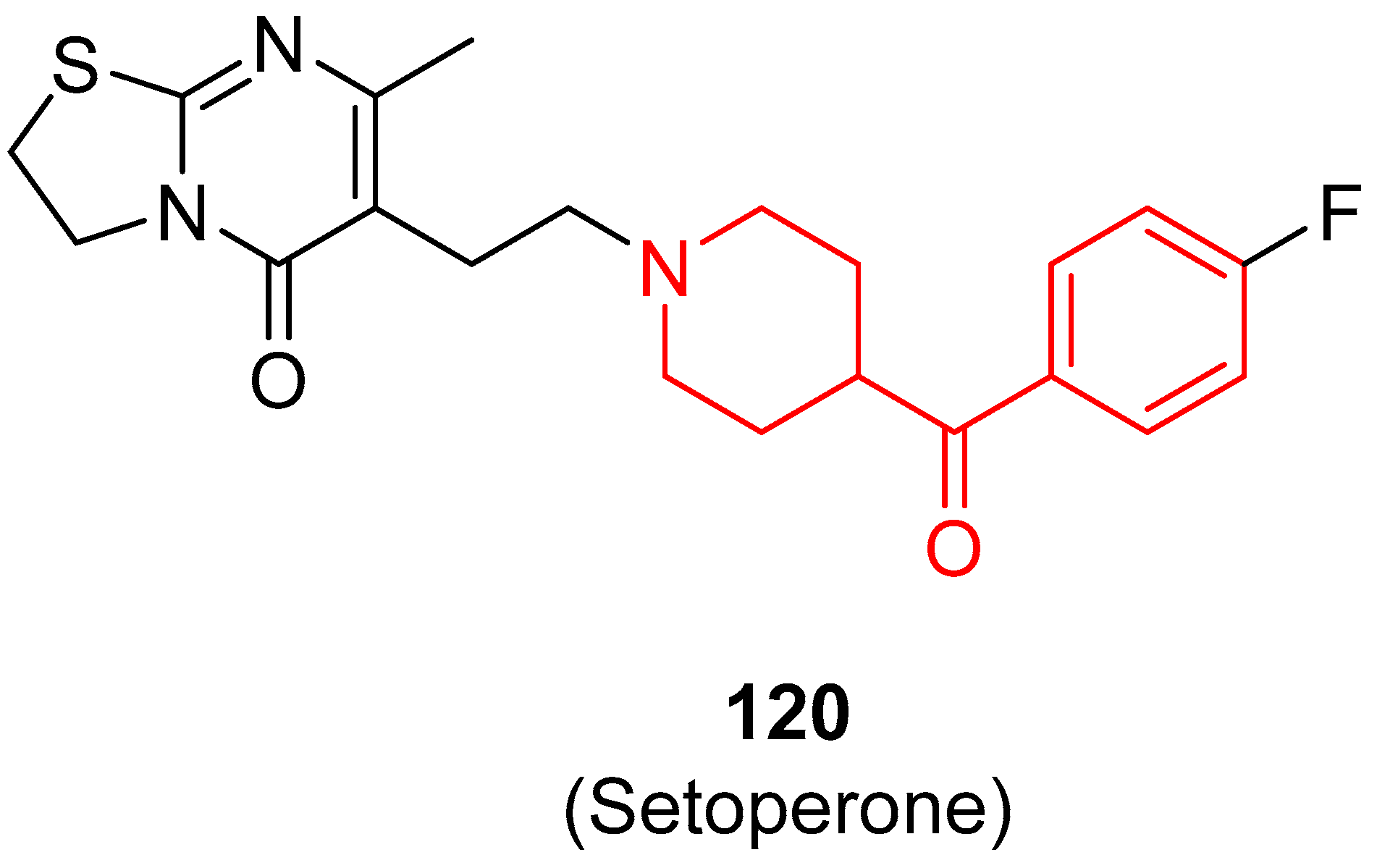
3.7.2. 5-HT2A Ligands for PET or SPECT Brain Imaging
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Guo, L.; Duan, H.; Zhang, L.; Jiang, N.; Zhen, X.; Shen, J. Discovery of 4-Benzoylpiperidine and 3-(Piperidin-4-Yl)Benzo[d]Isoxazole Derivatives as Potential and Selective GlyT1 Inhibitors. RSC Adv. 2015, 5, 40964–40977. [Google Scholar] [CrossRef]
- Yadav, V.D.; Boshoff, H.I.; Trifonov, L.; Roma, J.S.O.; Ioerger, T.R.; Barry, C.E.; Oh, S. Synthesis and Structure–Activity Relationships of a New Class of Oxadiazoles Targeting DprE1 as Antitubercular Agents. ACS Med. Chem. Lett. 2023, 14, 1275–1283. [Google Scholar] [CrossRef]
- Karmacharya, U.; Chaudhary, P.; Lim, D.; Dahal, S.; Awasthi, B.P.; Park, H.D.; Kim, J.-A.; Jeong, B.-S. Synthesis and Anticancer Evaluation of 6-Azacyclonol-2,4,6-Trimethylpyridin-3-Ol Derivatives: M3 Muscarinic Acetylcholine Receptor-Mediated Anticancer Activity of a Cyclohexyl Derivative in Androgen-Refractory Prostate Cancer. Bioorg. Chem. 2021, 110, 104805. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, K.; Dou, F.; Hao, C.; Zhang, Y.; Cao, X.; Gao, L.; Xiong, J.; Liu, X.; Liu, B.-F.; et al. Isoquinolinone Derivatives as Potent CNS Multi-Receptor D2/5-HT1A/5-HT2A/5-HT6/5-HT7 Agents: Synthesis and Pharmacological Evaluation. Eur. J. Med. Chem. 2020, 207, 112709. [Google Scholar] [CrossRef]
- Rolfe, A.; Yao, S.; Nguyen, T.-V.; Omoto, K.; Colombo, F.; Virrankoski, M.; Vaillancourt, F.H.; Yu, L.; Cook, A.; Reynolds, D.; et al. Discovery of 2,6-Dimethylpiperazines as Allosteric Inhibitors of CPS1. ACS Med. Chem. Lett. 2020, 11, 1305–1309. [Google Scholar] [CrossRef]
- Fukuda, T.; Ishiyama, T.; Katagiri, T.; Ueda, K.; Muramatsu, S.; Hashimoto, M.; Aki, A.; Baba, D.; Watanabe, K.; Tanaka, N. Discovery of DS42450411 as a Potent Orally Active Hepcidin Production Inhibitor: Design and Optimization of Novel 4-Aminopyrimidine Derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 3333–3337. [Google Scholar] [CrossRef]
- Migliore, M.; Pontis, S.; Fuentes de Arriba, A.L.; Realini, N.; Torrente, E.; Armirotti, A.; Romeo, E.; Di Martino, S.; Russo, D.; Pizzirani, D.; et al. Second-Generation Non-Covalent NAAA Inhibitors Are Protective in a Model of Multiple Sclerosis. Angew. Chem. Int. Ed. 2016, 55, 11193–11197. [Google Scholar] [CrossRef]
- Jones, A.M. Privileged Structures and Motifs (Synthetic and Natural Scaffolds). In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2–8, pp. 116–152. ISBN 9780128032008. [Google Scholar]
- Duncan, R.L.; Helsley, G.C.; Welstead, W.J.; DaVanzo, J.P.; Funderburk, W.H.; Lunsford, C.D. Aroylpiperidines and Pyrrolidines. New Class of Potent Central Nervous System Depressants. J. Med. Chem. 1970, 13, 1–6. [Google Scholar] [CrossRef]
- Ismaiel, A.M.; Arruda, K.; Teitler, M.; Glennon, R.A. Ketanserin Analogs: The Effect of Structural Modification on 5-HT2 Serotonin Receptor Binding. J. Med. Chem. 1995, 38, 1196–1202. [Google Scholar] [CrossRef]
- Masaguer, C. Butyrophenone Analogues in the Carbazole Series as Potential Atypical Antipsychotics: Synthesis and Determination of Affinities at D2, 5-HT2A, 5-HT2B and 5-HT2C Receptors. Eur. J. Med. Chem. 2000, 35, 83–95. [Google Scholar] [CrossRef]
- Herndon, J.L.; Ismaiel, A.; Ingher, S.P.; Teitler, M.; Glennon, R.A. Ketanserin Analogs: Structure-Affinity Relationships for 5-HT2 and 5-HT1C Serotonin Receptor Binding. J. Med. Chem. 1992, 35, 4903–4910. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Granchi, C.; Rizzolio, F.; Caligiuri, I.; Battistello, V.; Toffoli, G.; Minutolo, F.; Macchia, M.; Martinelli, A. Identification and Characterization of a New Reversible MAGL Inhibitor. Bioorg. Med. Chem. 2014, 22, 3285–3291. [Google Scholar] [CrossRef]
- Carro, L.; Raviña, E.; Domínguez, E.; Brea, J.; Loza, M.I.; Masaguer, C.F. Synthesis and Binding Affinity of Potential Atypical Antipsychotics with the Tetrahydroquinazolinone Motif. Bioorg. Med. Chem. Lett. 2009, 19, 6059–6062. [Google Scholar] [CrossRef]
- Kramer, V.; Herth, M.M.; Santini, M.A.; Palner, M.; Knudsen, G.M.; Rösch, F. Research Letter: Structural Combination of Established 5-HT2A Receptor Ligands: New Aspects of the Binding Mode. Chem. Biol. Drug Des. 2010, 76, 361–366. [Google Scholar] [CrossRef]
- Barceló, M.; Raviña, E.; Varela, M.J.; Brea, J.; Loza, M.I.; Masaguer, C.F. Potential Atypical Antipsychotics: Synthesis, Binding Affinity and SAR of New Heterocyclic Bioisosteric Butyrophenone Analogues as Multitarget Ligands. Medchemcomm 2011, 2, 1194. [Google Scholar] [CrossRef]
- Carro, L.; Torrado, M.; Raviña, E.; Masaguer, C.F.; Lage, S.; Brea, J.; Loza, M.I. Synthesis and Biological Evaluation of a Series of Aminoalkyl-Tetralones and Tetralols as Dual Dopamine/Serotonin Ligands. Eur. J. Med. Chem. 2014, 71, 237–249. [Google Scholar] [CrossRef]
