
Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
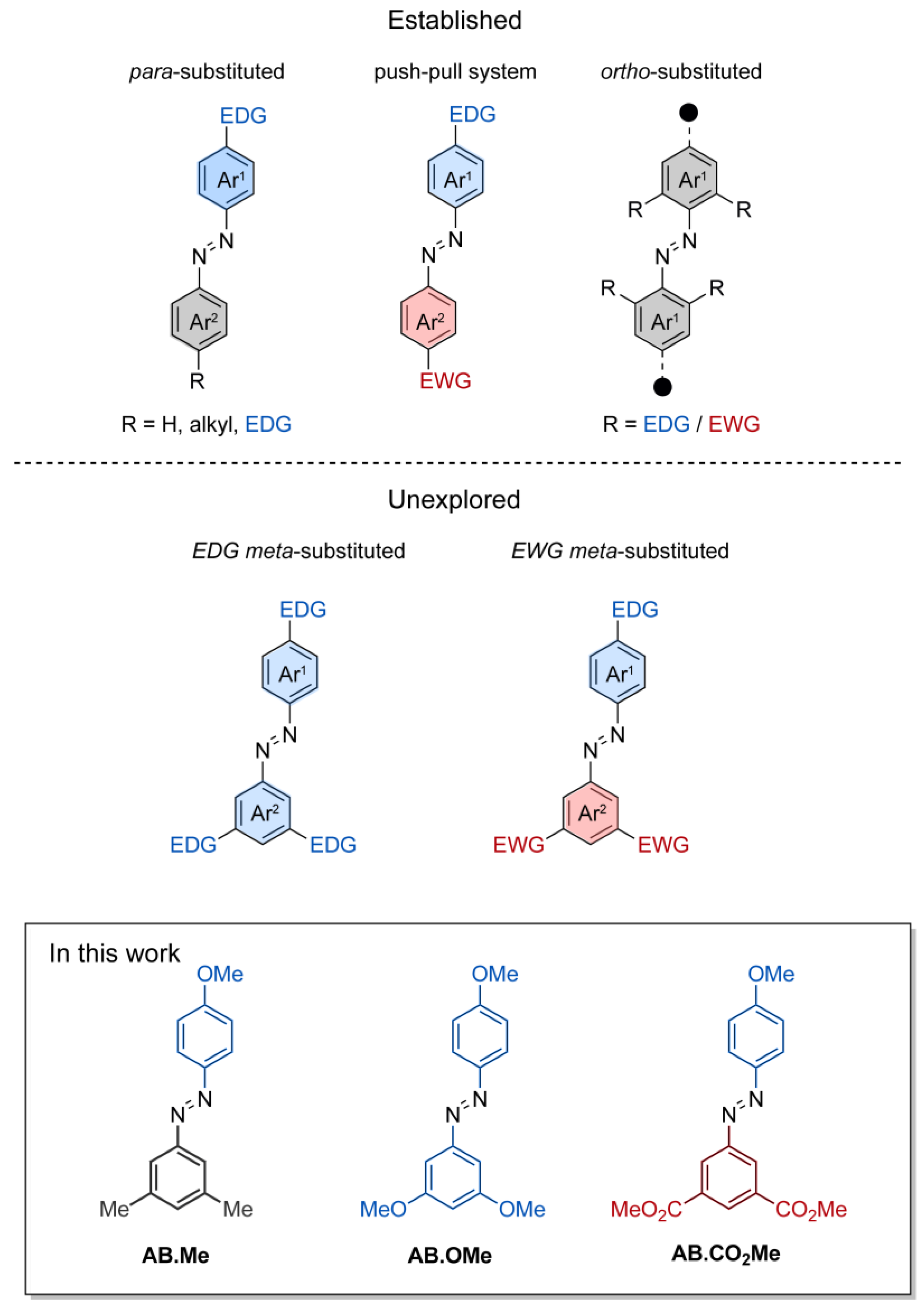
2.1. Design and Synthesis
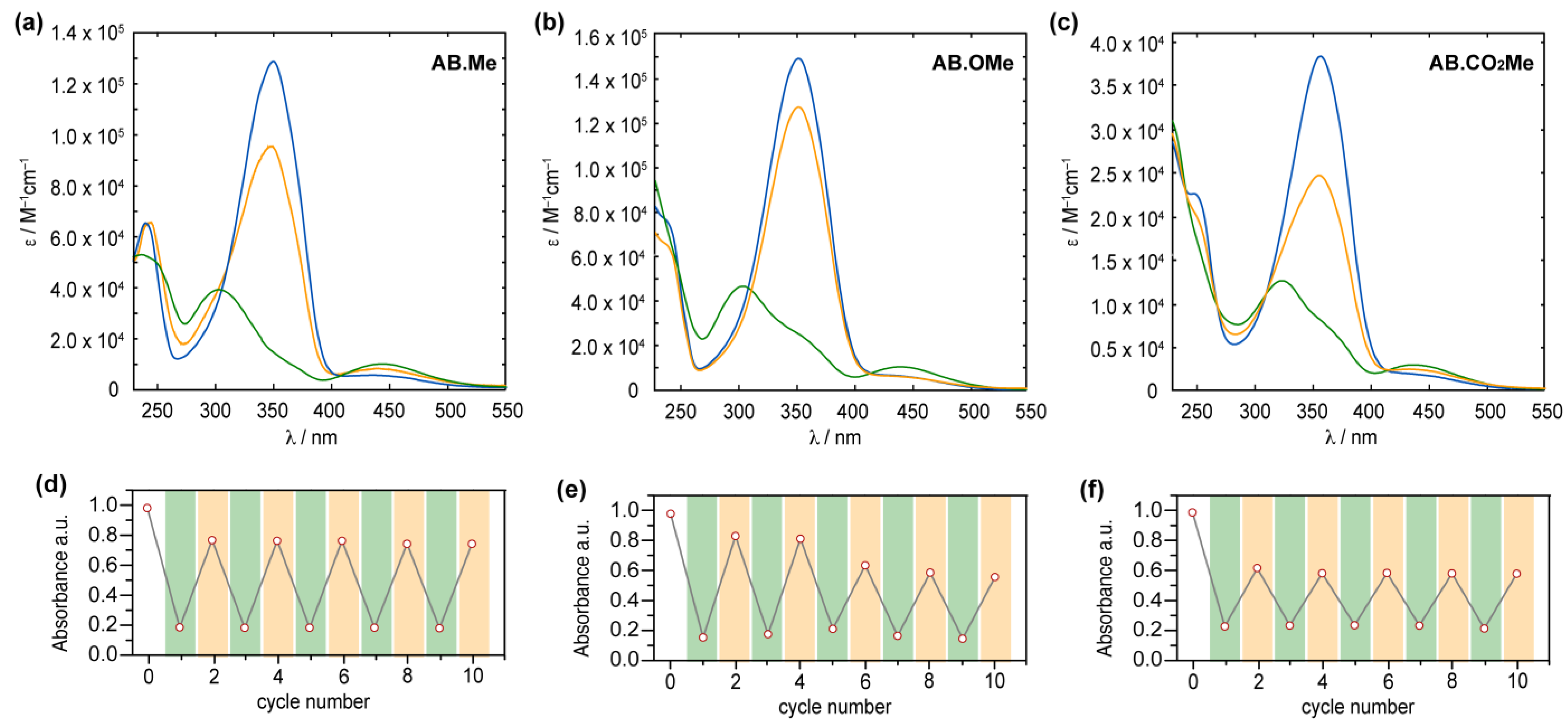
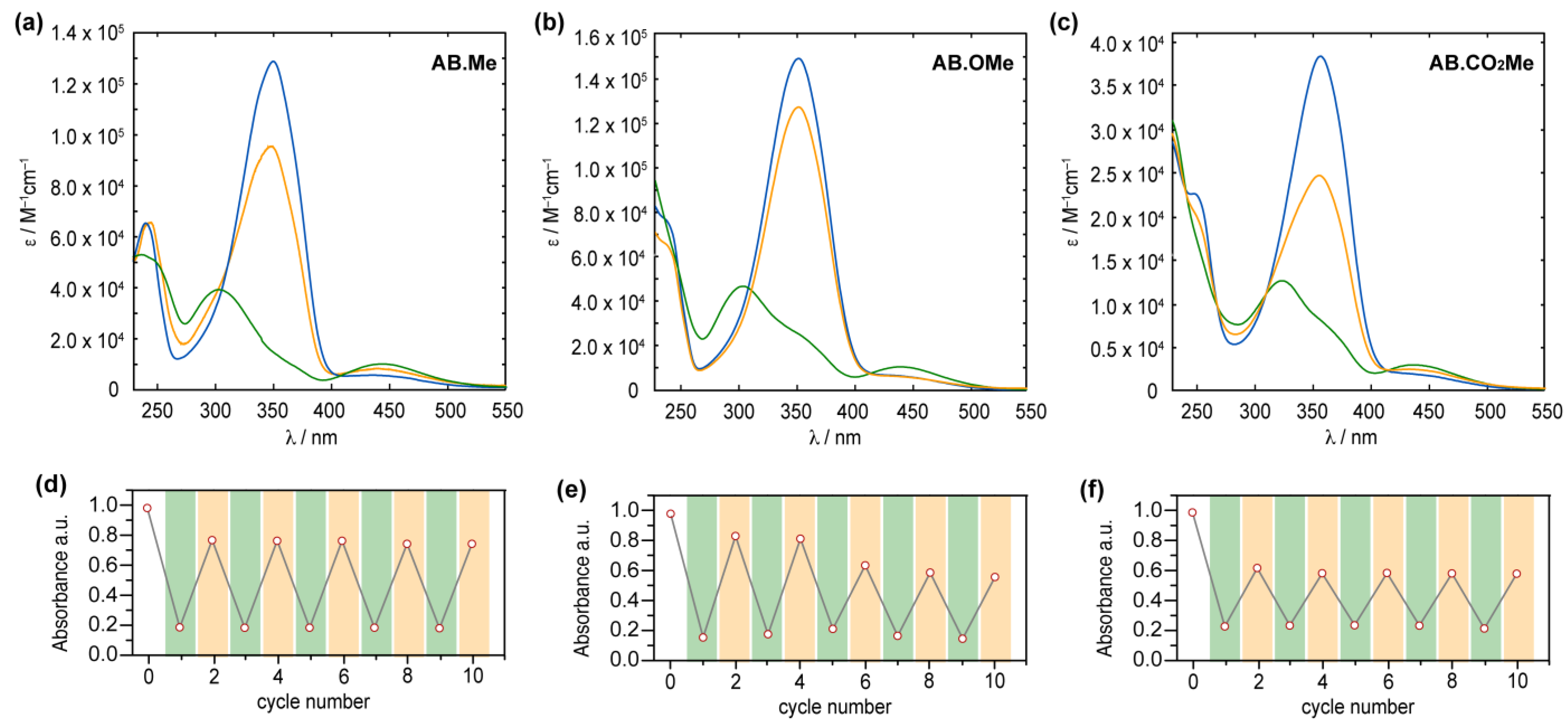
2.2. UV–vis Characterization
2.3. Thermal Relaxation
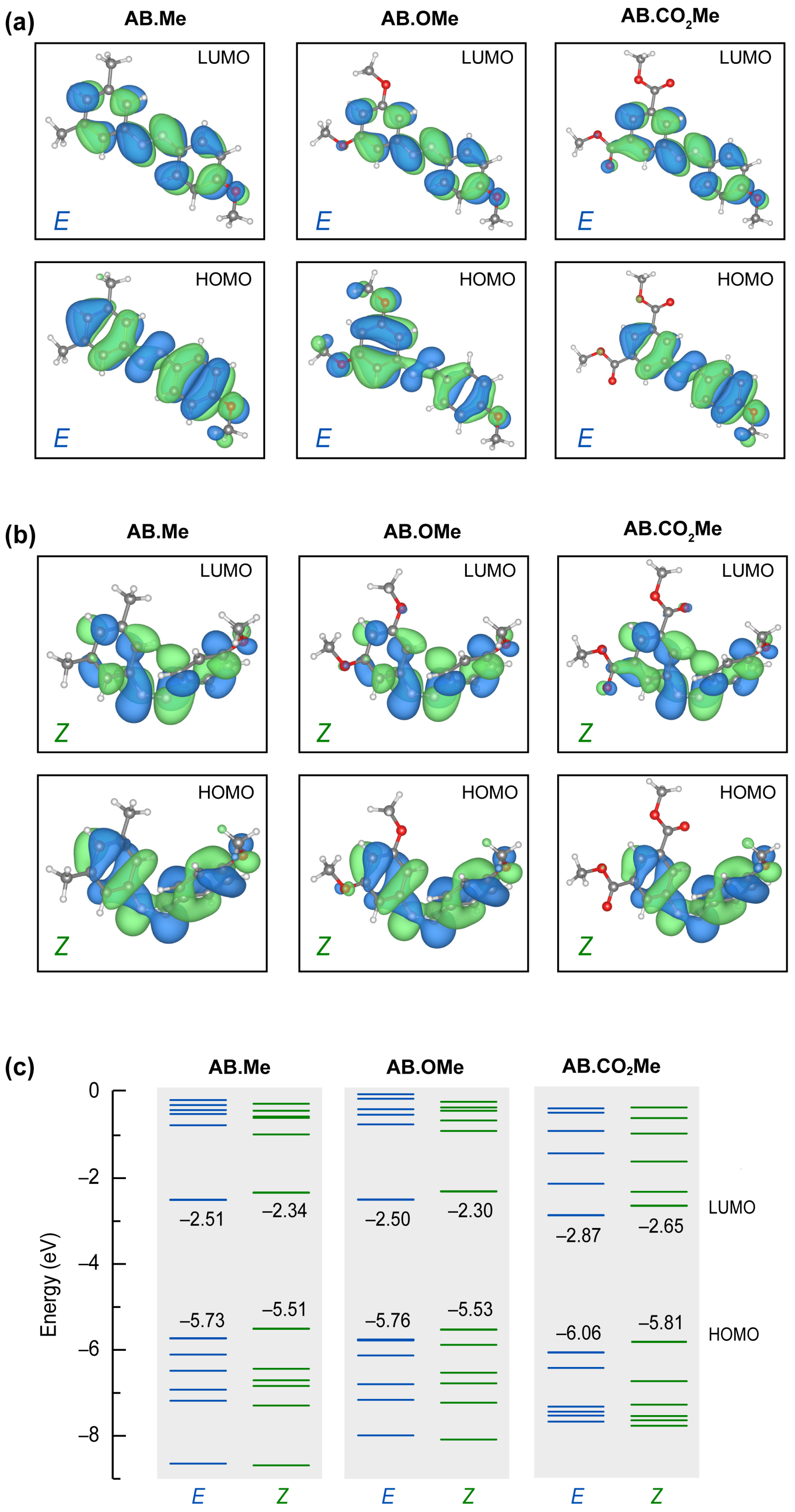
2.4. Computational Studies
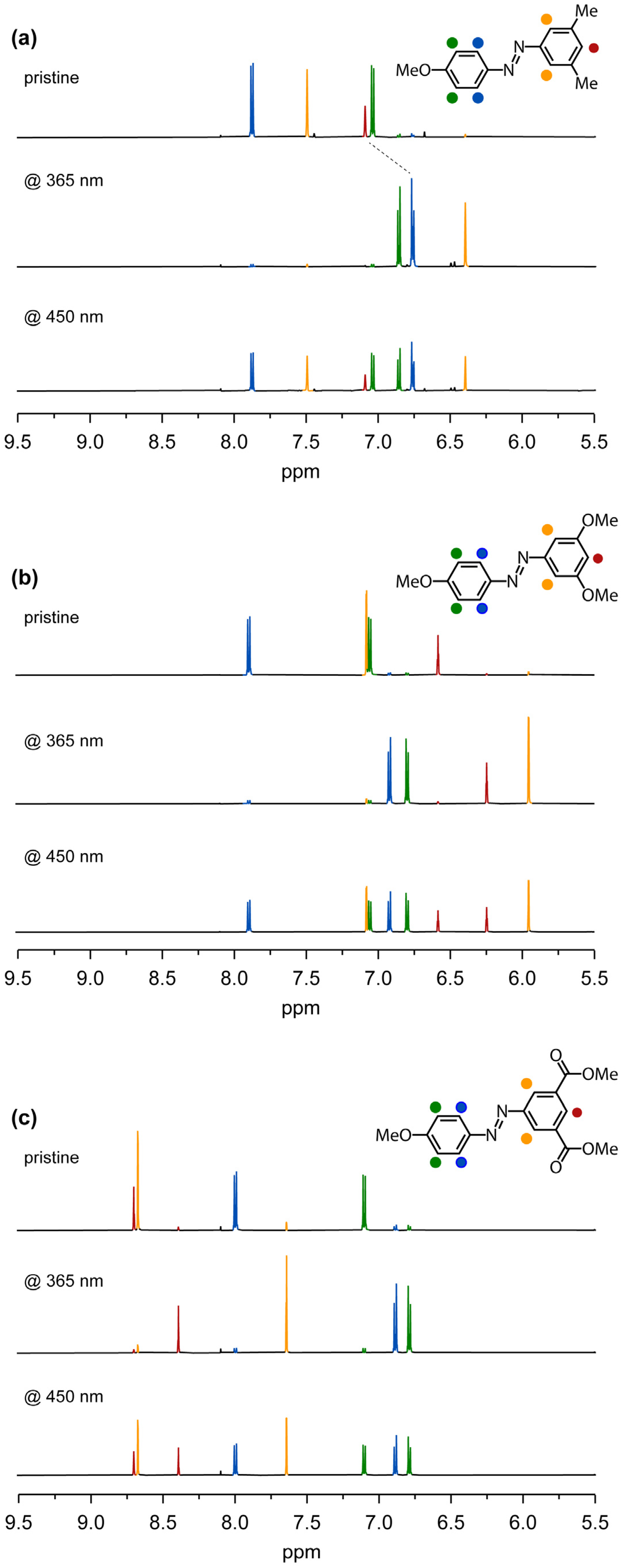
2.5. 1H-NMR Analysis
3. Materials and Methods
