
A Novel IRAK4 Inhibitor DW18134 Ameliorates Peritonitis and Inflammatory Bowel Disease

Abstract
1. Introduction
2. Results
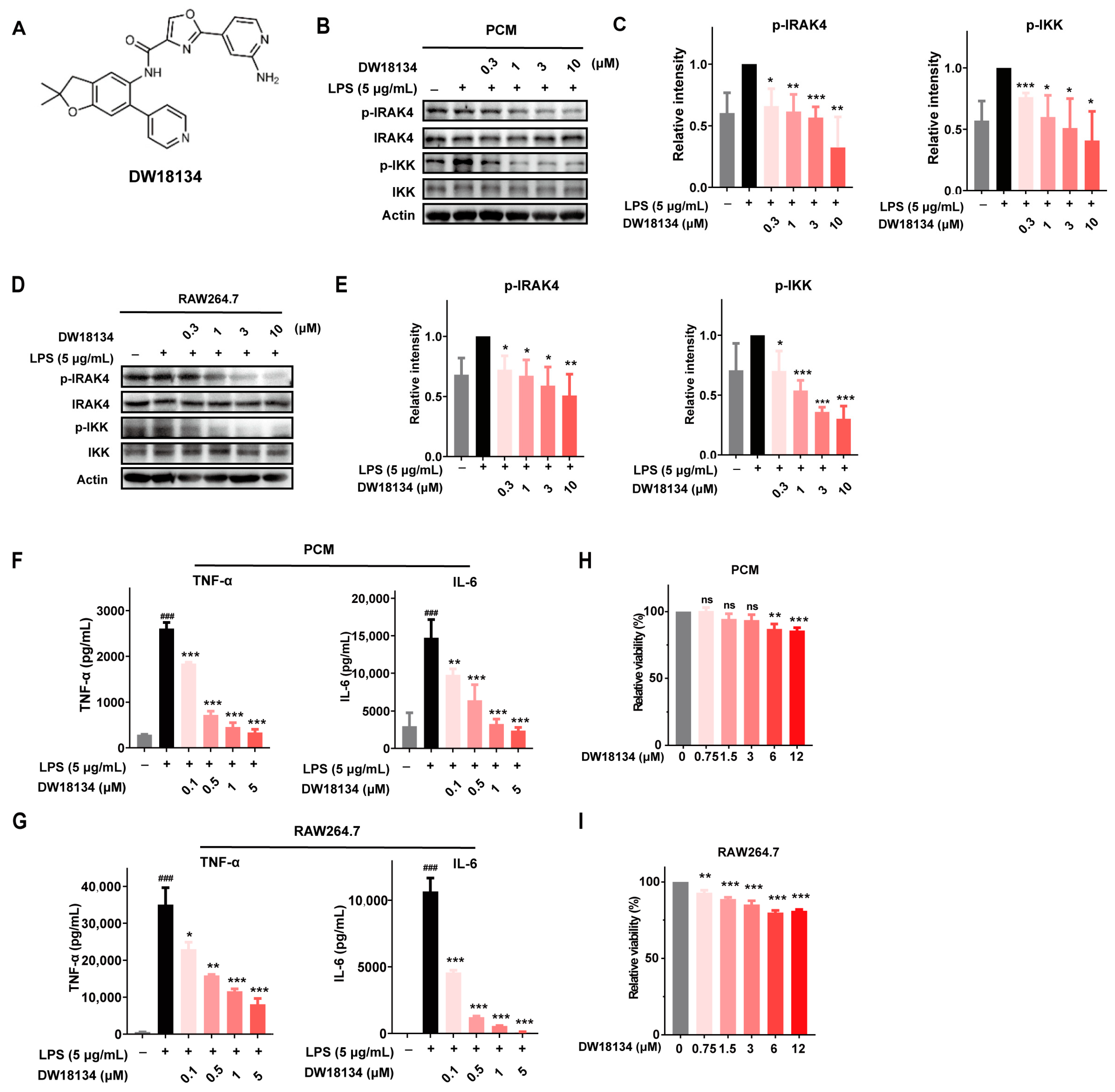
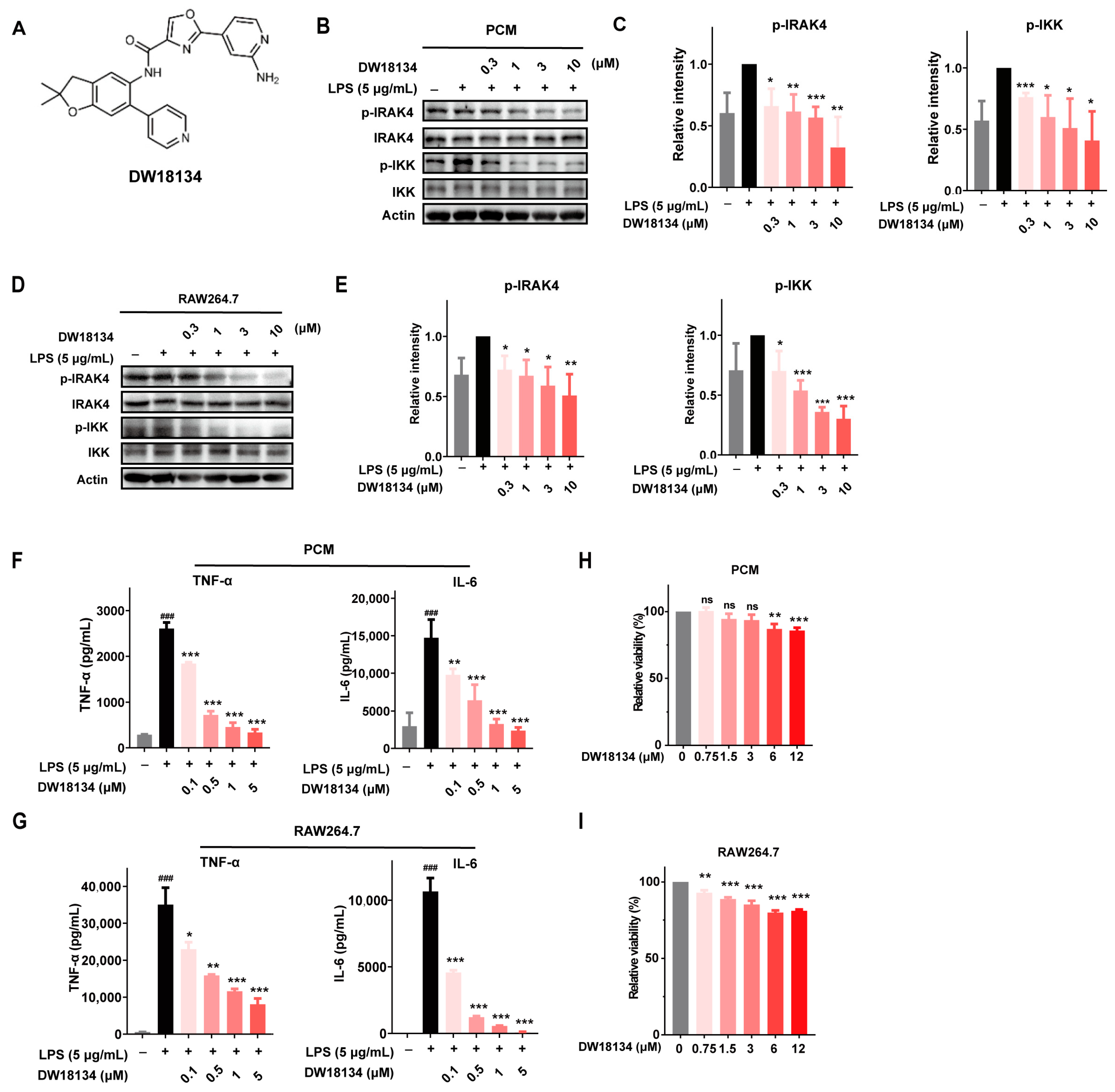
2.1. DW18134 Potently Inhibited IRAK4 Kinase Activity and Attenuated the LPS-Induced IRAK4 Signaling Transduction and Secretion of Cytokines in Macrophages
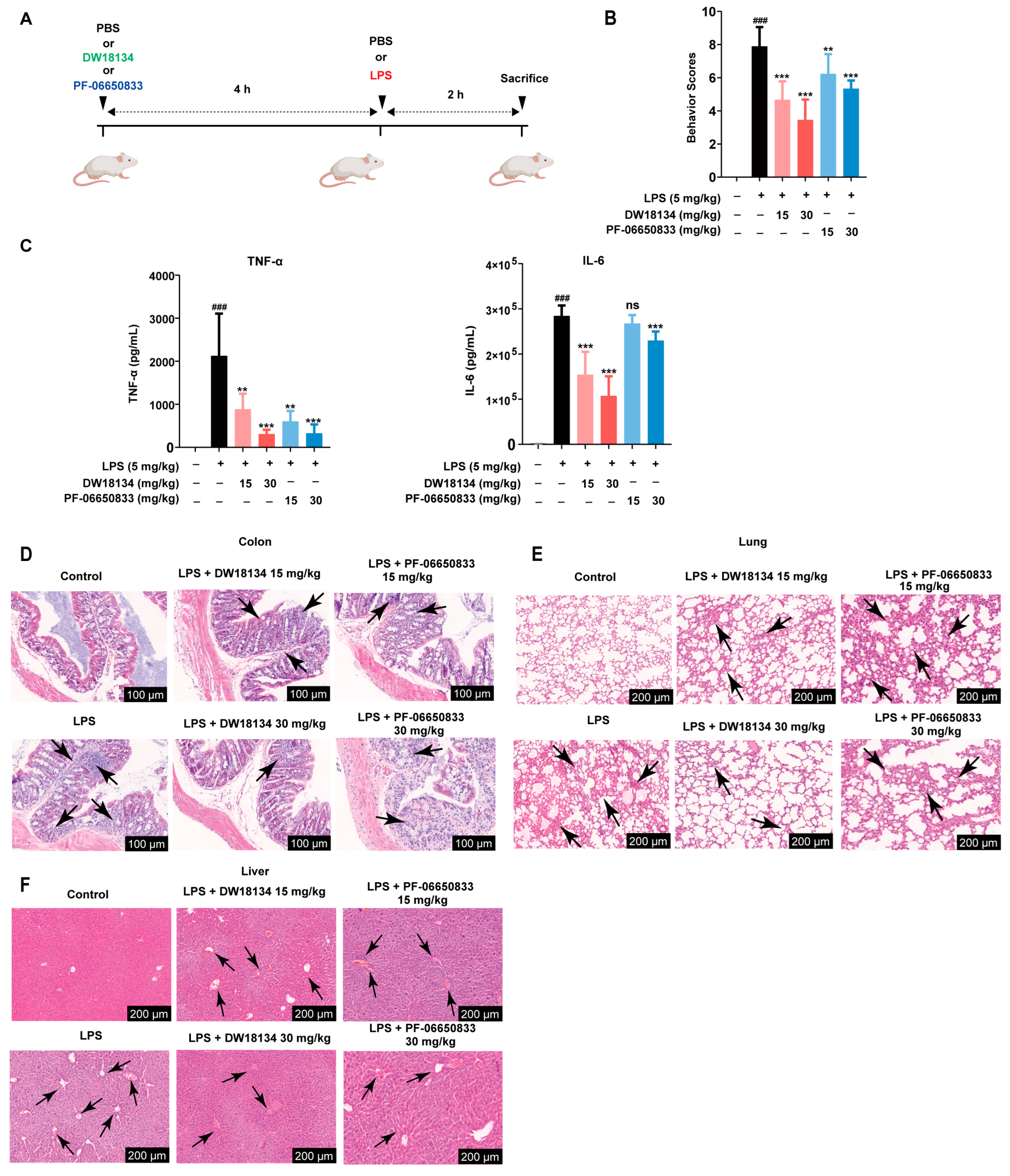
2.2. DW18134 Significantly Decreased LPS-Induced Inflammatory and Pathological Injury in the Mice Peritonitis Model
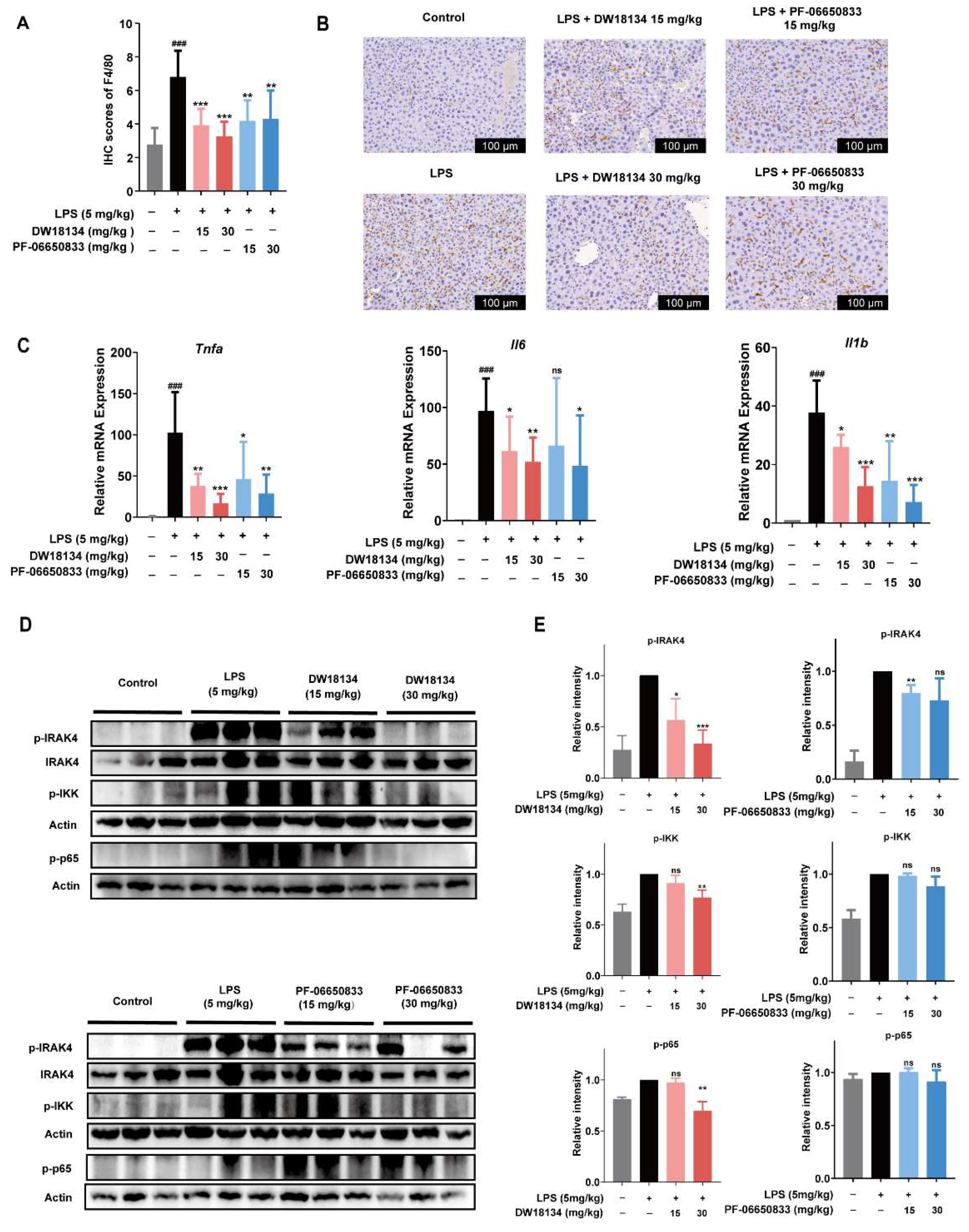
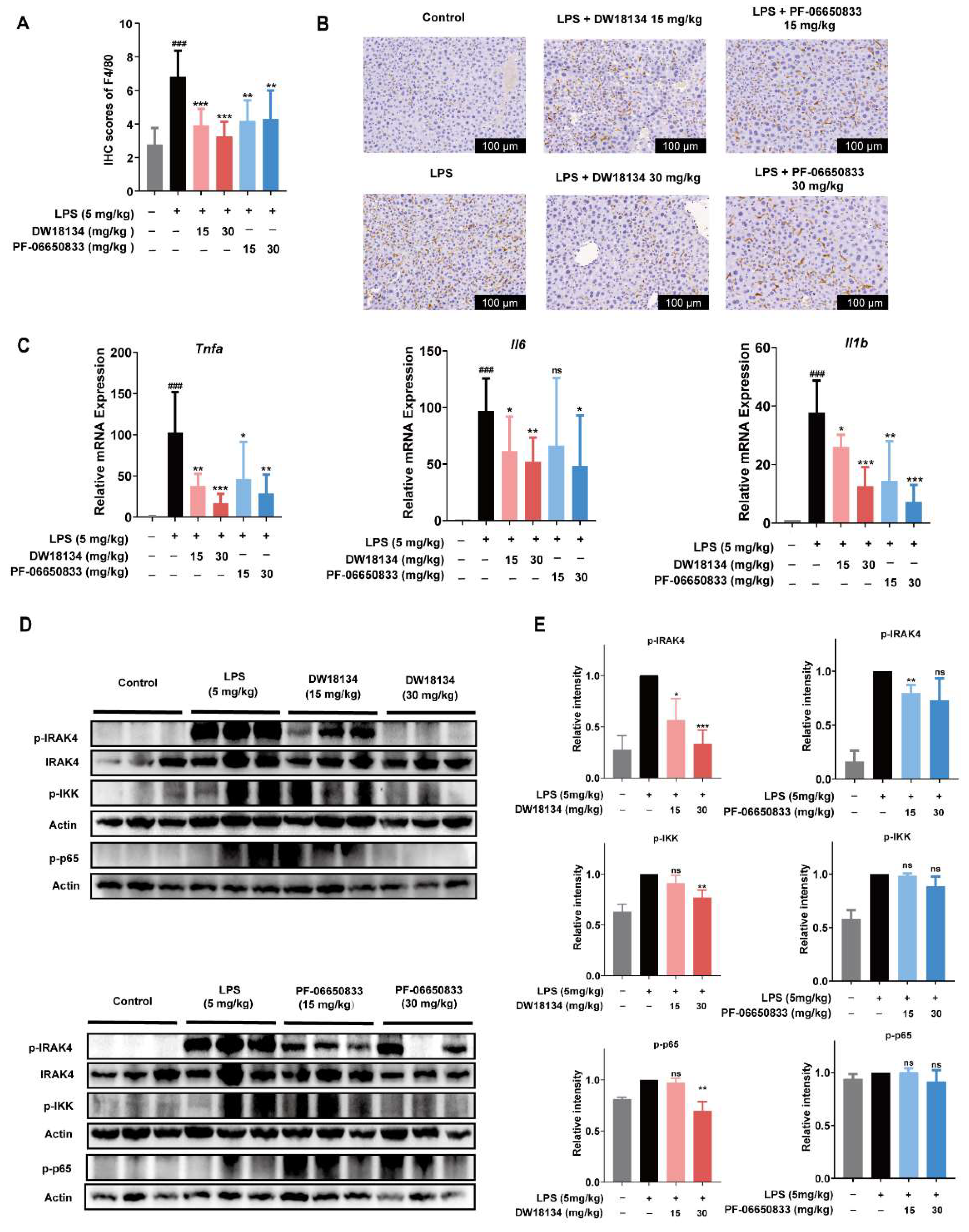
2.3. DW18134 Reduced Macrophage Infiltration and Pro-Inflammatory Gene Expression and Regulated IRAK4 Signaling Pathway Activation in LPS-Induced Peritonitis Mice
