
Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning


Abstract
1. Introduction
2. Results and Discussion
2.1. Difference in Contacts of Structural Domains Revealed by Deep Learning
2.2. Free Energy Profiles and Structural Dynamics of BRD4 and BRD9
2.3. Structural Property of BRD4 and BRD9
2.4. MM-GBSA Calculations
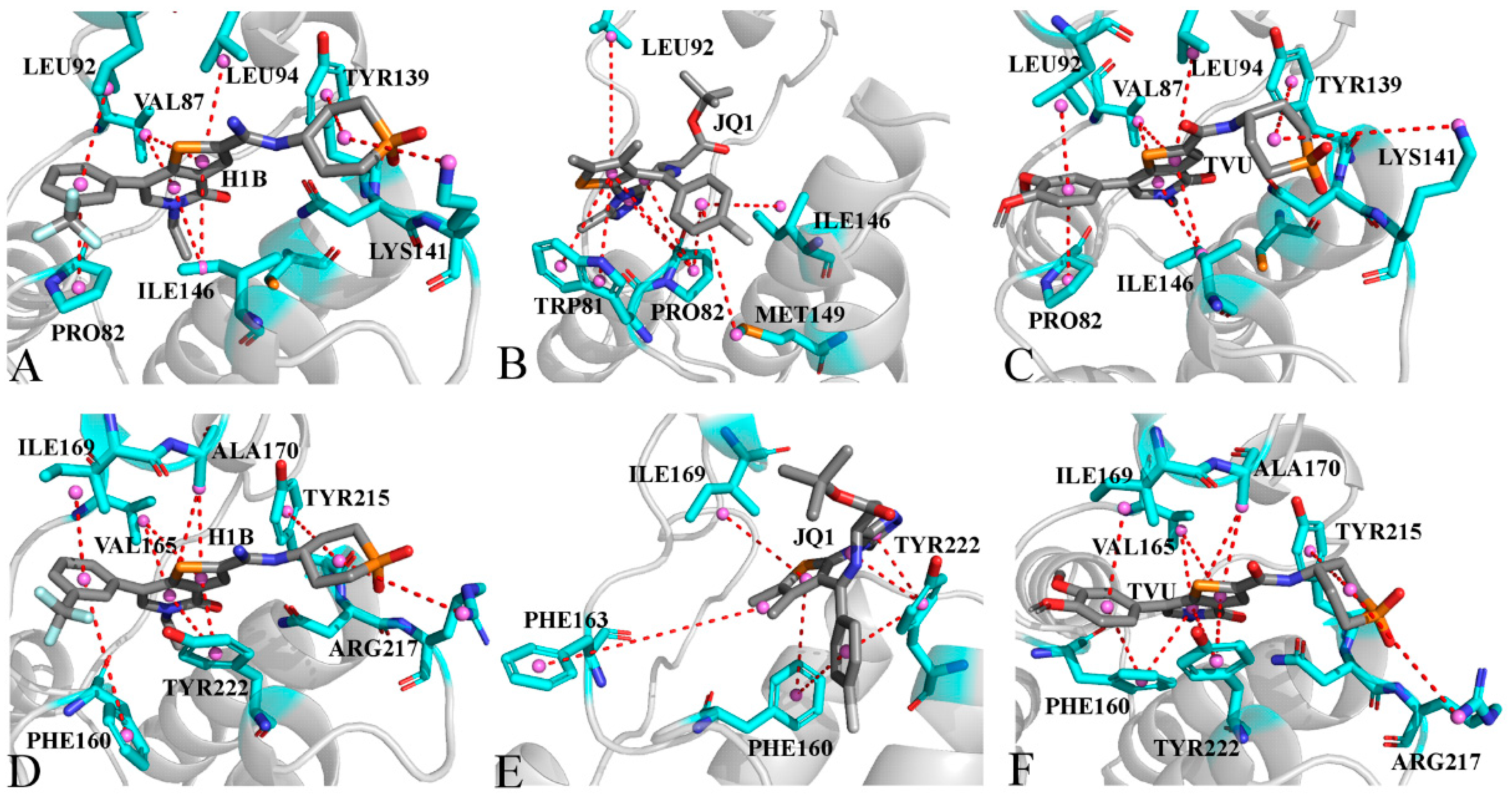
2.5. Interaction Network of Inhibitors with BRD9 and BRD4
3. Materials and Methods
3.1. Preparation of Simulation Systems
3.2. Multiple Independent Molecular Dynamics
3.3. Deep Learning
3.4. MM-GBSA Calculations
3.5. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.E.; Allis, C.D.; Strahl, B.D. OPERating ON Chromatin, a Colorful Language where Context Matters. J. Mol. Biol. 2011, 409, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.-W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-W. Small Molecule Bromodomain Inhibitors: Extending the Druggable Genome; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 51, pp. 1–55. [Google Scholar]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Rooney, T.P.C.; Jennings, L.E.; Hay, D.A.; Schofield, C.J.; Brennan, P.E.; Knapp, S.; Conway, S.J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413. [Google Scholar] [CrossRef] [PubMed]
- Jahagirdar, R.; Zhang, H.; Azhar, S.; Tobin, J.; Attwell, S.; Yu, R.; Wu, J.; McLure, K.G.; Hansen, H.C.; Wagner, G.S.; et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100. [Google Scholar] [CrossRef]
- Czerwinska, P.; Mackiewicz, A.A. Bromodomain (BrD) Family Members as Regulators of Cancer Stemness & mdash;A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 995. [Google Scholar]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Sabnis, R.W. BRD9 Bifunctional Degraders for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1879–1880. [Google Scholar]
- Karim, R.M.; Bikowitz, M.J.; Chan, A.; Zhu, J.-Y.; Grassie, D.; Becker, A.; Berndt, N.; Gunawan, S.; Lawrence, N.J.; Schönbrunn, E. Differential BET Bromodomain Inhibition by Dihydropteridinone and Pyrimidodiazepinone Kinase Inhibitors. J. Med. Chem. 2021, 64, 15772–15786. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Cheryala, N.; Karim, R.M.; Chan, A.; Berndt, N.; Qi, J.; Georg, G.I.; Schönbrunn, E. Bivalent BET Bromodomain Inhibitors Confer Increased Potency and Selectivity for BRDT via Protein Conformational Plasticity. J. Med. Chem. 2022, 65, 10441–10458. [Google Scholar] [CrossRef] [PubMed]
- Davison, G.; Martin, M.P.; Turberville, S.; Dormen, S.; Heath, R.; Heptinstall, A.B.; Lawson, M.; Miller, D.C.; Ng, Y.M.; Sanderson, J.N.; et al. Mapping Ligand Interactions of Bromodomains BRD4 and ATAD2 with FragLites and PepLites—Halogenated Probes of Druglike and Peptide-like Molecular Interactions. J. Med. Chem. 2022, 65, 15416–15432. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Duffy, B.C.; Liu, S.; Martin, G.S.; Wang, R.; Hsia, M.M.; Zhao, H.; Guo, C.; Ellis, M.; Quinn, J.F.; Kharenko, O.A.; et al. Discovery of a new chemical series of BRD4(1) inhibitors using protein-ligand docking and structure-guided design. Bioorg. Med. Chem. Lett. 2015, 25, 2818–2823. [Google Scholar] [CrossRef]
