
A Systematic Review of Statins for the Treatment of Nonalcoholic Steatohepatitis: Safety, Efficacy, and Mechanism of Action


Abstract
1. Introduction
2. Hypolipidemic and Pleiotropy of Statins
2.1. Hypolipidemic
2.2. Pleiotropy
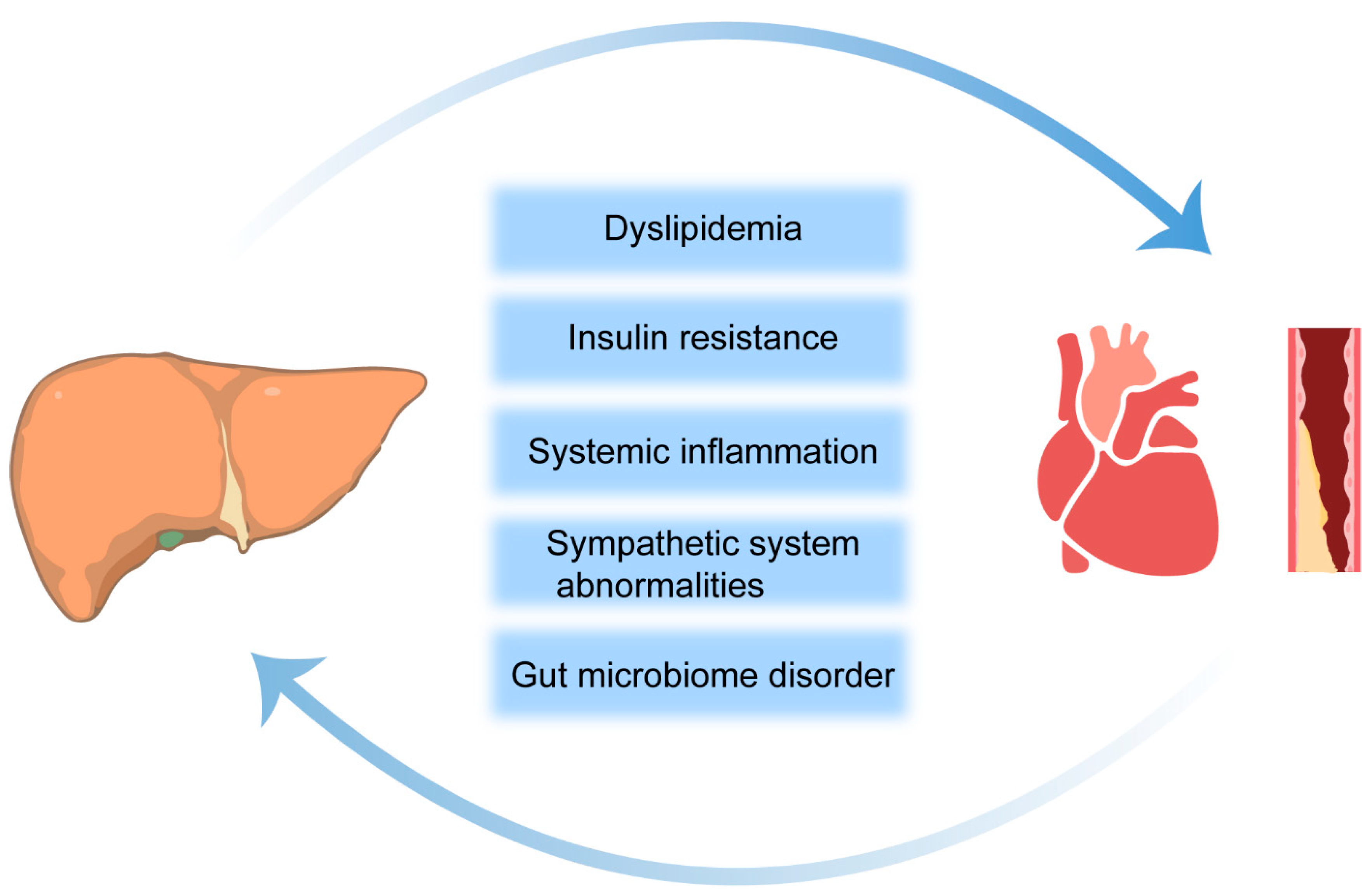
3. Mechanisms Linking NAFLD and CVD
3.1. Insulin Resistance
3.2. Dyslipidemia
3.3. Localized and Systemic Inflammation
4. Statin Therapy in Patients with NAFLD: Safety and Efficacy
4.1. Effects of Statins on NAFLD Patients with CVD
4.2. Safety Assessment of Statins in the Treatment of NASH/NAFLD
5. Effects of Statins on Liver Histology—From Animal Models to Human Studies
5.1. Improvement of Steatosis
5.2. Reduction of Inflammation
5.3. Improvement of Fibrosis
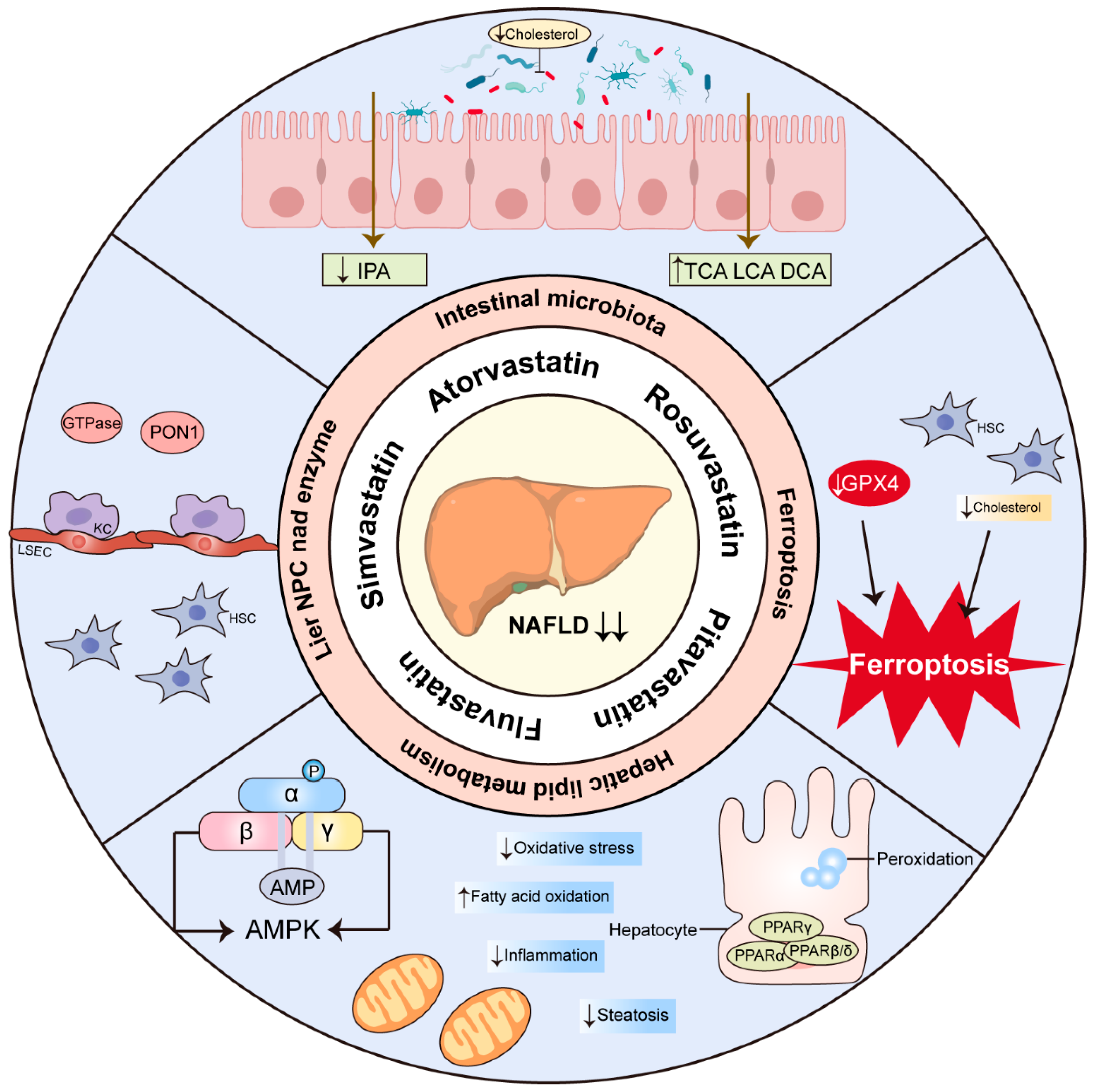
6. Mechanisms of Statins in the Treatment of NASH/NAFLD
6.1. KCs, HSCs and LSECs
6.2. PON1
6.3. GTPases
6.4. PPARs
6.5. AMPK
6.6. Ferroptosis
6.7. Intestinal Microbiota
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Paternostro, R.; Trauner, M. Current treatment of non-alcoholic fatty liver disease. J. Intern. Med. 2022, 292, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Ekstedt, M.; Wong, G.L.-H.; Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Gastroenterology, H. Resmetirom for NASH: Balancing promise and prudence. Lancet Gastroenterol. Hepatol. 2024, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, Q.; Wang, W.; Ling, Y.; Yan, Y.; Xia, P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 2020, 72, 156–166. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Linking liver metabolic and vascular disease via bile acid signaling. Trends Mol. Med. 2022, 28, 51–66. [Google Scholar] [CrossRef]
- Harrison, S.A.; Allen, A.M.; Dubourg, J.; Noureddin, M.; Alkhouri, N. Challenges and opportunities in NASH drug development. Nat. Med. 2023, 29, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Maretti-Mira, A.C.; Salomon, M.P.; Hsu, A.M.; Kanel, G.C.; Golden-Mason, L. Hepatic damage caused by long-term high cholesterol intake induces a dysfunctional restorative macrophage population in experimental NASH. Front. Immunol. 2022, 13, 968366. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Verheyen, F.; Guichot, Y.D.; Walenbergh, S.M.; Jeurissen, M.L.; Gijbels, M.; Rensen, S.S.; Bast, A.; et al. The cholesterol derivative 27-hydroxycholesterol reduces steatohepatitis in mice. Gastroenterology 2013, 144, 167–178.e161. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, X.H.; Ou, X.; Ouyang, X.P.; Tang, C.K. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Sun, B.; Li, L.; Zhou, X. Comparative analysis of the gut microbiota in distinct statin response patients in East China. J. Microbiol. 2018, 56, 886–892. [Google Scholar] [CrossRef]
- Yoo, D.-H.; Kim, I.S.; Van Le, T.K.; Jung, I.-H.; Yoo, H.H.; Kim, D.-H. Gut Microbiota-Mediated Drug Interactions between Lovastatin and Antibiotics. Drug Metab. Dispos. 2014, 42, 1508–1513. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists, C.; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative, G. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Awad, K.; Serban, M.C.; Penson, P.; Mikhailidis, D.P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Howard, G.; Lip, G.Y.H.; Banach, M.; et al. Effects of morning vs evening statin administration on lipid profile: A systematic review and meta-analysis. J. Clin. Lipidol. 2017, 11, 972–985 e979. [Google Scholar] [CrossRef]
- Mora-Rodriguez, R.; Ortega, J.F.; Morales-Palomo, F.; Ramirez-Jimenez, M.; Moreno-Cabanas, A. Effects of statin therapy and exercise on postprandial triglycerides in overweight individuals with hypercholesterolaemia. Br. J. Clin. Pharmacol. 2020, 86, 1089–1099. [Google Scholar] [CrossRef]
- Rosenson, R.S. Low HDL-C: A secondary target of dyslipidemia therapy. Am. J. Med. 2005, 118, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Liao, J.K. Emerging views of statin pleiotropy and cholesterol lowering. Cardiovasc. Res. 2022, 118, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Kitagawa, K.; Hosomi, N.; Nagai, Y.; Kagimura, T.; Ohtsuki, T.; Origasa, H.; Minematsu, K.; Uchiyama, S.; Nakamura, M.; Matsumoto, M.; et al. Reduction in High-Sensitivity C-Reactive Protein Levels in Patients with Ischemic Stroke by Statin Treatment: Hs-CRP Sub-Study in J-STARS. J. Atheroscler. Thromb. 2017, 24, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Wolters, A.A.B.; Dullaart, R.P.F.; Moshage, H.; Zurakowski, D.; de Meijer, V.E.; Blokzijl, H. Prescription of statins in suspected non-alcoholic fatty liver disease and high cardiovascular risk, a population-based study. Liver Int. 2019, 39, 1343–1354. [Google Scholar] [CrossRef]