- Wang, W.; Cui, J.; Lu, X.; Padakanti, P.K.; Xu, J.; Parsons, S.M.; Luedtke, R.R.; Rath, N.P.; Tu, Z. Synthesis and In Vitro Biological Evaluation of Carbonyl Group-Containing Analogues for Σ1 Receptors. J. Med. Chem. 2011, 54, 5362–5372. [Google Scholar] [CrossRef]
- Ikome, H.N.; Ntie-Kang, F.; Ngemenya, M.N.; Tu, Z.; Mach, R.H.; Efange, S.M.N. 4-Aroylpiperidines and 4-(α-Hydroxyphenyl)Piperidines as Selective Sigma-1 Receptor Ligands: Synthesis, Preliminary Pharmacological Evaluation and Computational Studies. Chem. Cent. J. 2016, 10, 53. [Google Scholar] [CrossRef]
- Gupta, M.; Ojha, M.; Yadav, D.; Pant, S.; Yadav, R. Novel Benzylated (Pyrrolidin-2-One)/(Imidazolidin-2-One) Derivatives as Potential Anti-Alzheimer’s Agents: Synthesis and Pharmacological Investigations. ACS Chem. Neurosci. 2020, 11, 2849–2860. [Google Scholar] [CrossRef]
- Zheng, Y.; Müller, J.; Kunz, S.; Siderius, M.; Maes, L.; Caljon, G.; Müller, N.; Hemphill, A.; Sterk, G.J.; Leurs, R. 3-Nitroimidazo[1,2-b]Pyridazine as a Novel Scaffold for Antiparasitics with Sub-Nanomolar Anti-Giardia Lamblia Activity. Int. J. Parasitol. Drugs Drug Resist. 2022, 19, 47–55. [Google Scholar] [CrossRef]
- Jenkins, Y.; Sun, T.-Q.; Markovtsov, V.; Foretz, M.; Li, W.; Nguyen, H.; Li, Y.; Pan, A.; Uy, G.; Gross, L.; et al. AMPK Activation through Mitochondrial Regulation Results in Increased Substrate Oxidation and Improved Metabolic Parameters in Models of Diabetes. PLoS ONE 2013, 8, e81870. [Google Scholar] [CrossRef] [PubMed]
- Louvel, J.; Carvalho, J.F.S.; Yu, Z.; Soethoudt, M.; Lenselink, E.B.; Klaasse, E.; Brussee, J.; IJzerman, A.P. Removal of Human Ether-à-Go-Go Related Gene (HERG) K + Channel Affinity through Rigidity: A Case of Clofilium Analogues. J. Med. Chem. 2013, 56, 9427–9440. [Google Scholar] [CrossRef]
- Granchi, C.; Rizzolio, F.; Palazzolo, S.; Carmignani, S.; Macchia, M.; Saccomanni, G.; Manera, C.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Structural Optimization of 4-Chlorobenzoylpiperidine Derivatives for the Development of Potent, Reversible, and Selective Monoacylglycerol Lipase (MAGL) Inhibitors. J. Med. Chem. 2016, 59, 10299–10314. [Google Scholar] [CrossRef]
- Li, Q.; Chai, L.; Dong, G.; Zhang, X.; Du, L. NBD-Based Environment-Sensitive Fluorescent Probes for the Human Ether-a-Go-Go–Related Gene Potassium Channel. Front. Mol. Biosci. 2021, 8, 666605. [Google Scholar] [CrossRef]
- Liebeschuetz, J.W.; Jones, S.D.; Morgan, P.J.; Murray, C.W.; Rimmer, A.D.; Roscoe, J.M.E.; Waszkowycz, B.; Welsh, P.M.; Wylie, W.A.; Young, S.C.; et al. PRO_SELECT: Combining Structure-Based Drug Design and Array-Based Chemistry for Rapid Lead Discovery. 2. The Development of a Series of Highly Potent and Selective Factor Xa Inhibitors. J. Med. Chem. 2002, 45, 1221–1232. [Google Scholar] [CrossRef]
- Barthel, C.; Sorger, D.; Deuther-Conrad, W.; Scheunemann, M.; Schweiger, S.; Jäckel, P.; Roghani, A.; Steinbach, J.; Schüürmann, G.; Sabri, O.; et al. New Systematically Modified Vesamicol Analogs and Their Affinity and Selectivity for the Vesicular Acetylcholine Transporter—A Critical Examination of the Lead Structure. Eur. J. Med. Chem. 2015, 100, 50–67. [Google Scholar] [CrossRef]
- Fu, X.; Tan, P.-Z.; Kula, N.S.; Baldessarini, R.; Tamagnan, G.; Innis, R.B.; Baldwin, R.M. Synthesis, Receptor Potency, and Selectivity of Halogenated Diphenylpiperidines as Serotonin 5-HT 2A Ligands for PET or SPECT Brain Imaging. J. Med. Chem. 2002, 45, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Blanckaert, P.; Vandecapelle, M.; Staelens, L.; Burvenich, I.; Dierckx, R.A.; Slegers, G. Synthesis, Radiosynthesis and Preliminaryin Vivo Evaluation of[123I]-(4-Fluorophenyl){1-[2-(2-Iodophenyl)Ethyl]Piperidin-4-Yl}methanone, a Potential 5-HT2A-Antagonist for SPECT Brain Imaging. J. Label. Compd. Radiopharm. 2004, 47, 591–598. [Google Scholar] [CrossRef]
- Blanckaert, P.; Burvenich, I.; Staelens, L.; Dierckx, R.A.; Slegers, G. Synthesis, Radiosynthesis and in Vivo Evaluation in Mice of [123I]-(4-Fluorophenyl) {1-[2-(4-Iodophenyl)Ethyl]Piperidin-4-Yl}methanone for Visualization of the 5-HT2A Receptor with SPECT. Appl. Radiat. Isot. 2005, 62, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Blanckaert, P.; Burvenich, I.; Devos, F.; Slegers, G. Synthesis Andin Vivo Evaluation in Mice of [123I]-(4-Fluorophenyl)[1-(3-Iodophenethyl)Piperidin-4-Yl]Methanone as a Potential SPECT-Tracer for the Serotonin 5-HT2A Receptor. J. Label. Compd. Radiopharm. 2007, 50, 183–188. [Google Scholar] [CrossRef]
- Shultz, M.D.; Cheung, A.K.; Kirby, C.A.; Firestone, B.; Fan, J.; Chen, C.H.-T.; Chen, Z.; Chin, D.N.; DiPietro, L.; Fazal, A.; et al. Identification of NVP-TNKS656: The Use of Structure–Efficiency Relationships to Generate a Highly Potent, Selective, and Orally Active Tankyrase Inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [Google Scholar] [CrossRef] [PubMed]
- Comoy, C.; Guérin, V.; Pfeiffer, B.; Rettori, M.-C.; Renard, P.; Guillaumet, G. Substituted 3-Amino and/or 3-Aminomethyl-3,4-Dihydro-2H-1-Benzopyrans: Synthesis and Biological Activity. Bioorg. Med. Chem. 2000, 8, 483–495. [Google Scholar] [CrossRef]