- Myers, S.M.; Miller, D.C.; Molyneux, L.; Arasta, M.; Bawn, R.H.; Blackburn, T.J.; Cook, S.J.; Edwards, N.; Endicott, J.A.; Golding, B.T.; et al. Identification of a novel orally bioavailable ERK5 inhibitor with selectivity over p38α and BRD4. Eur. J. Med. Chem. 2019, 178, 530–543. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, D.; Chen, T.; Wang, Y.; Miao, Z.; Xu, Y.; Chen, W.; Wang, X.; Li, Y.; Du, Z.; et al. Fragment-Based Drug Discovery of 2-Thiazolidinones as Inhibitors of the Histone Reader BRD4 Bromodomain. J. Med. Chem. 2013, 56, 3833–3851. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liu, X.; Zhang, S.; Yan, F.; Zhang, Q.; Chen, J. A computational insight into binding modes of inhibitors XD29, XD35, and XD28 to bromodomain-containing protein 4 based on molecular dynamics simulations. J. Biomol. Struct. Dyn. 2018, 36, 1212–1224. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- Yin, M.; Guo, Y.; Hu, R.; Cai, W.L.; Li, Y.; Pei, S.; Sun, H.; Peng, C.; Li, J.; Ye, R.; et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020, 11, 1833. [Google Scholar] [CrossRef]
- Andrieu, G.; Tran, A.H.; Strissel, K.J.; Denis, G.V. BRD4 Regulates Breast Cancer Dissemination through Jagged1/Notch1 Signaling. Cancer Res. 2016, 76, 6555–6567. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Walser, R.; Kaderli, J.; Rossi, E.; Bobby, R.; Packer, M.J.; Sarda, S.; Walker, G.; Hitchin, J.R.; Milbradt, A.G.; et al. NMR Molecular Replacement Provides New Insights into Binding Modes to Bromodomains of BRD4 and TRIM24. J. Med. Chem. 2022, 65, 5565–5574. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.M.; Chan, A.; Zhu, J.-Y.; Schönbrunn, E. Structural Basis of Inhibitor Selectivity in the BRD7/9 Subfamily of Bromodomains. J. Med. Chem. 2020, 63, 3227–3237. [Google Scholar] [CrossRef] [PubMed]
- Middeljans, E.; Wan, X.; Jansen, P.W.; Sharma, V.; Stunnenberg, H.G.; Logie, C. SS18 Together with Animal-Specific Factors Defines Human BAF-Type SWI/SNF Complexes. PLoS ONE 2012, 7, e33834. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.G.K.; Vieira, L.C.C.; Tallant, C.; Fedorov, O.; Singleton, D.C.; Rogers, C.M.; Monteiro, O.P.; Bennett, J.M.; Baronio, R.; Müller, S.; et al. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem. Int. Ed. 2015, 54, 6217–6221. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.D.; Vartanian, S.; Côté, A.; Bellon, S.; Duplessis, M.; Flynn, E.M.; Hewitt, M.; Huang, H.-R.; Kiefer, J.R.; Murray, J.; et al. Inhibition of bromodomain-containing protein 9 for the prevention of epigenetically-defined drug resistance. Bioorg. Med. Chem. Lett. 2017, 27, 3534–3541. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.-L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Picaud, S.; Strocchia, M.; Terracciano, S.; Lauro, G.; Mendez, J.; Daniels, D.L.; Riccio, R.; Bifulco, G.; Bruno, I.; Filippakopoulos, P. 9H-Purine Scaffold Reveals Induced-Fit Pocket Plasticity of the BRD9 Bromodomain. J. Med. Chem. 2015, 58, 2718–2736. [Google Scholar] [CrossRef]