- Almeida, S.O.; Budoff, M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, P.; Taccardi, A.A.; Grilli, A.; De Lutiis, M.A.; Barsotti, A.; Felaco, M.; De Caterina, R. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc. Res. 2005, 66, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Liu, Y.W.; Lin, L.J.; Chen, J.H.; Liao, J.K. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation 2009, 119, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liu, P.Y.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, H.; Zhang, X.J.; Cai, J.; Li, H. NAFLD: An Emerging Causal Factor for Cardiovascular Disease. Physiology 2023, 38, 255–265. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Lechner, K.; McKenzie, A.L.; Krankel, N.; Von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingartner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185. [Google Scholar] [CrossRef]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus–Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D. NAFLD, and cardiovascular and cardiac diseases: Factors influencing risk, prediction and treatment. Diabetes Metab. 2021, 47, 101215. [Google Scholar] [CrossRef] [PubMed]
- Kopin, L.; Lowenstein, C. Dyslipidemia. Ann. Intern. Med. 2017, 167, Itc81–Itc95. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.W.; Yu, J.H.; Jin, Y.J.; Suh, Y.J.; Lee, J.W. Correlation between the small dense LDL level and nonalcoholic fatty liver disease: Possibility of a new biomarker. Medicine 2020, 99, e21162. [Google Scholar] [CrossRef] [PubMed]
- Duran, E.K.; Aday, A.W.; Cook, N.R.; Buring, J.E.; Ridker, P.M.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Cholesterol, Small Dense LDL Cholesterol, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 75, 2122–2135. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Abdallah, L.R.; de Matos, R.C.; YPDM, E.S.; Vieira-Soares, D.; Muller-Machado, G.; Pollo-Flores, P. Non-alcoholic Fatty Liver Disease and Its Links with Inflammation and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 7. [Google Scholar] [CrossRef]
- Braza-Boils, A.; Mari-Alexandre, J.; Molina, P.; Arnau, M.A.; Barcelo-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martinez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef]
- Targher, G.; Marra, F.; Marchesini, G. Increased risk of cardiovascular disease in non-alcoholic fatty liver disease: Causal effect or epiphenomenon? Diabetologia 2008, 51, 1947–1953. [Google Scholar] [CrossRef]
- Johnston, M.P.; Patel, J.; Byrne, C.D. Causes of Mortality in Non-Alcoholic Fatty Liver Disease (NAFLD) and Alcohol Related Fatty Liver Disease (AFLD). Curr. Pharm. Des. 2020, 26, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.A.; Hafeez, H. Biochemistry of Statins. Adv. Clin. Chem. 2016, 73, 127–168. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Athyros, V.G.; Boutari, C.; Stavropoulos, K.; Anagnostis, P.; Imprialos, K.P.; Doumas, M.; Karagiannis, A. Statins: An Under-Appreciated Asset for the Prevention and the Treatment of NAFLD or NASH and the Related Cardiovascular Risk. Curr. Vasc. Pharmacol. 2018, 16, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Yebyo, H.G.; Aschmann, H.E.; Kaufmann, M.; Puhan, M.A. Comparative effectiveness and safety of statins as a class and of specific statins for primary prevention of cardiovascular disease: A systematic review, meta-analysis, and network meta-analysis of randomized trials with 94,283 participants. Am. Heart J. 2019, 210, 18–28. [Google Scholar] [CrossRef] [PubMed]
- du Souich, P.; Roederer, G.; Dufour, R. Myotoxicity of statins: Mechanism of action. Pharmacol. Ther. 2017, 175, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N. Statins and diabetes: What are the connections? Best Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101749. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Baratta, F.; Polimeni, L.; Pastori, D.; Loffredo, L.; Averna, M.; Violi, F.; Angelico, F. Under-prescription of statins in patients with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 161–167. [Google Scholar] [CrossRef]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Abel, T.; Fehér, J.; Dinya, E.; Eldin, M.G.; Kovács, A. Safety and efficacy of combined ezetimibe/simvastatin treatment and simvastatin monotherapy in patients with non-alcoholic fatty liver disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2009, 15, MS6–MS11. [Google Scholar]