- Raviña, E.; Casariego, I.; Masaguer, C.F.; Fontenla, J.A.; Montenegro, G.Y.; Rivas, M.E.; Loza, M.I.; Enguix, M.J.; Villazon, M.; Cadavid, M.I.; et al. Conformationally Constrained Butyrophenones with Affinity for Dopamine (D1, D2, D4) and Serotonin (5-HT2A, 5-HT2B, 5-HT2C) Receptors: Synthesis of Aminomethylbenzo[b]Furanones and Their Evaluation as Antipsychotics. J. Med. Chem. 2000, 43, 4678–4693. [Google Scholar] [CrossRef]
- Caro, Y.; Torrado, M.; Masaguer, C.F.; Raviña, E.; Padín, F.; Brea, J.; Loza, M.I. Chemoenzymatic Synthesis and Binding Affinity of Novel (R)- and (S)-3-Aminomethyl-1-Tetralones, Potential Atypical Antipsychotics. Bioorg. Med. Chem. Lett. 2004, 14, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, M.; Barceló, M.; Carro, L.; Masaguer, C.F.; Raviña, E. Synthesis and Biological Evaluation of New Quinazoline and Cinnoline Derivatives as Potential Atypical Antipsychotics. Chem. Biodivers. 2006, 3, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Barceló, M.; Raviña, E.; Masaguer, C.F.; Domínguez, E.; Areias, F.M.; Brea, J.; Loza, M.I. Synthesis and Binding Affinity of New Pyrazole and Isoxazole Derivatives as Potential Atypical Antipsychotics. Bioorg. Med. Chem. Lett. 2007, 17, 4873–4877. [Google Scholar] [CrossRef] [PubMed]
- Aranda, R.; Villalba, K.; Raviña, E.; Masaguer, C.F.; Brea, J.; Areias, F.; Domínguez, E.; Selent, J.; López, L.; Sanz, F.; et al. Synthesis, Binding Affinity, and Molecular Docking Analysis of New Benzofuranone Derivatives as Potential Antipsychotics. J. Med. Chem. 2008, 51, 6085–6094. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Lapillo, M.; Glasmacher, S.; Bononi, G.; Licari, C.; Poli, G.; el Boustani, M.; Caligiuri, I.; Rizzolio, F.; Gertsch, J.; et al. Optimization of a Benzoylpiperidine Class Identifies a Highly Potent and Selective Reversible Monoacylglycerol Lipase (MAGL) Inhibitor. J. Med. Chem. 2019, 62, 1932–1958. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Bononi, G.; Ferrisi, R.; Gori, E.; Mantini, G.; Glasmacher, S.; Poli, G.; Palazzolo, S.; Caligiuri, I.; Rizzolio, F.; et al. Design, Synthesis and Biological Evaluation of Second-Generation Benzoylpiperidine Derivatives as Reversible Monoacylglycerol Lipase (MAGL) Inhibitors. Eur. J. Med. Chem. 2021, 209, 112857. [Google Scholar] [CrossRef]
- Bononi, G.; Tonarini, G.; Poli, G.; Barravecchia, I.; Caligiuri, I.; Macchia, M.; Rizzolio, F.; Demontis, G.C.; Minutolo, F.; Granchi, C.; et al. Monoacylglycerol Lipase (MAGL) Inhibitors Based on a Diphenylsulfide-Benzoylpiperidine Scaffold. Eur. J. Med. Chem. 2021, 223, 113679. [Google Scholar] [CrossRef]
- Zhong, Y.; Gao, Y.; Xu, Y.; Qi, C.; Wu, B. Synthesis of Novel Aryloxyethylamine Derivatives and Evaluation of Their In Vitro and In Vivo Neuroprotective Activities. Chem. Biodivers. 2020, 17, e2000431. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Gao, N.; Zhu, N.; Lin, Y.; Li, Y.; Chen, M.; You, X.; Lu, Y.; Wan, K.; Jiang, J.-D.; et al. Discovery of the Disubstituted Oxazole Analogues as a Novel Class Anti-Tuberculotic Agents against MDR- and XDR-MTB. Bioorg. Med. Chem. Lett. 2015, 25, 5178–5181. [Google Scholar] [CrossRef] [PubMed]
- Vilums, M.; Overman, J.; Klaasse, E.; Scheel, O.; Brussee, J.; IJzerman, A.P. Understanding of Molecular Substructures That Contribute to HERG K + Channel Blockade: Synthesis and Biological Evaluation of E-4031 Analogues. ChemMedChem 2012, 7, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Nahm, S.; Weinreb, S.M. N-Methoxy-n-Methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Shashack, M.J.; Cunningham, K.A.; Seitz, P.K.; McGinnis, A.; Smith, T.D.; Watson, C.S.; Gilbertson, S.R. Synthesis and Evaluation of Dimeric Derivatives of 5-HT2A Receptor (5-HT2AR) Antagonist M-100907. ACS Chem. Neurosci. 2011, 2, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.A.; Shashack, M.J.; Fox, R.G.; Bubar, M.J.; Rice, K.C.; Watson, C.S.; Cunningham, K.A.; Gilbertson, S.R.; Anastasio, N.C. Novel Bivalent 5-HT 2A Receptor Antagonists Exhibit High Affinity and Potency In Vitro and Efficacy In Vivo. ACS Chem. Neurosci. 2018, 9, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, S.R.; Chen, Y.-C.; Soto, C.A.; Yang, Y.; Rice, K.C.; Cunningham, K.A.; Anastasio, N.C. Synthesis and Activity of Functionalizable Derivatives of the Serotonin (5-HT) 5-HT2A Receptor (5-HT2AR) Antagonist M100907. Bioorg. Med. Chem. Lett. 2018, 28, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Poulie, C.B.M.; Liu, N.; Jensen, A.A.; Bunch, L. Design, Synthesis, and Pharmacological Characterization of Heterobivalent Ligands for the Putative 5-HT2A/MGlu2 Receptor Complex. J. Med. Chem. 2020, 63, 9928–9949. [Google Scholar] [CrossRef] [PubMed]