3.1. General Experimental Details
3.2. Synthetic Procedures and Characterization
- Compound AB-OH.Me, (E)-4-((3,5-dimethylphenyl)diazenyl)phenol. This compound was synthesized as reported in the literature [15]. Concentrated hydrochloric acid (2 mL) was added to a suspension of 3,5-dimethylaniline Ph-NH2.Me (0.606 g, 5.00 mmol, 1 eq) in water (20 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.380 g, 5.50 mmol, 1.1 eq) in water (3 mL) was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.999 g, 25.0 mmol, 5 eq) was added to a solution of phenol (0.565 g, 6.00 mmol, 1.2 eq) in water (2 mL), and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The resultant precipitate was filtered with vacuo and purified by silica chromatography (dichloromethane/ethyl acetate 9.5:0.5) to give an orange powder (0.404 g, 35.7%). 1H NMR (600 MHz, CDCl3): δ = 7.86 (d, J = 8.8 Hz, 2H), 7.49 (s, 2H), 7.09 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 5.08 (s, 1H, OH), 2.41 (s, 6H). 13C NMR (151 MHz, CDCl3): δ = 158.15, 153.06, 147.47, 138.85, 132.32, 125.00, 120.51, 115.90, 21.47. HRMS (ESI, positive mode) m/z calculated for C14H15N2O [M + H]+: 227.1174; found: 227.1179 [40].