- Gomez-Dominguez, E.; Gisbert, J.P.; Moreno-Monteagudo, J.A.; Garcia-Buey, L.; Moreno-Otero, R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment. Pharmacol. Ther. 2006, 23, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Rana, H.; Yadav, S.S.; Reddy, H.D.; Singhal, S.; Singh, D.K.; Usman, K. Comparative Effect of Insulin Sensitizers and Statin on Metabolic Profile and Ultrasonographical Score in Non Alcoholic Fatty Liver Disease. J. Clin. Diagn. Res. 2016, 10, OC19–OC23. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Pani, A.; Di Rocco, A.; Menichelli, D.; Gazzaniga, G.; Farcomeni, A.; D’Erasmo, L.; Angelico, F.; Del Ben, M.; Baratta, F. Statin liver safety in non-alcoholic fatty liver disease: A systematic review and metanalysis. Br. J. Clin. Pharmacol. 2021, 88, 441–451. [Google Scholar] [CrossRef]
- Boutari, C.; Pappas, P.D.; Anastasilakis, D.; Mantzoros, C.S. Statins’ efficacy in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhao, J.; Zhai, Y.; Zang, P.; Lv, Q.; Shang, D. A prospective study of hepatic safety of statins used in very elderly patients. BMC Geriatr. 2019, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, N.; Chapin, S.; Goldberg, D.S.; Reddy, K.R.; Taddei, T.H.; Kaplan, D.E. Statin exposure is associated with reduced development of acute-on-chronic liver failure in a Veterans Affairs cohort. J. Hepatol. 2022, 76, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jang, S.-Y.; Nam, C.M.; Kang, E.S. Statin use and the risk of hepatocellular carcinoma in patients at high risk: A nationwide nested case-control study. J. Hepatol. 2018, 68, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Zou, B.; Odden, M.C.; Nguyen, M.H. Statin Use and Reduced Hepatocellular Carcinoma Risk in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2022, 21, 435–444.e6. [Google Scholar] [CrossRef]
- Cohen, D.E.; Anania, F.A.; Chalasani, N. An assessment of statin safety by hepatologists. Am. J. Cardiol. 2006, 97, 77C–81C. [Google Scholar] [CrossRef]
- Tokushige, K.; Ikejima, K.; Ono, M.; Eguchi, Y.; Kamada, Y.; Itoh, Y.; Akuta, N.; Yoneda, M.; Iwasa, M.; Yoneda, M.; et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis 2020. J. Gastroenterol. 2021, 56, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Fatima, K.; Moeed, A.; Waqar, E.; Atif, A.R.; Kamran, A.; Rizvi, H.; Suri, N.F.; Haider, H.; Shuja, S.H.; Khalid, M.; et al. Efficacy of statins in treatment and development of non-alcoholic fatty liver disease and steatohepatitis: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2021, 46, 101816. [Google Scholar] [CrossRef] [PubMed]
- Krishan, S. Correlation between non-alcoholic fatty liver disease (NAFLD) and dyslipidemia in type 2 diabetes. Diabetes Metab. Syndr. 2016, 10, S77–S81. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhao, X.; Leng, L.; Liu, P.; Jiang, Z. Comparison of dietary control and atorvastatin on high fat diet induced hepatic steatosis and hyperlipidemia in rats. Lipids Health Dis. 2011, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.-W.; Hsu, Y.-C.; Lee, T.-F.; Lin, Y.; Chiu, Y.-T.; Yang, K.-C.; Wu, J.-C.; Huang, Y.-T. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-W.; Zhu, Y.-C.; Zhang, L.; Li, P.; Yang, J.; Wen, X.-D. Ilexgenin A enhances the effects of simvastatin on non-alcoholic fatty liver disease without changes in simvastatin pharmacokinetics. Chin. J. Nat. Med. 2018, 16, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Rhee, H.; Kim, Y.E.; Lee, M.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Choi, J.Y.; Lee, Y.H. Ezetimibe combination therapy with statin for non-alcoholic fatty liver disease: An open-label randomized controlled trial (ESSENTIAL study). BMC Med. 2022, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Rha, S.W.; Kang, H.J.; Bae, J.W.; Choi, B.J.; Choi, S.Y.; Gwon, H.C.; Bae, J.H.; Hong, B.K.; Choi, D.H.; et al. Evaluation of short-term safety and efficacy of HMG-CoA reductase inhibitors in hypercholesterolemic patients with elevated serum alanine transaminase concentrations: PITCH study (PITavastatin versus atorvastatin to evaluate the effect on patients with hypercholesterolemia and mild to moderate hepatic damage). J. Clin. Lipidol. 2012, 6, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, K. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J. Gastroenterol. 2015, 21, 7860–7868. [Google Scholar] [CrossRef]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.-i.