- Deau, E.; Robin, E.; Voinea, R.; Percina, N.; Satała, G.; Finaru, A.-L.; Chartier, A.; Tamagnan, G.; Alagille, D.; Bojarski, A.J.; et al. Rational Design, Pharmacomodulation, and Synthesis of Dual 5-Hydroxytryptamine 7 (5-HT7)/5-Hydroxytryptamine 2A (5-HT2A) Receptor Antagonists and Evaluation by [18F]-PET Imaging in a Primate Brain. J. Med. Chem. 2015, 58, 8066–8096. [Google Scholar] [CrossRef]
- Tu, Z.; Efange, S.M.N.; Xu, J.; Li, S.; Jones, L.A.; Parsons, S.M.; Mach, R.H. Synthesis and In Vitro and In Vivo Evaluation of 18F-Labeled Positron Emission Tomography (PET) Ligands for Imaging the Vesicular Acetylcholine Transporter. J. Med. Chem. 2009, 52, 1358–1369. [Google Scholar] [CrossRef]
- Yue, X.; Luo, Z.; Liu, H.; Kaneshige, K.; Parsons, S.M.; Perlmutter, J.S.; Tu, Z. Radiosynthesis and Evaluation of a Fluorine-18 Labeled Radioligand Targeting Vesicular Acetylcholine Transporter. Bioorg. Med. Chem. Lett. 2018, 28, 3425–3430. [Google Scholar] [CrossRef]
- Efange, S.M.N.; Khare, A.B.; von Hohenberg, K.; Mach, R.H.; Parsons, S.M.; Tu, Z. Synthesis and in Vitro Biological Evaluation of Carbonyl Group-Containing Inhibitors of Vesicular Acetylcholine Transporter. J. Med. Chem. 2010, 53, 2825–2835. [Google Scholar] [CrossRef]
- Tu, Z.; Wang, W.; Cui, J.; Zhang, X.; Lu, X.; Xu, J.; Parsons, S.M. Synthesis and Evaluation of In Vitro Bioactivity for Vesicular Acetylcholine Transporter Inhibitors Containing Two Carbonyl Groups. Bioorg. Med. Chem. 2012, 20, 4422–4429. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Zhang, X.; Zhang, Z.; Padakanti, P.K.; Jin, H.; Cui, J.; Li, A.; Zeng, D.; Rath, N.P.; Flores, H.; et al. Heteroaromatic and Aniline Derivatives of Piperidines As Potent Ligands for Vesicular Acetylcholine Transporter. J. Med. Chem. 2013, 56, 6216–6233. [Google Scholar] [CrossRef]
- Liu, H.; Jin, H.; Li, J.; Zhang, X.; Kaneshige, K.; Parsons, S.M.; Perlmutter, J.S.; Tu, Z. In Vitro and Ex Vivo Characterization of (−)-TZ659 as a Ligand for Imaging the Vesicular Acetylcholine Transporter. Eur. J. Pharmacol. 2015, 752, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Padakanti, P.K.; Zhang, X.; Jin, H.; Cui, J.; Wang, R.; Li, J.; Flores, H.P.; Parsons, S.M.; Perlmutter, J.S.; Tu, Z. In Vitro and In Vivo Characterization of Two C-11-Labeled PET Tracers for Vesicular Acetylcholine Transporter. Mol. Imaging Biol. 2014, 16, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Padakanti, P.K.; Zhang, X.; Li, J.; Parsons, S.M.; Perlmutter, J.S.; Tu, Z. Syntheses and Radiosyntheses of Two Carbon-11 Labeled Potent and Selective Radioligands for Imaging Vesicular Acetylcholine Transporter. Mol. Imaging Biol. 2014, 16, 765–772. [Google Scholar] [CrossRef]
- Tu, Z.; Zhang, X.; Jin, H.; Yue, X.; Padakanti, P.K.; Yu, L.; Liu, H.; Flores, H.P.; Kaneshige, K.; Parsons, S.M.; et al. Synthesis and Biological Characterization of a Promising F-18 PET Tracer for Vesicular Acetylcholine Transporter. Bioorg. Med. Chem. 2015, 23, 4699–4709. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Jin, H.; Liu, H.; Luo, Z.; Zhang, X.; Kaneshige, K.; Flores, H.P.; Perlmutter, J.S.; Parsons, S.M.; Tu, Z. Synthesis, Resolution, and in Vitro Evaluation of Three Vesicular Acetylcholine Transporter Ligands and Evaluation of the Lead Fluorine-18 Radioligand in a Nonhuman Primate. Org. Biomol. Chem. 2017, 15, 5197–5209. [Google Scholar] [CrossRef]
- Uto, Y.; Ogata, T.; Kiyotsuka, Y.; Ueno, Y.; Miyazawa, Y.; Kurata, H.; Deguchi, T.; Watanabe, N.; Konishi, M.; Okuyama, R.; et al. Novel Benzoylpiperidine-Based Stearoyl-CoA Desaturase-1 Inhibitors: Identification of 6-[4-(2-Methylbenzoyl)Piperidin-1-Yl]Pyridazine-3-Carboxylic Acid (2-Hydroxy-2-Pyridin-3-Ylethyl)Amide and Its Plasma Triglyceride-Lowering Effects in Zucker Fatty Rats. Bioorg. Med. Chem. Lett. 2010, 20, 341–345. [Google Scholar] [CrossRef]
- Sander, K.; Galante, E.; Gendron, T.; Yiannaki, E.; Patel, N.; Kalber, T.L.; Badar, A.; Robson, M.; Johnson, S.P.; Bauer, F.; et al. Development of Fluorine-18 Labeled Metabolically Activated Tracers for Imaging of Drug Efflux Transporters with Positron Emission Tomography. J. Med. Chem. 2015, 58, 6058–6080. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, M.M.; Nomura, D.K. Therapeutic Potential of Monoacylglycerol Lipase Inhibitors. Life Sci. 2013, 92, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic Monoacylglycerol Lipase Blockade Causes Functional Antagonism of the Endocannabinoid System. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Haikarainen, T.; Krauss, S.; Lehtio, L. Tankyrases: Structure, Function and Therapeutic Implications in Cancer. Curr. Pharm. Des. 2014, 20, 6472–6488. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase Inhibition Stabilizes Axin and Antagonizes Wnt Signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. The Many Ways of Wnt in Cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial Complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Pollak, M. Targeting Oxidative Phosphorylation: Why, When, and How. Cancer Cell 2013, 23, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, G.; Wang, P.; Shi, R.; Zhang, Y.; Wen, X.; Sun, H.; Chen, C. Synthesis and Biological Evaluation of Complex I Inhibitor R419 and Its Derivatives as Anticancer Agents in HepG2 Cells. Bioorg. Med. Chem. Lett. 2018, 28, 2957–2960. [Google Scholar] [CrossRef]