- Compound AB-OH.OMe, (E)-4-((3,5-dimethoxyphenyl)diazenyl)phenol. This compound was synthesized adapting a procedure from the literature [40]. Concentrated hydrochloric acid (1.3 mL) was added to a suspension of 3,5-dimethoxyanline Ph-NH2.OMe (0.500 g, 3.26 mmol, 1 eq) in tetrahydrofuran (5 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.247 g, 3.59 mmol, 1.1 eq) in 1.95 mL of water was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.652 g, 16.3 mmol, 5 eq) was added to a solution of phenol (0.368 g, 3.91 mmol, 1.2 eq) in 2.6 mL of water, and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The reaction mixture was extracted with dichloromethane, the organic layers collected, dried over Na2SO4, filtered, and solvent removed with vacuo. The crude product was purified by silica chromatography using a gradient eluent (dichloromethane 100% → ethyl acetate 100%) to obtain the desired product as a red dark powder (0.377 g, 44.8%). 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 8.8 Hz, 2H), 7.08 (s, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.57 (s, 1H), 5.17 (s, 1H, OH), 3.87 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 161.22, 158.49, 154.68, 147.16, 125.22, 115.96, 103.55, 100.81, 55.75. HRMS (ESI, positive mode) m/z calculated for C14H15N2O3 [M + H]+: 259.1083; found: 259.1082.
- Compound AB-OH.CO2Me, (E)-dimethyl 5-((4-hydroxyphenyl)diazenyl)isophthalate. This compound was synthesized adapting a procedure from the literature [40]. Concentrated hydrochloric acid (0.95 mL) was added to a suspension of dimethyl 5-aminoisophthalate Ph-NH2.CO2Me (0.500 g, 2.39 mmol, 1 eq) in tetrahydrofuran (5 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.182 g, 2.63 mmol, 1.1 eq) in 1.42 mL of water was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.476 g, 11.9 mmol, 5 eq) was added to a solution of phenol (0.270 g, 2.87 mmol, 1.2 eq) in 1.92 mL of water, and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The reaction mixture was extracted with ethyl acetate, the organic layers collected, dried over Na2SO4, filtered, and solvent removed with vacuo. The crude product was purified by silica chromatography using dichloromethane/ethyl acetate (9.5:0.5) as an eluent, to obtain the desired product as bright orange powder (0.515 g, 68.6%). 1H NMR (600 MHz, CDCl3) δ = 8.75 (s, 1H), 8.70 (s, 2H), 7.94 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 5.32 (s, 1 H, OH), 4.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ = 166.06, 159.13, 153.04, 147.11, 131.87, 131.82, 127.74, 125.66, 116.10, 52.72. HRMS (ESI, positive mode) m/z calculated for C16H15N2O5 [M + H]+: 315.0981; found: 315.0983.
- Compound AB.Me, (E)-1-(3,5-dimethylphenyl)-2-(4-methoxyphenyl)diazene. A solution of compound AB-OH.Me (0.100 g, 0.442 mmol, 1 eq), iodomethane (0.033 mL, 0.530 mmol, 1.2 eq), K2CO3 (0.091 g, 0.663 mmol, 1.5 eq), and dimethylformamide (2 mL) was stirred at 7 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica chromatography (petroleum ether/ethyl acetate = 8:2) to obtain the desired product as a bright orange oil (0.088 g, 83.0%). 1H NMR (600 MHz, CDCl3) δ 7.91 (d, J = 9.0 Hz, 2H), 7.50 (s, 2H), 7.09 (s, 1H), 7.02 (d, J = 9.0 Hz, 2H), 3.89 (s, 3H), 2.41 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 162.06, 153.11, 147.25, 138.81, 132.22, 124.75, 120.48, 114.33, 55.70, 21.40. HRMS (ESI, positive mode) m/z calculated for C15H16N2O [M + H]+: 241.1341; found: 241.1341.
- Compound AB.OMe, (E)-1-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)diazene. A solution of compound AB-OH.OMe (0.146 g, 0.566 mmol, 1 eq), iodomethane (0.042 mL, 0.680 mmol, 1.2 eq), K2CO3 (0.117 g, 0.849 mmol, 1.5 eq), and dimethylformamide (2.5 mL) was stirred at 70 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by silica chromatography (petroleum ether/ethyl acetate = 8:2) to obtain the desired product as a bright orange powder (0.084 g, 54.5%). 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 9.4 Hz, 2H), 7.09 (d, 2H), 7.03 (d, J = 9.4 Hz, 2H), 6.58 (s, 1H), 3.90 (s, 3H), 3.88 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 162.32, 161.22, 154.76, 146.99, 124.98, 114.38, 103.49, 100.77, 76.95, 55.74. HRMS (ESI, positive mode) m/z calculated for C15H16N2O3 [M + H]+: 273.1239; found: 273.1238.