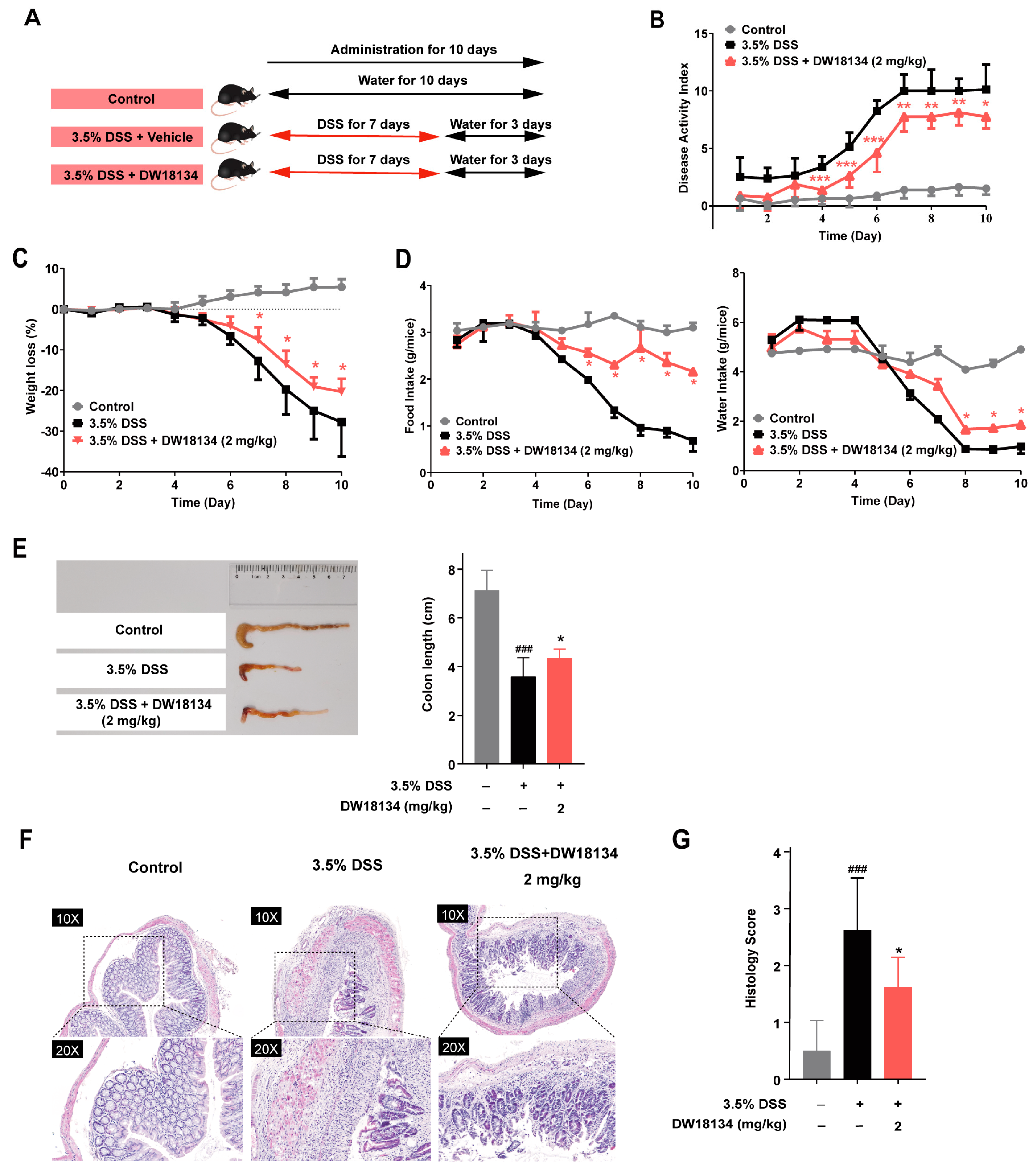
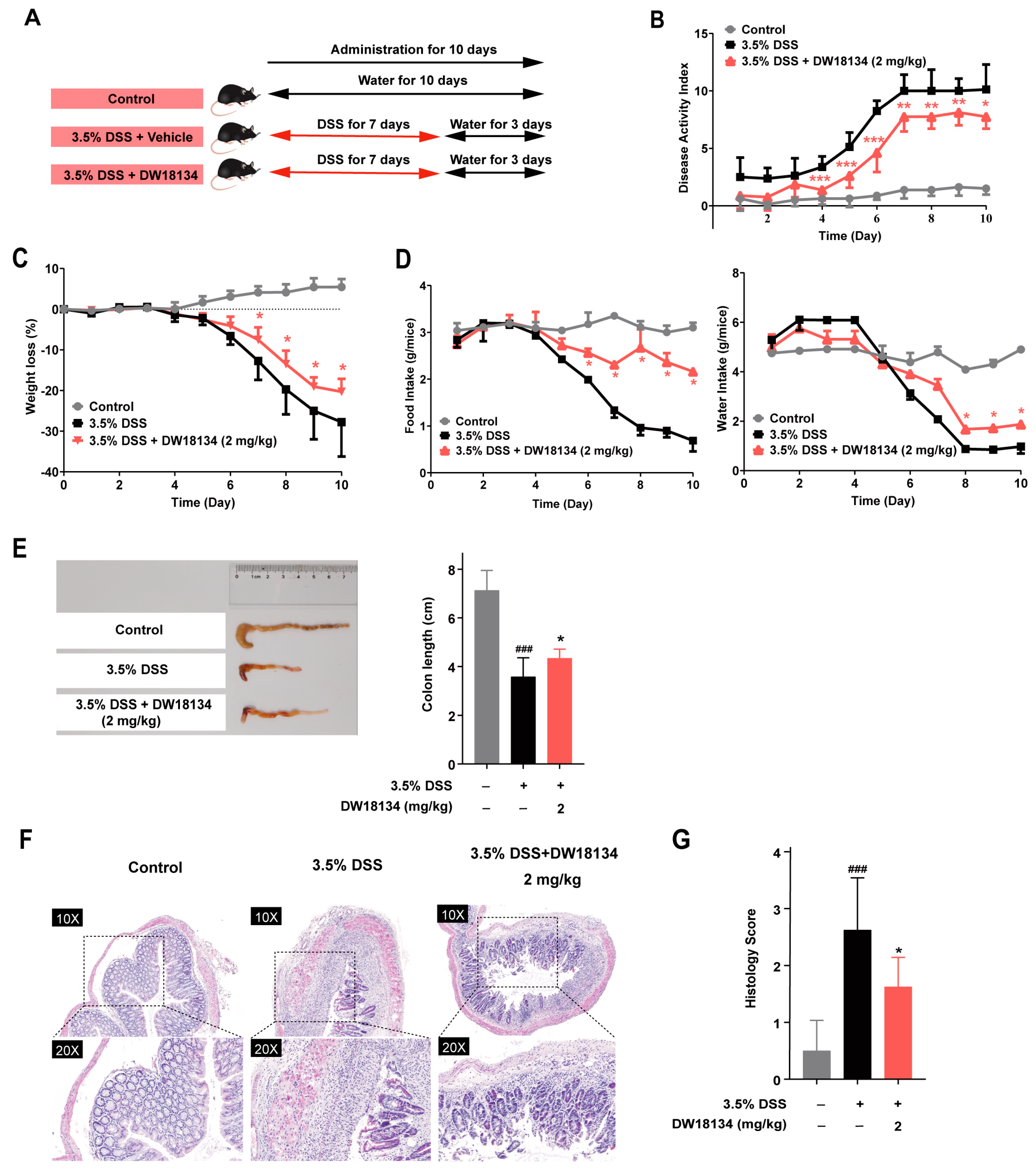
2.4. DW18134 Effectively Ameliorated Symptoms and Colonic Inflammatory Injury of DSS-Induced Colitis in Mice
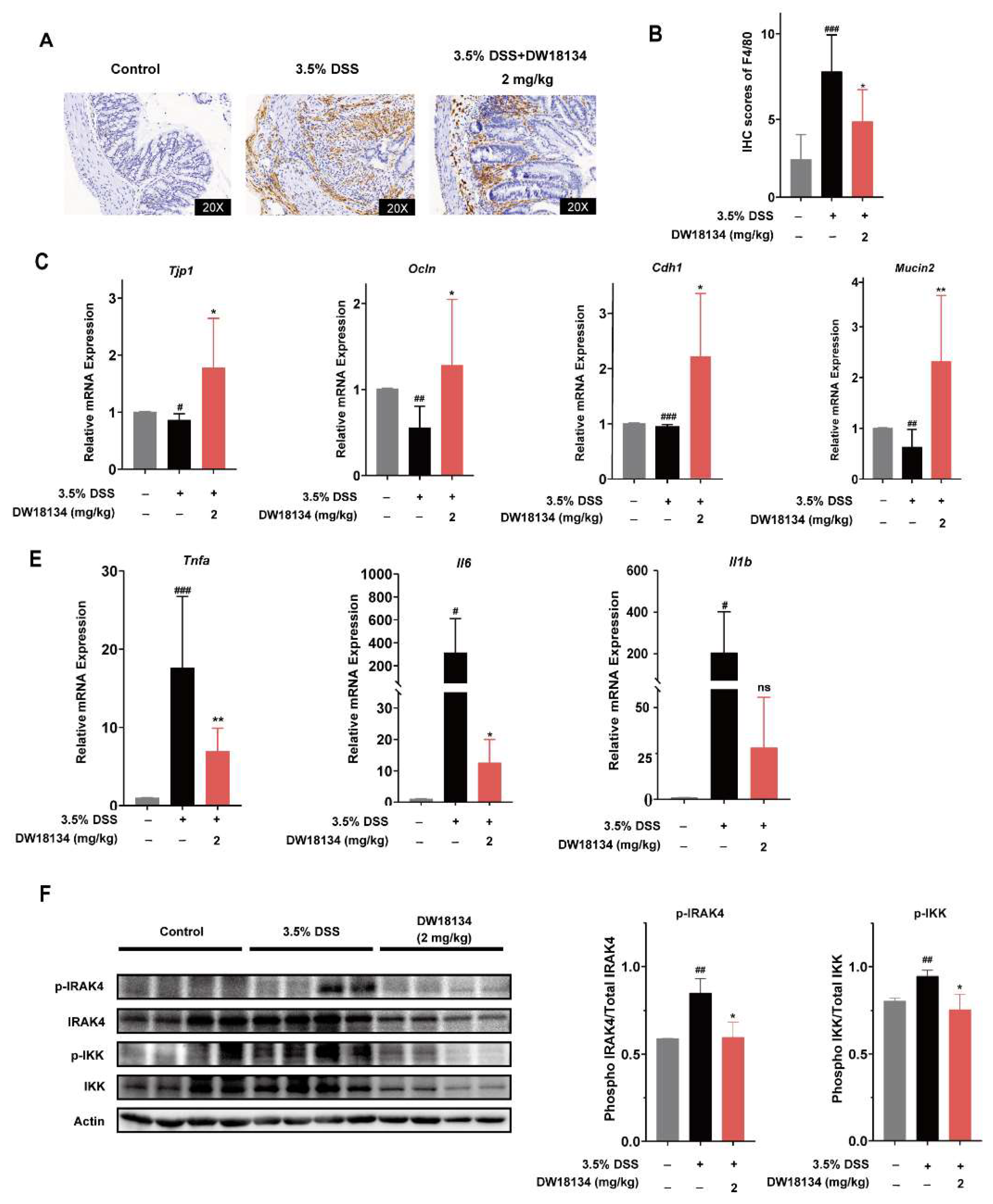
2.5. DW18134 Reduced Colonic Macrophage Infiltration and Inflammatory Factor Expression and Restored Intestinal Barrier in Mice with DSS-Induced Colitis
3. Materials and Methods
3.1. Compounds
3.2. Reagents and Antibodies
3.3. Cell Culture and Treatments
3.4. Cell Viability Assay
3.5. Western Blot Analysis and Antibodies
3.6. Real-Time Quantitative PCR
3.7. Enzyme-Linked Immunosorbent Assay (ELISA)
3.8. Animals and Treatments