- Lin, J.; Liu, W.; Guan, J.; Cui, J.; Shi, R.; Wang, L.; Chen, D.; Liu, Y. Latest Updates on the Serotonergic System in Depression and Anxiety. Front. Synaptic Neurosci. 2023, 15, 1124112. [Google Scholar] [CrossRef] [PubMed]
- Brasso, C.; Colli, G.; Sgro, R.; Bellino, S.; Bozzatello, P.; Montemagni, C.; Villari, V.; Rocca, P. Efficacy of Serotonin and Dopamine Activity Modulators in the Treatment of Negative Symptoms in Schizophrenia: A Rapid Review. Biomedicines 2023, 11, 921. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, S.F. The Role of Serotonin in Eating Disorders. Drugs 1990, 39, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Sinopoli, V.M.; Burton, C.L.; Kronenberg, S.; Arnold, P.D. A Review of the Role of Serotonin System Genes in Obsessive-Compulsive Disorder. Neurosci. Biobehav. Rev. 2017, 80, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.W.; Phebus, L.A.; Cohen, M.L. Serotonin in Migraine: Theories, Animal Models and Emerging Therapies. In Progress in Drug Research; Birkhäuser Basel: Basel, Switzerland, 1998; Volume 51, pp. 219–244. [Google Scholar]
- Graeff, F.G. Translational Approach to the Pathophysiology of Panic Disorder: Focus on Serotonin and Endogenous Opioids. Neurosci. Biobehav. Rev. 2017, 76, 48–55. [Google Scholar] [CrossRef] [PubMed]
- McCorvy, J.D.; Roth, B.L. Structure and Function of Serotonin G Protein-Coupled Receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Staroń, J.; Bugno, R.; Hogendorf, A.S.; Bojarski, A.J. 5-HT1A Receptor Ligands and Their Therapeutic Applications: Review of New Patents. Expert Opin. Ther. Pat. 2018, 28, 679–689. [Google Scholar] [CrossRef]
- Nagatomo, T.; Rashid, M.; Abul Muntasir, H.; Komiyama, T. Functions of 5-HT2A Receptor and Its Antagonists in the Cardiovascular System. Pharmacol. Ther. 2004, 104, 59–81. [Google Scholar] [CrossRef]
- van Rossum, J.M. The Significance of Dopamine-Receptor Blockade for the Mechanism of Action of Neuroleptic Drugs. Arch. Int. Pharmacodyn. Ther. 1966, 160, 492–494. [Google Scholar]
- Brea, J.; Rodrigo, J.; Carrieri, A.; Sanz, F.; Cadavid, M.I.; Enguix, M.J.; Villazón, M.; Mengod, G.; Caro, Y.; Masaguer, C.F.; et al. New Serotonin 5-HT2A, 5-HT2B, and 5-HT2C Receptor Antagonists: Synthesis, Pharmacology, 3D-QSAR, and Molecular Modeling of (Aminoalkyl)Benzo and Heterocycloalkanones. J. Med. Chem. 2002, 45, 54–71. [Google Scholar] [CrossRef]
- Fontenla, J.A.; Osuna, J.; Rosa, E.; Castro, M.E.; G.-Ferreiro, T.; Loza-Garcia, I.; Calleja, J.M.; Sanz, F.; Rodriguez, J. Synthesis and Atypical Antipsychotic Profile of Some 2-(2-Piperidinoethyl)Benzocycloalkanones as Analogs of Butyrophenone. J. Med. Chem. 1994, 37, 2564–2573. [Google Scholar] [CrossRef] [PubMed]
- Masaguer, C.F.; Casariego, I.; Ravina, E. Conformationally Restricted Butyrophenones with Mixed Dopaminergic (D2) and Serotoninergic (5-HT2A) Affinities. Synthesis of 5-Aminoethyl and 6-Aminomethyl-4-Oxotetrahydroindoles as Potential Atypical Antipsychotics. Chem. Pharm. Bull. 1999, 47, 621–632. [Google Scholar] [CrossRef][Green Version]
- Herrick-Davis, K.; Grinde, E.; Harrigan, T.J.; Mazurkiewicz, J.E. Inhibition of Serotonin 5-Hydroxytryptamine2C Receptor Function through Heterodimerization. J. Biol. Chem. 2005, 280, 40144–40151. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, L.P.; Barthet, G.; Gaven, F.; Cassier, E.; Trinquet, E.; Pin, J.-P.; Marin, P.; Dumuis, A.; Bockaert, J.; Banères, J.-L.; et al. G Protein Activation by Serotonin Type 4 Receptor Dimers. J. Biol. Chem. 2011, 286, 9985–9997. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Sharp, T. A Review of Central 5-HT Receptors and Their Function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, P.B. The 5-HT7 Receptor and Disorders of the Nervous System: An Overview. Psychopharmacology 2009, 206, 345–354. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Nativio, P.; Fabbri, A.; Ricceri, L.; Adriani, W.; Lacivita, E.; Leopoldo, M.; Passarelli, F.; Fuso, A.; Laviola, G. Pharmacological Stimulation of the Brain Serotonin Receptor 7 as a Novel Therapeutic Approach for Rett Syndrome. Neuropsychopharmacology 2014, 39, 2506–2518. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, A.; Chaki, S. Metabotropic Glutamate Receptors: Potential Drug Targets for Psychiatric Disorders. Open Med. Chem. J. 2010, 4, 20–36. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a Serotonin/Glutamate Receptor Complex Implicated in Psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef]
- Baki, L.; Fribourg, M.; Younkin, J.; Eltit, J.M.; Moreno, J.L.; Park, G.; Vysotskaya, Z.; Narahari, A.; Sealfon, S.C.; Gonzalez-Maeso, J.; et al. Cross-Signaling in Metabotropic Glutamate 2 and Serotonin 2A Receptor Heteromers in Mammalian Cells. Pflügers Arch.—Eur. J. Physiol. 2016, 468, 775–793. [Google Scholar] [CrossRef]