- Compound AB.CO2Me, (E)-dimethyl 5-((4-methoxyphenyl)diazenyl)isophthalate. A solution of compound AB-OH.CO2Me (0.100 g, 0.318 mmol, 1 eq), iodomethane (0.024 mL, 0.382 mmol, 1.2 eq), K2CO3 (0.066 g, 0.477 mmol, 1.5 eq) and dimethylformamide (1.5 mL) was stirred at 70 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica chromatography (dichloromethane 100%) to obtain the desired product as a bright yellow powder (0.033 g, 31.7%). 1H NMR (600 MHz, CDCl3) δ = 8.75 (s, 1H), 8.70 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 3.99 (s, 6H), 3.91 (s, 3H). 13C NMR (151 MHz, CDCl3) δ = 166.00, 162.90, 153.04, 146.86, 131.75, 127.67, 125.39, 114.48, 55.76, 52.67. HRMS (ESI, positive mode) m/z calculated for C17H16N2O5 [M + H]+: 329.1137; found: 329.1130.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jerca, F.A.; Jerca, V.V.; Hoogenboom, R. Advances and opportunities in the exciting world of azobenzenes. Nat. Rev. Chem. 2022, 6, 51–69. [Google Scholar] [CrossRef]
- Mukherjee, A.; Seyfried, M.D.; Ravoon, B.J. Azoheteroarene and Diazocine Molecular Photoswitches: Self-Assembly, Responsive Materials and Photopharmacology. Angew. Chem. Int. Ed. 2023, 62, e202304437. [Google Scholar] [CrossRef]
- Fedele, C.; Ruoko, T.; Kuntze, K.; Virkki, M.; Priimagi, A. New tricks and emerging applications from contemporary azobenzene research. Photochem. Photobiol. Sci. 2022, 21, 1719–1734. [Google Scholar] [CrossRef]
- Crespi, S.; Simeth, N.A.; König, B. Heteroaryl azo dyes as molecular photoswitches. Nat. Rev. Chem. 2019, 3, 33–146. [Google Scholar] [CrossRef]
- García-Amorós, J.; Velasco, D. Recent advances towards azobenzene-based light-driven real-time information-transmitting materials. Beilstein J. Org. Chem. 2012, 8, 1003–1017. [Google Scholar] [CrossRef]
- Chen, H.; Chen, W.; Lin, Y.; Xie, Y.; Liu, S.H.; Yin, J. Visible and near-infrared light activated azo dyes. Chin. Chem. Lett. 2021, 32, 2359–2368. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, T.; Wang, Z.; Zhao, Y. Triggered azobenzene-based prodrugs and drug delivery systems. J. Control. Release 2022, 325, 475–493. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, Y.; Chen, T.; Lu, D.; Zhao, Z.; Zhang, X.; Li, Z.; Yan, C.; Tan, W. Photon-Manipulated Drug Release from a Mesoporous Nanocontainer Controlled by Azobenzene-Modified Nucleic Acid. ACS Nano 2012, 6, 6337–6344. [Google Scholar] [CrossRef]
- Yadav, S.; Deka, S.R.; Verma, G.; Sharma, A.K.; Kumar, P. Photoresponsive amphiphilic azobenzene–PEG self-assembles to form supramolecular nanostructures for drug delivery applications. RSC Adv. 2016, 6, 8103. [Google Scholar] [CrossRef]
- Meng, X.; Gui, B.; Yuan, D.; Zeller, M.; Wang, C. Mechanized azobenzene-functionalized zirconium metal-organic framework for on-command cargo release. Sci. Adv. 2016, 2, e1600480. [Google Scholar] [CrossRef]
- Kryshenik, V.M.; Azhniuk, Y.M.; Kovtunenko, V.S. All-optical patterning in azobenzene polymers and amorphous chalcogenides. J. Non Cryst. Solids 2019, 512, 112–131. [Google Scholar] [CrossRef]
- Bakker, S.; Korver, E.; Fransen, M.; Kamer, E.; Metselaar, G.; Catarina, A.; Esteves, C.; Schenning, A.P.H.J. Optical Patterning in Photoresponsive Azobenzene-Based Waterborne Coatings. ACS Appl. Opt. Mater. 2023, 1, 403–411. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Kulikovsky, L.; Kulikovska, O.; Stumpe, J. Extremely high patterning efficiency in easily made azobenzene-containing polymer films. J. Mater. Chem. 2009, 19, 6103–6105. [Google Scholar] [CrossRef]
- Carroll, G.T.; Lee, K.M.; Mc Conney, M.E.; Hall, H.J. Optical control of alignment and patterning in an azobenzene liquid crystal photoresist. J. Mater. Chem. C 2023, 11, 2177–2185. [Google Scholar] [CrossRef]