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.R.; Feldpausch, M.N.; Czerwonka, N.; Weiss, J.; Branch, K.; Lee, H.; Martinez-Salazar, E.L.; Torriani, M.; Sponseller, C.A.; Grinspoon, S.K.; et al. Effects of Pitavastatin on Insulin Sensitivity and Liver Fat: A Randomized Clinical Trial. J. Clin. Endocrinol. Metab. 2018, 103, 4176–4186. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Portillo Sanchez, P.; Lomonaco, R.; Orsak, B.; Hecht, J.; Tio, F.; Cusi, K. Liver Safety of Statins in Prediabetes or T2DM and Nonalcoholic Steatohepatitis: Post Hoc Analysis of a Randomized Trial. J. Clin. Endocrinol. Metab. 2017, 102, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Inia, J.A.; Stokman, G.; Pieterman, E.J.; Morrison, M.C.; Menke, A.L.; Verschuren, L.; Caspers, M.P.M.; Giera, M.; Jukema, J.W.; van den Hoek, A.M.; et al. Atorvastatin Attenuates Diet-Induced Non-Alcoholic Steatohepatitis in APOE*3-Leiden Mice by Reducing Hepatic Inflammation. Int. J. Mol. Sci. 2023, 24, 7818. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.; Araujo, B.P.; Rodrigues, K.L.; Silvares, R.R.; Martins, C.S.M.; Flores, E.E.I.; Fernandes-Santos, C.; Daliry, A. Simvastatin Improves Microcirculatory Function in Nonalcoholic Fatty Liver Disease and Downregulates Oxidative and ALE-RAGE Stress. Nutrients 2022, 14, 716. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.-X.; Tarrio, M.; Grabie, N.; Zhang, Y.; Yamazaki, H.; Stavrakis, G.; Maganto-Garcia, E.; Pepper-Cunningham, Z.; Jarolim, P.; Aikawa, M.; et al. Statin-induced Krüppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J. Clin. Investig. 2010, 120, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S.-I.; Maeda, S.; Kimura, Y.; Ishitobi, T.; Chayama, K. Atorvastatin improves disease activity of nonalcoholic steatohepatitis partly through its tumour necrosis factor-α-lowering property. Dig. Liver Dis. 2012, 44, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kechagias, S.; Tsochatzis, E.A. Review article: Non-alcoholic fatty liver disease and cardiovascular diseases: Associations and treatment considerations. Aliment. Pharmacol. Ther. 2021, 54, 1013–1025. [Google Scholar] [CrossRef]
- Yang, J.I.; Yoon, J.H.; Bang, Y.J.; Lee, S.H.; Lee, S.M.; Byun, H.J.; Myung, S.J.; Kim, W.; Lee, H.S. Synergistic antifibrotic efficacy of statin and protein kinase C inhibitor in hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G126–G132. [Google Scholar] [CrossRef]
- Bosch, J.; Gracia-Sancho, J.; Abraldes, J.G. Cirrhosis as new indication for statins. Gut 2020, 69, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortigosa, C.M.; Wang, W.; Zhao, C.; Zhou, J.; Zhen, Z.; Wang, Y.; Shen, C. Simvastatin Ameliorates Liver Fibrosis via Mediating Nitric Oxide Synthase in Rats with Non-Alcoholic Steatohepatitis-Related Liver Fibrosis. PLoS ONE 2013, 8, e76538. [Google Scholar] [CrossRef]
- Ciardullo, S.; Perseghin, G. Statin use is associated with lower prevalence of advanced liver fibrosis in patients with type 2 diabetes. Metabolism 2021, 121, 154752. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Aron-Wisnewsky, J.; Pais, R.; Tordjman, J.; Poitou, C.; Charlotte, F.; Bedossa, P.; Poynard, T.; Clément, K.; Ratziu, V. Statins, antidiabetic medications and liver histology in patients with diabetes with non-alcoholic fatty liver disease. BMJ Open Gastroenterol. 2016, 3, e000075. [Google Scholar] [CrossRef]
- Sfikas, G.; Psallas, M.; Koumaras, C.; Imprialos, K.; Perdikakis, E.; Doumas, M.; Giouleme, O.; Karagiannis, A.; Athyros, V.G. Prevalence, Diagnosis, and Treatment with 3 Different Statins of Non-alcoholic Fatty Liver Disease/Non-alcoholic Steatohepatitis in Military Personnel. Do Genetics Play a Role? Curr. Vasc. Pharmacol. 2021, 19, 572–581. [Google Scholar] [CrossRef]
- Nelson, A.; Torres, D.M.; Morgan, A.E.; Fincke, C.; Harrison, S.A. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenterol. 2009, 43, 990–994. [Google Scholar] [CrossRef]
- Oni, E.T.; Sinha, P.; Karim, A.; Martin, S.S.; Blaha, M.J.; Agatston, A.S.; Blumenthal, R.S.; Meneghelo, R.S.; Conceiçao, R.D.; Santos, R.D.; et al. Statin Use Is Not Associated with Presence of and Severity of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2014, 45, 52–57. [Google Scholar] [CrossRef]
- Lee, J.I.; Lee, H.W.; Lee, K.S.; Lee, H.S.; Park, J.-Y. Effects of Statin Use on the Development and Progression of Nonalcoholic Fatty Liver Disease: A Nationwide Nested Case-Control Study. Am. J. Gastroenterol. 2021, 116, 116–124. [Google Scholar] [CrossRef]