3.9. Induction of DSS-Induced Experimental Colitis in Mice and Drug Treatment
3.10. Disease Activity Index (DAI)
3.11. Induction of LPS-Induced Peritonitis in Mice and Drug Treatment
3.12. Hematoxylin and Eosin
3.13. Immunohistochemistry Analysis
3.14. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BTK | Bruton’s tyrosine kinase |
| CD | Crohn’s disease |
| COPD | chronic obstructive pulmonary disease |
| DSS | dextran sulfate sodium salt |
| DAI | disease activity index |
| ELISA | enzyme linked immunosorbent assay |
| DLBCL | diffuse large B-cell lymphoma |
| IL-1β | interleukin 1 beta |
| IBD | inflammatory bowel disease |
| IL-6 | interleukin-6 |
| JAK | Janus kinase |
| LPS | lipopolysaccharide |
| IRAK4 | interleukin receptor-associated kinase 4 |
| IKK | inhibitor of Kappa B kinase |
| IL-1R | interleukin-1 receptor |
| Myd88 | myeloid differentiation factor 88 |
| MZL | marginal zone lymphomas |
| MUC2 | mucoprotein |
| RT-qPCR | real-time quantitative PCR |
| RA | rheumatoid arthritis |
| SD | standard deviation |
| SBDD | Structure-based drug design |
| TNF-α | tumor necrosis factor alpha |
| TLR | toll-like receptor |
| UC | ulcerative colitis |
| ZO-1 | zonula Occludin protein 1 |
References
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Marx, W.; Veronese, N.; Kelly, J.T.; Smith, L.; Hockey, M.; Collins, S.; Trakman, G.L.; Hoare, E.; Teasdale, S.B.; Wade, A.; et al. The Dietary Inflammatory Index and Human Health: An Umbrella