- Maciejewski, J.; Sobczuk, A.; Claveau, A.; Nicolai, A.; Petraglia, R.; Cervini, L.; Baudat, E.; Miéville, P.; Fazzi, D.; Corminboeuf, C.; et al. Photochromic Torsional Switch (PTS): A Light-Driven Actuator for the Dynamic Tuning of π-Conjugation Extension. Chem. Sci. 2017, 8, 361–365. [Google Scholar] [CrossRef]
- Jozeliūnaitė, A.; Rahmanudin, A.; Gražulis, S.; Baudat, E.; Sivula, K.; Fazzi, D.; Orentas, E.; Sforazzini, G. Light-Responsive Oligothiophenes Incorporating Photochromic Torsional Switches (PTS). Chem. Eur. J. 2022, 28, e202202698. [Google Scholar] [CrossRef]
- Jousselme, B.; Blanchard, P.; Gallego-Planas, N.; Delaunay, J.; Allain, M.; Richomme, P.; Levillain, E.; Roncali, J. Photomechanical Actuation and Manipulation of the Electronic Properties of Linear π-Conjugated Systems. J. Am. Chem. Soc. 2003, 125, 2888–2889. [Google Scholar] [CrossRef]
- Jousselme, B.; Blanchard, P.; Allain, M.; Levillain, E.; Dias, M.; Roncali, J. Structural Control of the Electronic Properties of Photodynamic Azobenzene-Derivatized π-Conjugated Oligothiophenes. J. Phys. Chem. 2006, 110, 3488–3494. [Google Scholar] [CrossRef]
- Azov, V.A.; Cordes, J.; Schlüter, D.; Dülcks, T.; Böckmann, M.; Doltsinis, N.L. Light-Controlled Macrocyclization of Tetrathiafulvalene with Azobenzene: Designing an Optoelectronic Molecular Switch. J. Org. Chem. 2014, 79, 11714–11721. [Google Scholar] [CrossRef]
- Mart, R.J.; Allemann, R.K. Azobenzene photocontrol of peptides and proteins. Chem. Commun. 2016, 52, 12262. [Google Scholar] [CrossRef]
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Barrett, C.J. Photo-Control of Biological Systems with Azobenzene Polymers. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3058–3070. [Google Scholar] [CrossRef]
- Butler, C.S.G.; King, J.P.; Giles, L.W.; Marlow, J.B.; Vidallon, M.L.P.; Sokolova, A.; de Campo, L.; Tuck, K.L.; Tabor, R.F. Design and synthesis of an azobenzene–betaine surfactant for photo-rheological fluids. J. Colloid Interface Sci. 2021, 594, 669–680. [Google Scholar] [CrossRef]
- Wu, Z.; Li, X.; Jiang, X.; Xie, T.; Li, H.; Zhang, G.; Jiang, J. Photoswitchable de/adsorption of an azobenzene-derived surfactant on a silica surface. Phys. Chem. Chem. Phys. 2019, 21, 21030–21037. [Google Scholar] [CrossRef]
- Chu, Z.; Han, Y.; Bian, T.; De, S.; Král, P.; Klajn, R. Supramolecular Control of Azobenzene Switching on Nanoparticles. J. Am. Chem. Soc. 2019, 141, 1949–1960. [Google Scholar] [CrossRef]
- Moldt, T.; Przyrembel, D.; Schulze, M.; Bronsch, W.; Boie, L.; Brete, D.; Gahl, C.; Klajn, R.; Tegeder, P.; Weinelt, M. Differing Isomerization Kinetics of Azobenzene-Functionalized Self-Assembled Monolayers in Ambient Air and in Vacuum. Langmuir 2016, 32, 10795–10801. [Google Scholar] [CrossRef]
- Cuétara-Guadarrama, F.; Vonlanthen, V.; Sorroza-Martínez, K.; González-Méndez, I.; Rivera, E. Photoisomerizable azobenzene dyes incorporated into polymers and dendrimers. Influence of the molecular aggregation on the nonlinear optical properties. Dyes Pigm. 2021, 194, 109551. [Google Scholar] [CrossRef]
- Cunha, M.P.; Foelen, Y.; Engels, T.A.P.; Papamichou, K.; Hagenbeek, M.; Debije, M.G.; Schenning, P.H.J. On Untethered, Dual Magneto- and Photoresponsive Liquid Crystal Bilayer Actuators Showing Bending and Rotating Motion. Adv. Opt. Mater. 2019, 7, 1801604. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef]
- Beharry, A.A.; Sadovski, O.; Woolley, G.A. Azobenzene Photoswitching without Ultraviolet Light. J. Am. Chem. Soc. 2011, 133, 19684–19687. [Google Scholar] [CrossRef]
- Kerckhoffs, A.; Bo, Z.; Penty, S.E.; Duarte, F.; Langton, M.J. Red-shifted tetra-ortho-halo-azobenzenes for photo-regulated transmembrane anion transport. Org. Biomol. Chem. 2021, 19, 9058–9067. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Beharry, A.A.; OSadovski, O.; McCormick, T.M.; Babalhavaeji, A.; Tropepe, V.; Woolley, G.A. Photoswitching Azo Compounds in Vivo with Red Light. J. Am. Chem. Soc. 2013, 135, 9777–9784. [Google Scholar] [CrossRef] [PubMed]