- Pinyopornpanish, K.; Al-Yaman, W.; Butler, R.S.; Carey, W.; McCullough, A.; Romero-Marrero, C. Chemopreventive Effect of Statin on Hepatocellular Carcinoma in Patients with Nonalcoholic Steatohepatitis Cirrhosis. Am. J. Gastroenterol. 2021, 116, 2258–2269. [Google Scholar] [CrossRef]
- German, M.N.; Lutz, M.K.; Pickhardt, P.J.; Bruce, R.J.; Said, A. Statin Use is Protective Against Hepatocellular Carcinoma in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Gastroenterol. 2020, 54, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Walenbergh, S.M.A.; Hofker, M.H.; Shiri-Sverdlov, R. Lysosomal cholesterol accumulation: Driver on the road to inflammation during atherosclerosis and non-alcoholic steatohepatitis. Obes. Rev. 2014, 15, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Vinué, Á.; Herrero-Cervera, A.; González-Navarro, H. Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models. Nutrients 2018, 10, 939. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Lastuvkova, H.; Faradonbeh, F.A.; Schreiberova, J.; Hroch, M.; Mokry, J.; Faistova, H.; Nova, Z.; Hyspler, R.; Igreja Sa, I.C.; Nachtigal, P.; et al. Atorvastatin Modulates Bile Acid Homeostasis in Mice with Diet-Induced Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2021, 22, 6468. [Google Scholar] [CrossRef]
- Kaminsky-Kolesnikov, Y.; Rauchbach, E.; Abu-Halaka, D.; Hahn, M.; García-Ruiz, C.; Fernandez-Checa, J.C.; Madar, Z.; Tirosh, O. Cholesterol Induces Nrf-2- and HIF-1α-Dependent Hepatocyte Proliferation and Liver Regeneration to Ameliorate Bile Acid Toxicity in Mouse Models of NASH and Fibrosis. Oxid. Med. Cell. Longev. 2020, 2020, 5393761. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Gan, L.T.; Yeh, M.M.; Haigh, W.G.; Larter, C.Z.; Ioannou, G.; Teoh, N.C.; Farrell, G.C. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J. Hepatol. 2013, 59, 144–152. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Van Rooyen, D.M.; Savard, C.; Haigh, W.G.; Yeh, M.M.; Teoh, N.C.; Farrell, G.C. Cholesterol-lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J. Lipid Res. 2015, 56, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Bravo, M.; Raurell, I.; Hide, D.; Fernández-Iglesias, A.; Gil, M.; Barberá, A.; Salcedo, M.T.; Augustin, S.; Genescà, J.; Martell, M. Restoration of liver sinusoidal cell phenotypes by statins improves portal hypertension and histology in rats with NASH. Sci. Rep. 2019, 9, 20183. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 2005, 44, 6371–6382. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N.; Bobryshev, Y.V. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie 2017, 132, 19–27. [Google Scholar] [CrossRef]
- Soran, H.; Younis, N.N.; Charlton-Menys, V.; Durrington, P. Variation in paraoxonase-1 activity and atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Watanabe, J.; Miura, K.; Gugliucci, A. Paraoxonase 1 and Non-Alcoholic Fatty Liver Disease: A Meta-Analysis. Molecules 2021, 26, 2323. [Google Scholar] [CrossRef] [PubMed]
- Samy, W.; Hassanian, M.A. Paraoxonase-1 activity, malondialdehyde and glutathione peroxidase in non-alcoholic fatty liver disease and the effect of atorvastatin. Arab. J. Gastroenterol. 2011, 12, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Goud, B.; Liu, S.; Storrie, B. Rab proteins as major determinants of the Golgi complex structure. Small GTPases 2018, 9, 66–75. [Google Scholar] [CrossRef]
- Laufs, U.; Wassmann, S.; Schackmann, S.; Heeschen, C.; Böhm, M.; Nickenig, G. Beneficial effects of statins in patients with non-ischemic heart failure. Z. Kardiol. 2004, 93, 103–108. [Google Scholar] [CrossRef]
- Rikitake, Y.; Hirata, K. Inhibition of RhoA or Rac1? Mechanism of cholesterol-independent beneficial effects of statins. Circ. J. 2009, 73, 231–232. [Google Scholar] [CrossRef]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Kilter, H.; Konkol, C.; Wassmann, S.; Böhm, M.; Nickenig, G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc. Res. 2002, 53, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Maybüchen, L.; Hittatiya, K.; Klein, S.; Uschner, F.E.; Braga, T.T.; Franklin, B.S.; Nickenig, G.; Strassburg, C.P.; Plat, J.; et al. Statins improve NASH via inhibition of RhoA and Ras. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G724–G733. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Roglans, N.; Sanguino, E.; Peris, C.; Alegret, M.; Vázquez, M.; Adzet, T.; Díaz, C.; Hernández, G.; Laguna, J.C.; Sánchez, R.M. Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J. Pharmacol. Exp. Ther. 2002, 302, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Marinho, T.d.S.; Kawasaki, A.; Bryntesson, M.; Souza-Mello, V.; Barbosa-da-Silva, S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Rosuvastatin limits the activation of hepatic stellate cells in diet-induced obese mice. Hepatol. Res. 2017, 47, 928–940. [Google Scholar] [CrossRef]