- Clegg, M.A.; Bamborough, P.; Chung, C.-W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Leveridge, M.; Lindon, M.; Liwicki, G.M.; Michon, A.-M.; et al. Application of Atypical Acetyl-lysine Methyl Mimetics in the Development of Selective Inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J. Med. Chem. 2020, 63, 5816–5840. [Google Scholar] [CrossRef]
- Cui, H.; Divakaran, A.; Hoell, Z.J.; Ellingson, M.O.; Scholtz, C.R.; Zahid, H.; Johnson, J.A.; Griffith, E.C.; Gee, C.T.; Lee, A.L.; et al. A Structure-based Design Approach for Generating High Affinity BRD4 D1-Selective Chemical Probes. J. Med. Chem. 2022, 65, 2342–2360. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-B.; Xiang, Q.-P.; Wang, C.; Wu, C.; Zhang, C.; Zhang, M.-F.; Liu, Z.-X.; Zhang, Y.; Xiao, L.-J.; Xu, Y. Y06014 is a selective BET inhibitor for the treatment of prostate cancer. Acta Pharmacol. Sin. 2021, 42, 2120–2131. [Google Scholar] [CrossRef]
- Su, J.; Liu, X.; Zhang, S.; Yan, F.; Zhang, Q.; Chen, J. A theoretical insight into selectivity of inhibitors toward two domains of bromodomain-containing protein 4 using molecular dynamics simulations. Chem. Biol. Drug Des. 2018, 91, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Yang, Z.; Xu, S.; Li, H. Binding Selectivity of Inhibitors toward Bromodomains BAZ2A and BAZ2B Uncovered by Multiple Short Molecular Dynamics Simulations and MM-GBSA Calculations. ACS Omega 2021, 6, 12036–12049. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Jiang, F.; Hu, Q.; Zhang, Z.; Li, H.; Li, H.; Zhang, D.; Li, H.; Ma, Y.; Xu, J.; Chen, H.; et al. Discovery of Benzo[cd]indol-2(1H)-ones and Pyrrolo[4,3,2-de]quinolin-2(1H)-ones as Bromodomain and Extra-Terminal Domain (BET) Inhibitors with Selectivity for the First Bromodomain with Potential High Efficiency against Acute Gouty Arthritis. J. Med. Chem. 2019, 62, 11080–11107. [Google Scholar] [CrossRef]
- Su, J.; Liu, X.; Zhang, S.; Yan, F.; Zhang, Q.; Chen, J. Insight into selective mechanism of class of I-BRD9 inhibitors toward BRD9 based on molecular dynamics simulations. Chem. Biol. Drug Des. 2019, 93, 163–176. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Yu, Y.; Liu, D.; Zhao, J.; Zhang, L. Deciphering Selectivity Mechanism of BRD9 and TAF1(2) toward Inhibitors Based on Multiple Short Molecular Dynamics Simulations and MM-GBSA Calculations. Molecules 2023, 28, 2583. [Google Scholar] [CrossRef]
- Sun, J.; Liu, X.; Zhang, S.; Li, M.; Zhang, Q.; Chen, J. Molecular insights and optimization strategies for the competitive binding of engineered ACE2 proteins: A multiple replica molecular dynamics study. Phys. Chem. Chem. Phys. 2023, 25, 28479–28496. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, T.; Chen, J. Exploring drug-resistant mechanisms of I84V mutation in HIV-1 protease toward different inhibitors by thermodynamics integration and solvated interaction energy method. Chem. Phys. Lett. 2018, 706, 400–408. [Google Scholar] [CrossRef]