- Moreno, J.L.; Miranda-Azpiazu, P.; García-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric Signaling through an MGlu2 and 5-HT2A Heteromeric Receptor Complex and Its Potential Contribution to Schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Glycine and N-Methyl-D-Aspartate Receptors: Physiological Significance and Possible Therapeutic Applications. Pharmacol. Rev. 1998, 50, 597–664. [Google Scholar] [PubMed]
- Kantrowitz, J.; Javitt, D.C. Glutamatergic Transmission in Schizophrenia. Curr. Opin. Psychiatry 2012, 25, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.J.; Knutsen, L.J.S.; Williams, M. Emerging Opportunities for Antipsychotic Drug Discovery in the Postgenomic Era. J. Med. Chem. 2008, 51, 1077–1107. [Google Scholar] [CrossRef] [PubMed]
- Sur, C.; Kinney, G. Glycine Transporter 1 Inhibitors and Modulation of NMDA Receptor-Mediated Excitatory Neurotransmission. Curr. Drug Targets 2007, 8, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Raiteri, M. Functional ‘Glial’ GLYT1 Glycine Transporters Expressed in Neurons. J. Neurochem. 2010, 114, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Pinard, E.; Alanine, A.; Alberati, D.; Bender, M.; Borroni, E.; Bourdeaux, P.; Brom, V.; Burner, S.; Fischer, H.; Hainzl, D.; et al. Selective GlyT1 Inhibitors: Discovery of [4-(3-Fluoro-5-Trifluoromethylpyridin-2-Yl)Piperazin-1-Yl][5-Methanesulfonyl-2-((S)-2,2,2-Trifluoro-1-Methylethoxy)Phenyl]Methanone (RG1678), a Promising Novel Medicine To Treat Schizophrenia. J. Med. Chem. 2010, 53, 4603–4614. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhen, X.; Duan, H.; Guo, L.; Zhang, L.; Zhu, L. Piperidine Compounds, and Preparation Method, Pharmaceutical Compositions and Use Thereof. U.S. Patent No. CN104211635A, 17 December 2014. [Google Scholar]
- Bourrie, B.; Bribes, E.; Derocq, J.-M.; Vidal, H.; Casellas, P. Sigma Receptor Ligands: Applications in Inflammation and Oncology. Curr. Opin. Investig. Drugs 2004, 5, 1158–1163. [Google Scholar]
- Bowen, W.D. Sigma Receptors: Recent Advances and New Clinical Potentials. Pharm. Acta Helv. 2000, 74, 211–218. [Google Scholar] [CrossRef]
- Ogawa, K.; Shiba, K.; Akhter, N.; Yoshimoto, M.; Washiyama, K.; Kinuya, S.; Kawai, K.; Mori, H. Evaluation of Radioiodinated Vesamicol Analogs for Sigma Receptor Imaging in Tumor and Radionuclide Receptor Therapy. Cancer Sci. 2009, 100, 2188–2192. [Google Scholar] [CrossRef]
- John, C.S.; Bowen, W.D.; Varma, V.M.; McAfee, J.G.; Moody, T.W. Sigma Receptors Are Expressed in Human Non-Small Cell Lung Carcinoma. Life Sci. 1995, 56, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Vilner, B.J.; John, C.S.; Bowen, W.D. Sigma-1 and Sigma-2 Receptors Are Expressed in a Wide Variety of Human and Rodent Tumor Cell Lines. Cancer Res. 1995, 55, 408–413. [Google Scholar] [PubMed]
- John, C.S.; Gulden, M.E.; Li, J.; Bowen, W.D.; McAfee, J.G.; Thakur, M.L. Synthesis, In Vitro Binding, and Tissue Distribution of Radioiodinated 2-[125I]N-(N-Benzylpiperidin-4-Yl)-2-Iodo Benzamide, 2-[125I]BP: A Potential σ Receptor Marker for Human Prostate Tumors. Nucl. Med. Biol. 1998, 25, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, W.; Giannella, M.; Piergentili, A.; Pigini, M.; Brasili, L.; Di Toro, R.; Rossetti, L.; Spampinato, S.; Melchiorre, C. 1′-Benzyl-3,4-Dihydrospiro[2H-1-Benzothiopyran-2,4′-Piperidine] (Spipethiane), a Potent and Highly Selective σ 1 Ligand. J. Med. Chem. 1998, 41, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Kumar, A.; Prasun, C.; Nair, M.S.; Kini, S.G.; Yadav, D.; Nain, S. Design, Synthesis, Extra-Precision Docking, and Molecular Dynamics Simulation Studies of Pyrrolidin-2-One Derivatives as Potential Acetylcholinesterase Inhibitors. J. Biomol. Struct. Dyn. 2023, 41, 6282–6294. [Google Scholar] [CrossRef] [PubMed]
- Dye, C.; Williams, B.G. The Population Dynamics and Control of Tuberculosis. Science 2010, 328, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, G.; Raviglione, M.C.; Maher, D. Scale up: Meeting Targets in Global Tuberculosis Control. Lancet 2004, 363, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Maitre, T.; Baulard, A.; Aubry, A.; Veziris, N. Optimizing the Use of Current Antituberculosis Drugs to Overcome Drug Resistance in Mycobacterium Tuberculosis. Infect. Dis. Now 2023, 54, 104807. [Google Scholar] [CrossRef]
- Thompson, R.C.A. Giardiasis as a Re-Emerging Infectious Disease and Its Zoonotic Potential. Int. J. Parasitol. 2000, 30, 1259–1267. [Google Scholar] [CrossRef]
- el-Sayad, M.H.; Lotfy, W.M.; El-Kholy, S.M.; Yehia, M.A.H. Efficacy of Praziquantel against Giardia Lamblia in Rats: Parasitological, Pathological and Therapeutic Study. J. Egypt. Soc. Parasitol. 2002, 32, 201–218. [Google Scholar] [PubMed]