- Park, H.S.; Jang, J.E.; Ko, M.S.; Woo, S.H.; Kim, B.J.; Kim, H.S.; Park, H.S.; Park, I.S.; Koh, E.H.; Lee, K.U. Statins Increase Mitochondrial and Peroxisomal Fatty Acid Oxidation in the Liver and Prevent Non-Alcoholic Steatohepatitis in Mice. Diabetes Metab. J. 2016, 40, 376–385. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2022, 24, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lee, J.M.; Kim, J.-H.; Kim, K.R. Fluvastatin activates sirtuin 6 to regulate sterol regulatory element-binding proteins and AMP-activated protein kinase in HepG2 cells. Biochem. Biophys. Res. Commun. 2018, 503, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, C.; Zhang, X.; Li, N.; Dong, Z.; Sun, G.; Sun, X. Atorvastatin promotes AMPK signaling to protect against high fat diet-induced non-alcoholic fatty liver in golden hamsters. Exp. Ther. Med. 2020, 19, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-T.; Ho, H.J.; Lin, J.-T.; Shieh, J.-J.; Wu, C.-Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef]
- Protchenko, O.; Baratz, E.; Jadhav, S.; Li, F.; Shakoury-Elizeh, M.; Gavrilova, O.; Ghosh, M.C.; Cox, J.E.; Maschek, J.A.; Tyurin, V.A.; et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver from Lipid Peroxidation and Steatosis. Hepatology 2021, 73, 1176–1193. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Yao, X.; Xie, R.; Cao, Y.; Tang, J.; Men, Y.; Peng, H.; Yang, W. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J. Nanobiotechnol. 2021, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Kitsugi, K.; Noritake, H.; Matsumoto, M.; Hanaoka, T.; Umemura, M.; Yamashita, M.; Takatori, S.; Ito, J.; Ohta, K.; Chida, T.; et al. Simvastatin inhibits hepatic stellate cells activation by regulating the ferroptosis signaling pathway. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2023, 1869, 166750. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Foroutan, Z.; Katsiki, N.; Jamialahmadi, T.; Mantzoros, C.S. Ferroptosis, a new pathogenetic mechanism in cardiometabolic diseases and cancer: Is there a role for statin therapy? Metabolism 2023, 146, 155659. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2017, 3, 8–16. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef]
- Khan, T.J.; Ahmed, Y.M.; Zamzami, M.A.; Mohamed, S.A.; Khan, I.; Baothman, O.A.S.; Mehanna, M.G.; Yasir, M. Effect of atorvastatin on the gut microbiota of high fat diet-induced hypercholesterolemic rats. Sci. Rep. 2018, 8, 662. [Google Scholar] [CrossRef]
- Khan, T.J.; Ahmed, Y.M.; Zamzami, M.A.; Siddiqui, A.M.; Khan, I.; Baothman, O.A.S.; Mehanna, M.G.; Kuerban, A.; Kaleemuddin, M.; Yasir, M. Atorvastatin Treatment Modulates the Gut Microbiota of the Hypercholesterolemic Patients. OMICS A J. Integr. Biol. 2018, 22, 154–163. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X.; Huang, Y.; Chen, J.; Shang, W.; Shi, G.; Zhang, L.; Zhang, C.; Chen, R. Atorvastatin alleviates microglia-mediated neuroinflammation via modulating the microbial composition and the intestinal barrier function in ischemic stroke mice. Free Radic. Biol. Med. 2021, 162, 104–117. [Google Scholar] [CrossRef]
- Hu, X.; Li, H.; Zhao, X.; Zhou, R.; Liu, H.; Sun, Y.; Fan, Y.; Shi, Y.; Qiao, S.; Liu, S.; et al. Multi-omics study reveals that statin therapy is associated with restoration of gut microbiota homeostasis and improvement in outcomes in patients with acute coronary syndrome. Theranostics 2021, 11, 5778–5793. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.; An, J.; Song, Y.; Lee, C.-K.; Kim, K.; Kong, H. Alterations in Gut Microbiota by Statin Therapy and Possible Intermediate Effects on Hyperglycemia and Hyperlipidemia. Front. Microbiol. 2019, 10, 1947. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.A.; Skuse, P.; Govindarajan, K.; Patterson, E.; Konstantinidou, N.; Casey, P.G.; MacSharry, J.; Shanahan, F.; Stanton, C.; Hill, C.; et al. The influence of rosuvastatin on the gastrointestinal microbiota and host gene expression profiles. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G488–G497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.H.; Fu, K.; Lau, H.C.H.; Wang, Y.-X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Marchianò, S.; Biagioli, M.; Roselli, R.; Zampella, A.; Di Giorgio, C.; Bordoni, M.; Bellini, R.; Morretta, E.; Monti, M.C.; Distrutti, E.; et al. Atorvastatin protects against liver and vascular damage in a model of diet induced steatohepatitis by resetting FXR and GPBAR1 signaling. FASEB J. 2021, 36, e22060. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2021, 12, 411–425. [Google Scholar] [CrossRef]