Review of Meta-Analyses of Observational Studies. Adv. Nutr. 2021, 12, 1681–1690. [Google Scholar] [CrossRef]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Nathens, A.B.; Rotstein, O.D.; Marshall, J.C. Tertiary peritonitis: Clinical features of a complex nosocomial infection. World J. Surg. 1998, 22, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cao, X.; Liu, W.; Zhao, D.; Pan, S.; Sun, X.; Cai, G.; Zhou, J.; Chen, X. Peritoneal Dialysis Care in Mainland China: Nationwide Survey. JMIR Public Health Surveill. 2023, 9, e39568. [Google Scholar] [CrossRef] [PubMed]
- Spalding, D.R.; Williamson, R.C. Peritonitis. Br. J. Hosp. Med. 2008, 69, M12–M15. [Google Scholar] [CrossRef]
- Capobianco, A.; Cottone, L.; Monno, A.; Manfredi, A.A.; Rovere-Querini, P. The peritoneum: Healing, immunity, and diseases. J. Pathol. 2017, 243, 137–147. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Buijk, S.E.; Bruining, H.A. Future directions in the management of tertiary peritonitis. Intensive Care Med. 2002, 28, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C. Emerging leadership lecture: Inflammatory bowel disease in Asia: Emergence of a “Western” disease. J. Gastroenterol. Hepatol. 2015, 30, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L. The burden of inflammatory bowel disease in Europe. J. Crohns Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Pedersen, N.; Čuković-Čavka, S.; Brinar, M.; Kaimakliotis, I.; Duricova, D.; Shonová, O.; Vind, I.; Avnstrøm, S.; Thorsgaard, N.; et