- Rosenblatt, J.E. Antiparasitic Agents. Mayo Clin. Proc. 1999, 74, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Vanelle, P.; Maldonado, J.; Gasquet, M.; Delmas, F.; Timon-David, P.; Jentzer, O.; Crozet, M.P. Studies on Antiparasitic Agents: Effect of the Lactam Nucleus Substitution in the 2-Position on the In-Vitro Activity of New 5-Nitroimidazoles. J. Pharm. Pharmacol. 2011, 43, 735–736. [Google Scholar] [CrossRef] [PubMed]
- Upcroft, J.A.; Campbell, R.W.; Benakli, K.; Upcroft, P.; Vanelle, P. Efficacy of New 5-Nitroimidazoles against Metronidazole-Susceptible and -Resistant Giardia, Trichomonas, and Entamoeba Spp. Antimicrob. Agents Chemother. 1999, 43, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Tomcufcik, A.; Izzo, P.; Fabio, P. 6-Substituted 3-nitroimidazo[1,2-b]pyridazines and Method of Preparing Same. U.S. Patent No. US3828041, 16 August 1974. [Google Scholar]
- Dobrzyn, A.; Ntambi, J.M. Stearoyl-CoA Desaturase as a New Drug Target for Obesity Treatment. Obes. Rev. 2005, 6, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Ntambi, J.M.; Miyazaki, M. Recent Insights into Stearoyl-CoA Desaturase-1. Curr. Opin. Lipidol. 2003, 14, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, L.; Schmidt, R.E.; Su, C.; Huang, X.; Gould, K.; Cao, G. Characterization of HSCD5, a Novel Human Stearoyl-CoA Desaturase Unique to Primates. Biochem. Biophys. Res. Commun. 2005, 332, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ge, L.; Parimoo, S.; Stenn, K.; Prouty, S.M. Human Stearoyl-CoA Desaturase: Alternative Transcripts Generated from a Single Gene by Usage of Tandem Polyadenylation Sites. Biochem. J. 1999, 340, 255. [Google Scholar] [CrossRef]
- Miyazaki, M.; Kim, Y.-C.; Gray-Keller, M.P.; Attie, A.D.; Ntambi, J.M. The Biosynthesis of Hepatic Cholesterol Esters and Triglycerides Is Impaired in Mice with a Disruption of the Gene for Stearoyl-CoA Desaturase 1. J. Biol. Chem. 2000, 275, 30132–30138. [Google Scholar] [CrossRef]
- Flowers, M.T.; Ntambi, J.M. Role of Stearoyl-Coenzyme A Desaturase in Regulating Lipid Metabolism. Curr. Opin. Lipidol. 2008, 19, 248–256. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An Emerging Drug Target for Diabetes and the Metabolic Syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef]
- Hitoshi, Y.; Jenkins, Y.; Markovtsov, V.; Kinsella, T.; Sun, T. Methods for Using and Biomarkers for AMPK-Activating Compounds. U.S. Patent No. US2015087673A1, 26 March 2015. [Google Scholar]
- Shaw, S.J.; Goff, D.A.; Carroll, D.C.; Singh, R.; Sweeny, D.J.; Park, G.; Jenkins, Y.; Markovtsov, V.; Sun, T.-Q.; Issakani, S.D.; et al. Structure Activity Relationships Leading to the Identification of the Indirect Activator of AMPK, R419. Bioorg. Med. Chem. 2022, 71, 116951. [Google Scholar] [CrossRef]
- Shaw, S.J.; Goff, D.A.; Boralsky, L.A.; Singh, R.; Sweeny, D.J.; Park, G.; Sun, T.-Q.; Jenkins, Y.; Markovtsov, V.; Issakani, S.D.; et al. Optimization of Pharmacokinetic and In Vitro Safety Profile of a Series of Pyridine Diamide Indirect AMPK Activators. J. Med. Chem. 2023, 66, 17086–17104. [Google Scholar] [CrossRef] [PubMed]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The HERG K+ Channel: Target and Antitarget Strategies in Drug Development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef]
- Yu, Z.; van Veldhoven, J.P.D.; Louvel, J.; ’t Hart, I.M.E.; Rook, M.B.; van der Heyden, M.A.G.; Heitman, L.H.; IJzerman, A.P. Structure–Affinity Relationships (SARs) and Structure–Kinetics Relationships (SKRs) of K v 11.1 Blockers. J. Med. Chem. 2015, 58, 5916–5929. [Google Scholar] [CrossRef]
- Jehle, J.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Novel Roles for HERG K+ Channels in Cell Proliferation and Apoptosis. Cell Death Dis. 2011, 2, e193. [Google Scholar] [CrossRef]
- Gross, P.L.; Weitz, J.I. New Antithrombotic Drugs. Clin. Pharmacol. Ther. 2009, 86, 139–146. [Google Scholar] [CrossRef]
- Pinto, D.J.P.; Smallheer, J.M.; Cheney, D.L.; Knabb, R.M.; Wexler, R.R. Factor Xa Inhibitors: Next-Generation Antithrombotic Agents. J. Med. Chem. 2010, 53, 6243–6274. [Google Scholar] [CrossRef]
- Jones, S.D.; Liebeschuetz, J.W.; Morgan, P.J.; Murray, C.W.; Rimmer, A.D.; Roscoe, J.M.; Waszkowycz, B.; Welsh, P.M.; Wylie, W.A.; Young, S.C.; et al. The Design of Phenylglycine Containing Benzamidine Carboxamides as Potent and Selective Inhibitors of Factor Xa. Bioorg. Med. Chem. Lett. 2001, 11, 733–736. [Google Scholar] [CrossRef]
- Hieble, J.P. Recent Advances in Identification and Characterization of β-Adrenoceptor Agonists and Antagonists. Curr. Top. Med. Chem. 2007, 7, 207–216. [Google Scholar] [CrossRef]