- Chen, J.; Thomsen, M.; Vitetta, L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J. Cell. Biochem. 2019, 120, 2713–2720. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Wang, H.; Zhou, X.; Wei, X.; Xie, Z.; Zhang, Z.; Wang, K.; Mu, J. The influence of the intestinal microflora to the efficacy of Rosuvastatin. Lipids Health Dis. 2018, 17, 151. [Google Scholar] [CrossRef]
- He, X.; Zheng, N.; He, J.; Liu, C.; Feng, J.; Jia, W.; Li, H. Gut Microbiota Modulation Attenuated the Hypolipidemic Effect of Simvastatin in High-Fat/Cholesterol-Diet Fed Mice. J. Proteome Res. 2017, 16, 1900–1910. [Google Scholar] [CrossRef]
- Chen, G.; Wang, Z.; Song, W.; Liao, Y.; Wang, X.; Chen, C.; Ming, J.; Cui, J.; Xu, K. Effects of long-term regular oral aspirin combined with atorvastatin to prevent ischemic stroke on human gut microbiota. Eur. J. Pharmacol. 2023, 951, 175800. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kubeck, R.; Bonet-Ripoll, C.; Hoffmann, C.; Walker, A.; Muller, V.M.; Schuppel, V.L.; Lagkouvardos, I.; Scholz, B.; Engel, K.H.; Daniel, H.; et al. Dietary fat and gut microbiota interactions determine diet-induced obesity in mice. Mol. Metab. 2016, 5, 1162–1174. [Google Scholar] [CrossRef]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257 e246. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study Title | Agent | Dose/Day (Statins) | Status | Major Inclusion Criteria | Estimated Completion | Trial Number |
|---|---|---|---|---|---|---|
| NAFLD pharmacological treatment: metformin versus atorvastatin | Metformin and atorvastatin | 20 mg | Unknown, N/A | Signs of simple liver steatosis at ultrasonography | 1 June 2015 | NCT01544751 |
| NAFLD influence of statin therapy | Rosuvastatin | / | Withdrawn, N/A | Cardiology patient fatty liver in a cohort of patients | 1 September 2009 | NCT00375349 |
| Combination of obeticholic acid (OCA) and statins for monitoring of lipids (control) [54] | Obeticholic acid and atorvastatin | 10 mg | Completed, phase 2 | Histologic evidence of NASH | 12 March 2018 | NCT02633956 |
| Atorvastatin versus vitamin E in treatment of NAFLD | Atorvastatin | 20 mg | Unknown, N/A | Sign informed consent before involvement in any trial-related activity | 1 December 2016 | NCT01720719 |
| Phase IV study to evaluate the effects of statin monotherapy or statin/ezetimibe combination therapy on hepatic steatosis in patients with hyperlipidemia and NAFLD | Rosuvastatin | 5 mg | Completed, phase 4 | Patients diagnosed with fatty liver or liver fibroscan | 11 September 2019 | NCT03434613 |
| Atorvastatin, L-Carnitine and NASH | Atorvastatin, carnitine and atoral | 20 mg | Unknown, N/A | NASH diagnosed on the basis of established criteria | 1 December 2019 | NCT01617772 |
| Assessment of endothelial function in patients with NAFLD and the impact of statin treatment | Atorvastatin | 20 mg | Withdrawn, N/A | Patients with fatty liver | 1 December 2015 | NCT01987310 |
| Statins for the treatment of NASH | Atorvastatin | 40 mg | Recruiting, phase 2 | NASH or fibrosis stage ≥ 2 | 1 December 2024 | NCT04679376 |
| Effects of pitavastatin on insulin sensitivity and liver fat | Pitavastatin | 4 mg | Completed, Unknown | BMI ≥ 27 kg/m2 and waist circumference ≥ 102 cm, high probability risk factors for NAFLD | 30 April 2018 | NCT02290106 |
| Liver cirrhosis network rosuvastatin efficacy and safety for cirrhosis in the United States | Rosuvastatin | 20 mg | Recruiting, phase 2 | Cirrhosis due to NASH, alcohol-associated liver disease, or chronic viral hepatitis or cryptogenic cirrhosis | 1 November 2026 | NCT05832229 |
| Clinical effects of new approach on patients with NASH | Rosuvastatin, vitamin E, and N-acetyl cysteine | 20 mg | Not yet recruiting, early phase 1 | NASH diagnosis using fibroscan detecting the degree of steatosis and fibrosis | 17 January 2025 | NCT06105060 |
| Comparative clinical study to evaluate the efficacy and safety of rosuvastatin vs CoQ10 on NASH | Coenzyme Q10 and rosuvastatin | 20 mg | Not yet recruiting, phase 3 | Patients have established diagnosis of NASH | 1 April 2024 | NCT05731596 |
| Type of Statin | Study Design | Diseases | Dose/Day | Results | Comments | Reference |
|---|---|---|---|---|---|---|