- Sun, Z.; Gong, Z.; Xia, F.; He, X. Ion dynamics and selectivity of Nav channels from molecular dynamics simulation. Chem. Phys. 2021, 548, 111245. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Yan, D.; Zeng, Y.; Wei, B.; Chen, J.; He, W. Identification Mechanism of BACE1 on Inhibitors Probed by Using Multiple Separate Molecular Dynamics Simulations and Comparative Calculations of Binding Free Energies. Molecules 2023, 28, 4773. [Google Scholar] [CrossRef]
- Xue, W.; Yang, F.; Wang, P.; Zheng, G.; Chen, Y.; Yao, X.; Zhu, F. What Contributes to Serotonin–Norepinephrine Reuptake Inhibitors’ Dual-Targeting Mechanism? The Key Role of Transmembrane Domain 6 in Human Serotonin and Norepinephrine Transporters Revealed by Molecular Dynamics Simulation. ACS Chem. Neurosci. 2018, 9, 1128–1140. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, D.; Fan, J.; Li, M.; Zhang, J.; Hua, C.; Nussinov, R.; Lu, S. Markov State Models and Molecular Dynamics Simulations Reveal the Conformational Transition of the Intrinsically Disordered Hypervariable Region of K-Ras4B to the Ordered Conformation. J. Chem. Inf. Model. 2022, 62, 4222–4231. [Google Scholar] [CrossRef]
- Şenol, H.; Ghaffari-Moghaddam, M.; Alim Toraman, G.Ö.; Güller, U. Novel chalcone derivatives of ursolic acid as acetylcholinesterase inhibitors: Synthesis, characterization, biological activity, ADME prediction, molecular docking and molecular dynamics studies. J. Mol. Struct. 2024, 1295, 136804. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Miao, Y. Peptide Gaussian accelerated molecular dynamics (Pep-GaMD): Enhanced sampling and free energy and kinetics calculations of peptide binding. J. Chem. Phys. 2020, 153, 154109. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.-M.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics: Principles and applications. WIREs Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Y.; Yan, D.; Liu, Z.; Wei, B.; Chen, J.; He, W. Binding Mechanism of Inhibitors to Heat Shock Protein 90 Investigated by Multiple Independent Molecular Dynamics Simulations and Prediction of Binding Free Energy. Molecules 2023, 28, 4792. [Google Scholar] [CrossRef]
- Sun, Z.; Huai, Z.; He, Q.; Liu, Z. A General Picture of Cucurbit[8]uril Host–Guest Binding. J. Chem. Inf. Model. 2021, 61, 6107–6134. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Pang, L.; Zhang, J.Z.H.; Zhu, T. Effect of mutations on binding of ligands to guanine riboswitch probed by free energy perturbation and molecular dynamics simulations. Nucleic Acids Res. 2019, 47, 6618–6631. [Google Scholar] [CrossRef]
- Xue, W.; Wang, P.; Tu, G.; Yang, F.; Zheng, G.; Li, X.; Li, X.; Chen, Y.; Yao, X.; Zhu, F. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys. Chem. Chem. Phys. 2018, 20, 6606–6616. [Google Scholar] [CrossRef]