- Kolb, P.; Rosenbaum, D.M.; Irwin, J.J.; Fung, J.J.; Kobilka, B.K.; Shoichet, B.K. Structure-Based Discovery of β2-Adrenergic Receptor Ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 6843–6848. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C.; Breuiller-Fouché, M.; Mercier, F.J.; Leroy, M.J.; Loustalot, C.; Naline, E.; Frydman, R.; Croci, T.; Morcillo, E.J.; Advenier, C.; et al. The Human Near-term Myometrial β3-adrenoceptor but Not the β2-adrenoceptor Is Resistant to Desensitisation after Sustained Agonist Stimulation. Br. J. Pharmacol. 2004, 141, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yoshizato, T.; Kawarabayashi, T. Investigation of β2-adrenoceptor Subtype Selectivity and Organ Specificity for Bedoradrine (KUR-1246), a Novel Tocolytic Beta-adrenergic Receptor Stimulant. J. Obstet. Gynaecol. Res. 2009, 35, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Doggrell, S.A. Recent Pharmacological Advances in the Treatment of Preterm Membrane Rupture, Labour and Delivery. Expert Opin. Pharmacother. 2004, 5, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Tasler, S.; Baumgartner, R.; Aschenbrenner, A.; Ammendola, A.; Wolf, K.; Wieber, T.; Schachtner, J.; Blisse, M.; Quotschalla, U.; Ney, P. A VHTS Approach for the Identification of β-Adrenoceptor Ligands. Bioorg. Med. Chem. Lett. 2010, 20, 3399–3404. [Google Scholar] [CrossRef]
- Bullock, R.; Touchon, J.; Bergman, H.; Gambina, G.; He, Y.; Rapatz, G.; Nagel, J.; Lane, R. Rivastigmine and Donepezil Treatment in Moderate to Moderately-Severe Alzheimer’s Disease over a 2-Year Period. Curr. Med. Res. Opin. 2005, 21, 1317–1327. [Google Scholar] [CrossRef]
- Bravo, D.T.; Kolmakova, N.G.; Parsons, S.M. Choline Is Transported by Vesicular Acetylcholine Transporter. J. Neurochem. 2004, 91, 766–768. [Google Scholar] [CrossRef]
- Lemaire, C.; Cantineau, R.; Guillaume, M.; Plenevaux, A.; Christiaens, L. Fluorine-18-Altanserin: A Radioligand for the Study of Serotonin Receptors with PET: Radiolabeling and in Vivo Biologic Behavior in Rats. J. Nucl. Med. 1991, 32, 2266–2272. [Google Scholar]
- Lundkvist, C.; Halldin, C.; Ginovart, N.; Nyberg, S.; Swahn, C.-G.; Carr, A.A.; Brunner, F.; Farde, L. [11C]MDL 100907, a Radioligand for Selective Imaging of 5-HT2A Receptors with Positron Emission Tomography. Life Sci. 1996, 58, 187–192. [Google Scholar] [CrossRef]
- Meyer, J.H.; Kapur, S.; Houle, S.; DaSilva, J.; Owczarek, B.; Brown, G.M.; Wilson, A.A.; Kennedy, S.H. Prefrontal Cortex 5-HT2 Receptors in Depression: An [18F]Setoperone PET Imaging Study. Am. J. Psychiatry 1999, 156, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Rosales Martínez, A.; Rodríguez-García, I.; López-Martínez, J.L. Divergent Strategy in Marine Tetracyclic Meroterpenoids Synthesis. Mar. Drugs 2021, 19, 273. [Google Scholar] [CrossRef] [PubMed]
- Rosales Martínez, A.; Rodríguez-Maecker, R.N.; Rodríguez-García, I. Unifying the Synthesis of a Whole Family of Marine Meroterpenoids through a Biosynthetically Inspired Sequence of 1,2-Hydride and Methyl Shifts as Key Step. Mar. Drugs 2023, 21, 118. [Google Scholar] [CrossRef] [PubMed]
































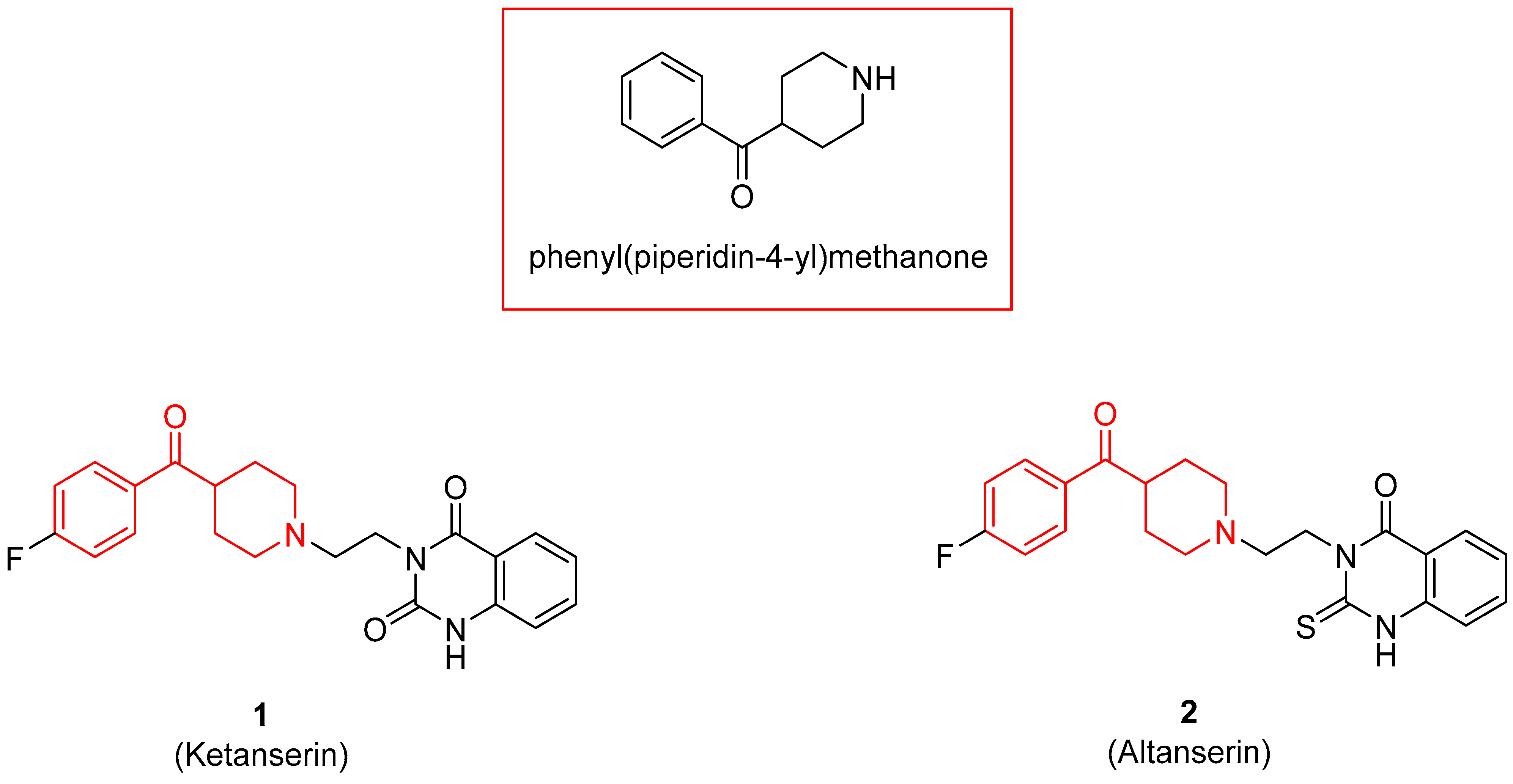{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
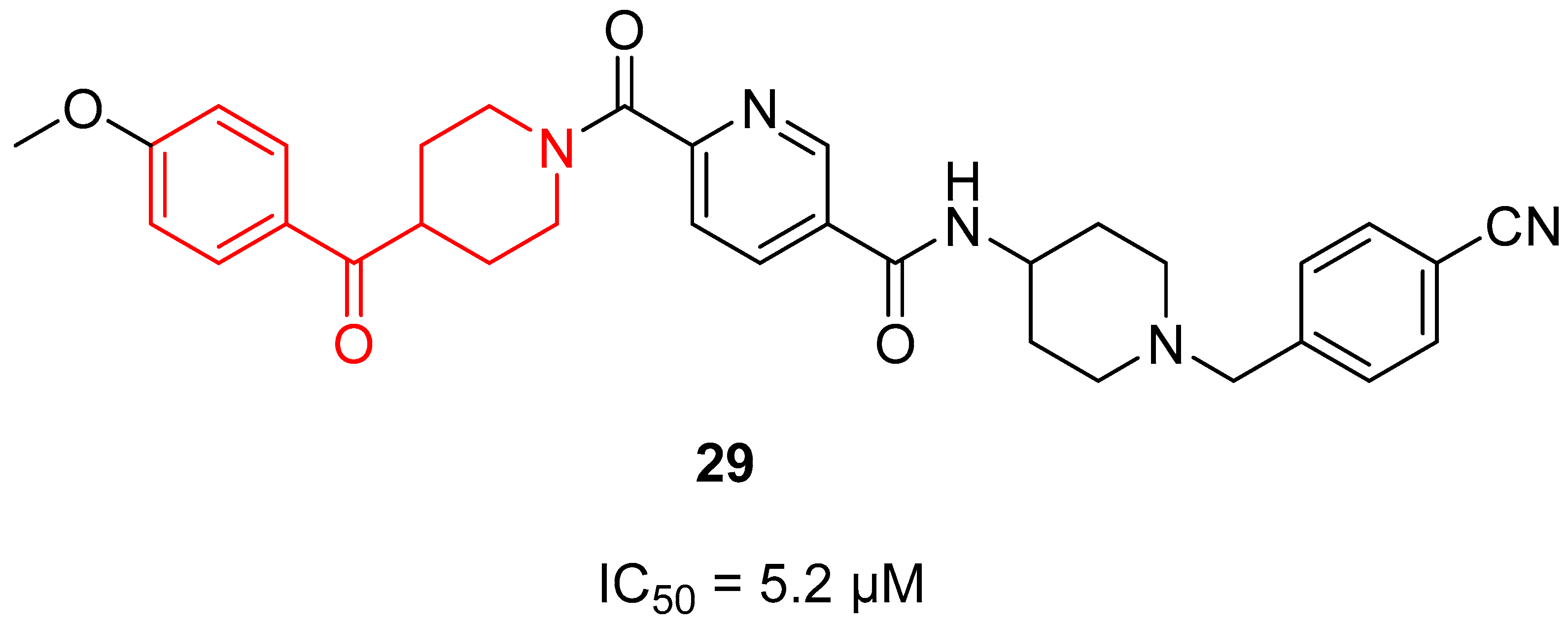{kind=link}
{kind=link}
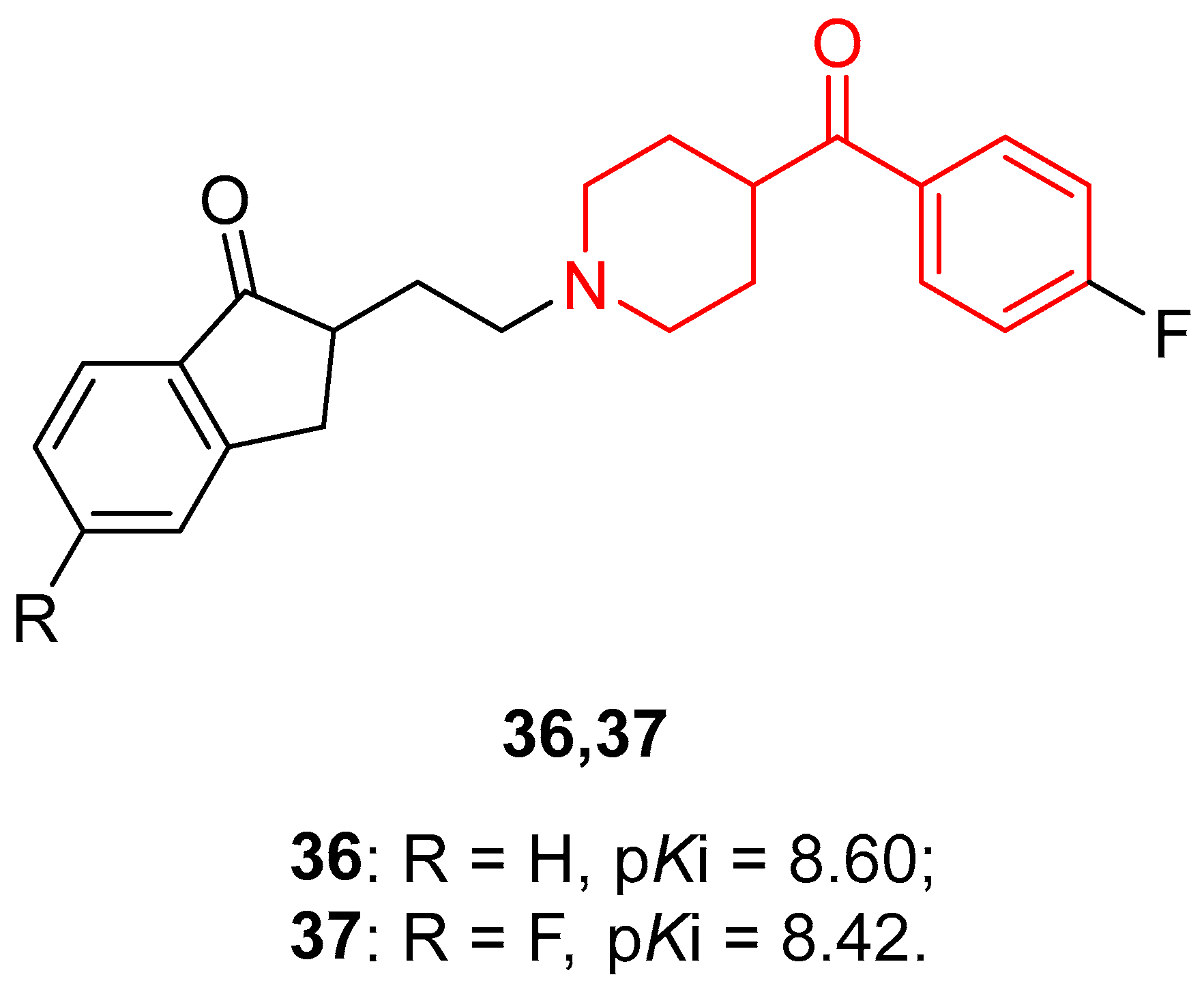{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
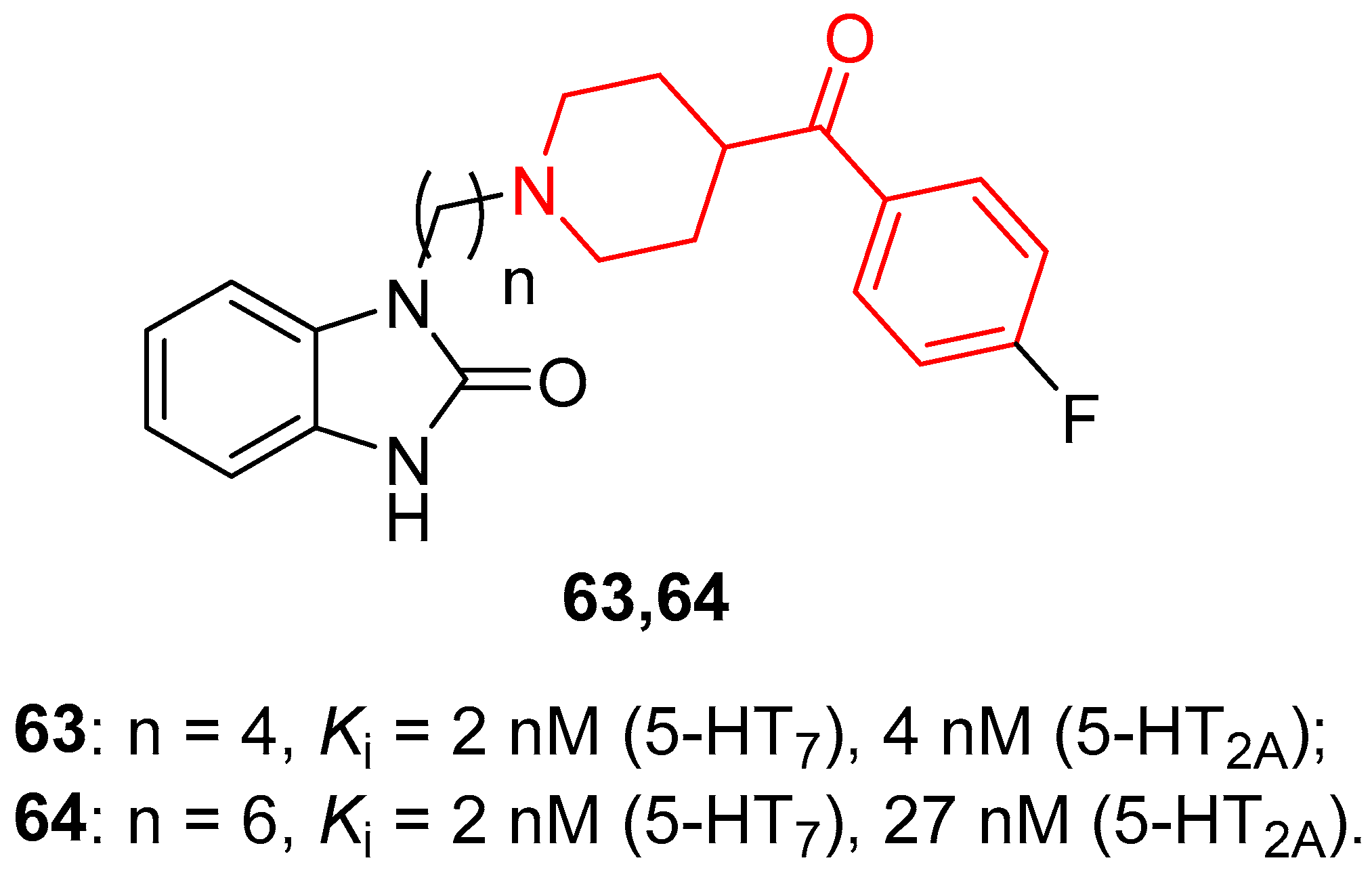{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Ar | Compound | Activity |
 | 34 | pKi = 8.04 (5-HT2A), 6.25 (D2) |
 | 38: X = Y = Z = C, R1 = R2 = H | pKi = 8.23 (5-HT2A), 6.98 (D2) * |
| 39: X = Y = Z = C, R1 = R2 = OCH3 | pKi = 8.25 (5-HT2A), 6.00 (D2) * | |
| 40: X = Z = N, Y = C, R2 = H | Ki = 32 nM (5-HT2A), 160 nM (D2) | |
| 41: X = C, Y = Z = N, R1 = CH3 | Ki = 9950 nM (5-HT2A), >10,000 nM (D2) | |
| 42: X = Z = N, Y = C, R2 = SCH3. | pKi = 6.99 (5-HT2A) | |
 | 35: X = C, Y = O, R3 = R4 = H | pKi = 7.29 (5-HT2A), 7.02 (D2) |
| 47: X = C, Y = O, R3 = COOCH3, R4 = H | pKi = 7.59 (5-HT2A), <5 (D2) | |
| 48: X = C, Y = O, R3 = H, R4 = Ph | pKi = 7.76 (5-HT2A), <5 (D2) | |
| 49: X = C, Y = O, R3 = CH3, R4 = COOCH2CH3 | pKi = 7.66 (5-HT2A), <5 (D2) | |
| 50: X = N, Y = O, R4 = CH3 | pKi = 6.90 (5-HT2A), <5 (D2) | |
 | 51 | pKi = 6.55 (5-HT2A), <5 (D2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bononi, G.; Lonzi, C.; Tuccinardi, T.; Minutolo, F.; Granchi, C. The Benzoylpiperidine Fragment as a Privileged Structure in Medicinal Chemistry: A Comprehensive Review. Molecules 2024, 29, 1930. https://doi.org/10.3390/molecules29091930
Bononi G, Lonzi C, Tuccinardi T, Minutolo F, Granchi C. The Benzoylpiperidine Fragment as a Privileged Structure in Medicinal Chemistry: A Comprehensive Review. Molecules. 2024; 29(9):1930. https://doi.org/10.3390/molecules29091930
Chicago/Turabian StyleBononi, Giulia, Chiara Lonzi, Tiziano Tuccinardi, Filippo Minutolo, and Carlotta Granchi. 2024. "The Benzoylpiperidine Fragment as a Privileged Structure in Medicinal Chemistry: A Comprehensive Review" Molecules 29, no. 9: 1930. https://doi.org/10.3390/molecules29091930
APA StyleBononi, G., Lonzi, C., Tuccinardi, T., Minutolo, F., & Granchi, C. (2024). The Benzoylpiperidine Fragment as a Privileged Structure in Medicinal Chemistry: A Comprehensive Review. Molecules, 29(9), 1930. https://doi.org/10.3390/molecules29091930