| Atorvastatin | Rat | NAFLD | 30 mg/kg | Atorvastatin up-regulated the expression of PPARα, liver fatty acid β-oxidation, and reduced the liver TG | Atorvastatin treatment effectively improved NAFLD-related hyperlipidemia and inhibited liver steatosis, accompanied by modulating the expression of genes for regulating lipid metabolism | [74] |
| Mice | NAFLD-NASH | 4.5 mg/kg | Atorvastatin prevents cholesterol crystal formation, thereby precluding NLRP3 inflammasome activation to prevent further development of NAFLD | Atorvastatin prevents development of hepatic steatosis, inflammation and fibrosis in mice | [85] | |
| Human | Hypercholesteremia | 10 mg | Atorvastatin reduced LDL-C concentrations and the severity of hepatic steatosis | Atorvastatin effectively and safely reduces elevated hepatic enzyme concentrations in hypercholesterolemic patients | [78] | |
| Fluvastatin | Rat | NASH | 5 mg/kg or 10 mg/kg | Fluvastatin reduced steatosis and fibrosis scores, α-SMA protein expression, mRNA expression of pro-inflammatory and pro-fibrogenic genes in livers | Fluvastatin alleviated steatosis-induced HSCs activation and hepatic fibrogenesis through mitigating inflammation and oxidative stress | [75] |
| Rosuvastatin | Human | NASH | 10 mg | Rosuvastatin resulted in complete resolution of NASH in 19 patients, and lipid values were normalized | Rosuvastatin could ameliorate biopsy-proven NASH and reduce the risk of vascular and liver morbidity and mortality in NASH patients | [79] |
| Simvastatin | Mice | NAFLD | 20 mg/kg | Simvastatin restored antioxidant enzyme activity and decreased lipid peroxidation and ALE-RAGE pathway activation | Simvastatin improved microcirculatory function in NAFLD by downregulating oxidative and ALE-RAGE stress and attenuated steatosis, inflammation and fibrosis. | [86] |
| Disease | Type of Statin | Study Design | Dose/Day | Intestinal Microbial Changes | Results | Reference |
|---|---|---|---|---|---|---|
| Hypercholesteremia | Atorvastatin | Rat | 10, 15, 20 mg/kg | Proteobacteria increased, Firmicutes decreased | Intestinal microbial diversity increased | [149] |
| Human | 20 mg | Faecalibacterium prausnitzii, Akkermansia muciniphilaa and Oscillospira increased Desufovibrio decreased | Reduced pro-inflammatory bacteria and taxa associated with atherosclerosis formation and CVD progression | [150] | ||
| Ischemic stroke | Atorvastatin | Mice | 20 mg/kg | Firmicutes and Lactobacillus increased, Bacteroidetes decreased | Increased fecal butyrate level, promoted intestinal barrier function | [151] |
| Acute coronary syndrome | Statins | Human | / | Parabacteroides merdae decreased Bifidobacterium longum subsp. longum, Anaerostipes hadrus and Ruminococcus obeum increased | Specific changes in bacterial taxa were associated with disease severity or outcomes either directly or by mediating metabolites such as fatty acids and prenol lipids | [152] |
| Obesity | Atorvastatin | Mice | 10 mg/kg | Bacteroides, Butyricimonas, and Mucispirillum increased | Statins improved the inflammation associated microbiota in elderly obese mice | [153] |
| Rosuvastatin | 3 mg/kg 9.3 mg/kg | Lachnospiraceae, Rikenella and Coprococcus increased Akkermansiaceae, Proteobacteria decreased | [153,154] | |||
| NASH/NAFLD | Atorvastatin | Mice | 20 mg/kg | Mucispirillum, Desulfovibrio, Anaerotruncus and Desulfovibrionaceae decreased | Increased serum TCA and depleted 3-IPA | [155] |
| 20 mg/kg | Ruminococcaceae increased | Promoted the formation of deoxycholic acid and lithocholic acid | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Ren, X.; Zhang, B.; Lan, T.; Liu, B. A Systematic Review of Statins for the Treatment of Nonalcoholic Steatohepatitis: Safety, Efficacy, and Mechanism of Action. Molecules 2024, 29, 1859. https://doi.org/10.3390/molecules29081859
Zhang S, Ren X, Zhang B, Lan T, Liu B. A Systematic Review of Statins for the Treatment of Nonalcoholic Steatohepatitis: Safety, Efficacy, and Mechanism of Action. Molecules. 2024; 29(8):1859. https://doi.org/10.3390/molecules29081859
Chicago/Turabian StyleZhang, Shiqin, Xiaoling Ren, Bingzheng Zhang, Tian Lan, and Bing Liu. 2024. "A Systematic Review of Statins for the Treatment of Nonalcoholic Steatohepatitis: Safety, Efficacy, and Mechanism of Action" Molecules 29, no. 8: 1859. https://doi.org/10.3390/molecules29081859
APA StyleZhang, S., Ren, X., Zhang, B., Lan, T., & Liu, B. (2024). A Systematic Review of Statins for the Treatment of Nonalcoholic Steatohepatitis: Safety, Efficacy, and Mechanism of Action. Molecules, 29(8), 1859. https://doi.org/10.3390/molecules29081859