- Jia, Z.-J.; Lan, X.-W.; Lu, K.; Meng, X.; Jing, W.-J.; Jia, S.-R.; Zhao, K.; Dai, Y.-J. Synthesis, molecular docking, and binding Gibbs free energy calculation of β-nitrostyrene derivatives: Potential inhibitors of SARS-CoV-2 3CL protease. J. Mol. Struct. 2023, 1284, 135409. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J.A. Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 12162–12167. [Google Scholar] [CrossRef]
- Wang, J.; Miao, Y. Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors. J. Phys. Chem. B 2019, 123, 6462–6473. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, Q.; Wang, W.; Sun, H.; Hu, G. Decoding the Identification Mechanism of an SAM-III Riboswitch on Ligands through Multiple Independent Gaussian-Accelerated Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 6118–6132. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J. Chem. Inf. Model. 2021, 61, 1954–1969. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Sun, H.; He, W. Roles of Accelerated Molecular Dynamics Simulations in Predictions of Binding Kinetic Parameters. Mini-Rev. Med. Chem. 2024, 24. [Google Scholar] [CrossRef]
- dos Santos Nascimento, J.I.; Santana Gomes, J.N.; de Oliveira Viana, J.; de Medeiros e Silva, M.S.Y.; Barbosa, G.E.; de Moura, O.R. The Power of Molecular Dynamics Simulations and Their Applications to Discover Cysteine Protease Inhibitors. Mini-Rev. Med. Chem. 2024, 24, 1125–1146. [Google Scholar] [CrossRef]
- Plante, A.; Shore, D.M.; Morra, G.; Khelashvili, G.; Weinstein, H. A Machine Learning Approach for the Discovery of Ligand-Specific Functional Mechanisms of GPCRs. Molecules 2019, 24, 2097. [Google Scholar] [CrossRef]
- Plante, A.; Weinstein, H. Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol. Molecules 2021, 26, 3059. [Google Scholar] [CrossRef]
- Do, H.N.; Wang, J.; Bhattarai, A.; Miao, Y. GLOW: A Workflow Integrating Gaussian-Accelerated Molecular Dynamics and Deep Learning for Free Energy Profiling. J. Chem. Theory Comput. 2022, 18, 1423–1436. [Google Scholar] [CrossRef]
- Do, H.N.; Wang, J.; Miao, Y. Deep Learning Dynamic Allostery of G-Protein-Coupled Receptors. JACS Au 2023, 3, 3165–3180. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Levy, R.M.; Srinivasan, A.R.; Olson, W.K.; McCammon, J.A. Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers 1984, 23, 1099–1112. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; Wang, W.; Sun, H.; Pang, L.; Bao, H. Conformational transformation of switch domains in GDP/K-Ras induced by G13 mutants: An investigation through Gaussian accelerated molecular dynamics simulations and principal component analysis. Comput. Biol. Med. 2021, 135, 104639. [Google Scholar] [CrossRef]
- Bao, H.; Wang, W.; Sun, H.; Chen, J. Probing mutation-induced conformational transformation of the GTP/M-RAS complex through Gaussian accelerated molecular dynamics simulations. J. Enzym. Inhib. Med. Chem. 2023, 38, 2195995. [Google Scholar] [CrossRef]