- Ohzono, T.; Koyama, E. Effects of photo-isomerizable side groups on the phase and mechanical properties of main-chain nematic elastomers. Polym. Chem. 2022, 13, 2694–2704. [Google Scholar] [CrossRef]
- Togari, Y.; Hirota, S.; Kitagawa, H.; Tsukamoto, Y.; Kobayashi, K. Hydrogen-bonded six-component assembly for capsule formation based on tetra(4-pyridyl) cavitand and isophthalic acid linker and its application to photoresponsive capsule. Org. Biomol. Chem. 2018, 16, 7626–7635. [Google Scholar] [CrossRef] [PubMed]
- Di Berardin, C.; Strauss, M.A.; Schatz, D.; Wegner, H.A. An Incremental System To Predict the Effect of Different London Dispersion Donors in All-meta-Substituted Azobenzenes. Chem. Eur. J. 2022, 28, e202104284. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Yadav, M.K.; Singla, L.; Kumar, A.; Karanam, M.; Dev, S.; Choudhury, A.R. Understanding of the Kinetic Stability of cis- Isomer of Azobenzenes through Kinetic and Computational Studies. ChemistrySelect 2020, 5, 13957–13962. [Google Scholar] [CrossRef]
- Singer, N.K.; Schlögl, K.; Zobel, J.P.; Mihovilovic, M.D.; González, L. Singlet and Triplet Pathways Determine the Thermal Z/E Isomerization of an Arylazopyrazole-Based Photoswitch. J. Phys. Chem. Lett. 2023, 14, 8956–8961. [Google Scholar] [CrossRef]
- Reimann, M.; Teichmann, E.; Hecht, S.; Kaupp, M. Solving the Azobenzene Entropy Puzzle: Direct Evidence for Multi-State Reactivity. J. Phys. Chem. Lett. 2022, 13, 10882–10888. [Google Scholar] [CrossRef]
- Winter, A.H.; Falvey, D.E.; Cramer, C.J. Effect of meta Electron-Donating Groups on the Electronic Structure of Substituted Phenyl Nitrenium Ions. J. Am. Chem. Soc. 2004, 126, 9661–9668. [Google Scholar] [CrossRef]
- Song, Y.M.; Ha, Y.M.; Kim, J.A.; Chung, K.W.; Uehara, Y.; Lee, K.J.; Chun, P.; Byun, Y.; Chung, H.Y.; Moon, H.R. Synthesis of novel azo-resveratrol, azo-oxyresveratrol and their derivatives as potent tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7451–7455. [Google Scholar] [CrossRef]
- Ślusarek, J.; Nowoświat, A.; Olechowska, M. Logistic Model of Phase Transformation of Hardening Concrete. Materials 2022, 15, 4403. [Google Scholar] [CrossRef] [PubMed]
- Jerca, V.V.; Jerca, F.A.; Rau, I.; Manea, A.M.; Vuluga, D.M.; Kajzar, F. Advances in understanding the photoresponsive behavior of azobenzenes substituted with strong electron withdrawing groups. Opt. Mater. 2015, 48, 160–164. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Berland, K.; Cooper, V.R.; Lee, K.; Schröder, E.; Thonhauser, T.; Hyldgaard, P.; Lundqvist, B.I. van der Waals forces in density functional theory: A review of the vdW-DF method. Rep. Prog. Phys. 2015, 78, 066501. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanna, A.L.; Pachova, T.; Catellani, A.; Calzolari, A.; Sforazzini, G. Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship. Molecules 2024, 29, 1929. https://doi.org/10.3390/molecules29091929
Sanna AL, Pachova T, Catellani A, Calzolari A, Sforazzini G. Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship. Molecules. 2024; 29(9):1929. https://doi.org/10.3390/molecules29091929
Chicago/Turabian StyleSanna, Anna Laura, Tatiana Pachova, Alessandra Catellani, Arrigo Calzolari, and Giuseppe Sforazzini. 2024. "Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship" Molecules 29, no. 9: 1929. https://doi.org/10.3390/molecules29091929
APA StyleSanna, A. L., Pachova, T., Catellani, A., Calzolari, A., & Sforazzini, G. (2024). Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship. Molecules, 29(9), 1929. https://doi.org/10.3390/molecules29091929