- Weisberg, E.; Chowdhury, B.; Meng, C.; Case, A.E.; Ni, W.; Garg, S.; Sattler, M.; Azab, A.K.; Sun, J.; Muz, B.; et al. BRD9 degraders as chemosensitizers in acute leukemia and multiple myeloma. Blood Cancer J. 2022, 12, 110. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000, 303, 567–582. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 14937–14942. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | H1B | JQ1 | TVU | |||
|---|---|---|---|---|---|---|
| Average | Std | Average | Std | Average | Std | |
| −29.97 | 9.39 | −22.91 | 15.93 | −34.45 | 9.84 | |
| −41.78 | 3.38 | −28.51 | 7.13 | −44.36 | 2.92 | |
| 42.71 | 7.61 | 33.53 | 15.38 | 48.59 | 7.69 | |
| −5.70 | 0.44 | −3.50 | 0.90 | −5.97 | 0.33 | |
| 12.74 | 12.09 | 10.62 | 22.14 | 14.14 | 12.49 | |
| 18.65 | 5.52 | 15.01 | 6.13 | 19.34 | 6.15 | |
| −16.09 | 5.81 | −6.38 | 6.54 | −16.85 | 6.44 | |
| −10.00 | ||||||
| Complex | H1B | JQ1 | TVU | |||
|---|---|---|---|---|---|---|
| Average | Std | Average | Std | Average | Std | |
| −43.82 | 7.69 | 15.43 | 9.03 | −39.46 | 4.91 | |
| −48.74 | 2.89 | −40.37 | 4.25 | −50.03 | 3.28 | |
| 52.35 | 6.15 | −2.66 | 8.85 | 50.06 | 4.22 | |
| −6.29 | 0.30 | −4.58 | 0.41 | −6.33 | 0.29 | |
| 3.62 | 6.80 | 12.77 | 12.65 | 10.60 | 6.48 | |
| 25.11 | 4.35 | 20.24 | 3.75 | 22.58 | 4.15 | |
| −21.38 | 6.08 | −11.94 | 5.55 | −23.17 | 5.26 | |
| −10.16 | −11.35 | |||||
| Inhibitor | Donor | Acceptor | a Distance(Å) | a Angle(°) | b Occupied(%) |
|---|---|---|---|---|---|
| H1B | ASN140:ND2-HD21 | H1B:O33 | 3.10 | 158.21 | 87.02 |
| H1B:N37-H13 | ASN:140:OD1 | 3.96 | 157.19 | 90.26 | |
| LYS141:N-H | H1B:O49 | 2.96 | 160.86 | 61.73 | |
| TVU | ASN140:ND2-HD21 | TVU:O2 | 2.94 | 158.70 | 99.62 |
| TVU:N1-H4 | ASN140:OD1 | 3.20 | 152.23 | 78.73 | |
| LYS141:N-H | TVU:O5 | 2.94 | 159.11 | 65.18 |
| Inhibitor | Donor | Acceptor | a Distance(Å) | a Angle(°) | b Occupied(%) |
|---|---|---|---|---|---|
| H1B | ASN216:ND2-HD21 | H1B:O33 | 2.96 | 155.69 | 99.16 |
| H1B:N37-H13 | ASN216:OD1 | 2.93 | 160.29 | 99.64 | |
| ARG217:N-H | H1B:O49 | 2.96 | 164.08 | 96.82 | |
| H1B:N35-H12 | ILE169:O | 3.00 | 153.78 | 54.80 | |
| TVU | ASN216:ND2-HD21 | TVU:O2 | 2.86 | 157.25 | 99.96 |
| TVU:N1-H10 | ASN216:OD1 | 3.26 | 152.05 | 69.30 | |
| ARG217:N-H | TVU:O5 | 2.96 | 160.72 | 96.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yang, W.; Zhao, L.; Wei, B.; Chen, J. Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning. Molecules 2024, 29, 1857. https://doi.org/10.3390/molecules29081857
Wang J, Yang W, Zhao L, Wei B, Chen J. Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning. Molecules. 2024; 29(8):1857. https://doi.org/10.3390/molecules29081857
Chicago/Turabian StyleWang, Jian, Wanchun Yang, Lu Zhao, Benzheng Wei, and Jianzhong Chen. 2024. "Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning" Molecules 29, no. 8: 1857. https://doi.org/10.3390/molecules29081857
APA StyleWang, J., Yang, W., Zhao, L., Wei, B., & Chen, J. (2024). Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning. Molecules, 29(8), 1857. https://doi.org/10.3390/molecules29081857