al. East-West gradient in the incidence of inflammatory bowel disease in Europe: The ECCO-EpiCom inception cohort. Gut 2014, 63, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, L.M.; Jones, G.R.; Bain, C.C. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 538–553. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Mahida, Y.R.; Rolfe, V.E. Host-bacterial interactions in inflammatory bowel disease. Clin. Sci. 2004, 107, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kotla, N.G.; Rochev, Y. IBD disease-modifying therapies: Insights from emerging therapeutics. Trends Mol. Med. 2023, 29, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Zoubek, M.E.; Pinazo-Bandera, J.; Ortega-Alonso, A.; Hernández, N.; Crespo, J.; Contreras, F.; Medina-Cáliz, I.; Sanabria-Cabrera, J.; Sanjuan-Jiménez, R.; González-Jiménez, A.; et al. Liver injury after methylprednisolone pulses: A disputable cause of hepatotoxicity. A case series and literature review. United Eur. Gastroenterol. J. 2019, 7, 825–837. [Google Scholar] [CrossRef]
- De, S.; Karim, F.; Kiessu, E.; Cushing, L.; Lin, L.L.; Ghandil, P.; Hoarau, C.; Casanova, J.L.; Puel, A.; Rao, V.R. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J. Biol. Chem. 2018, 293, 15208–15220. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Staschke, K.; Bulek, K.; Yao, J.; Peters, K.; Oh, K.H.; Vandenburg, Y.; Xiao, H.; Qian, W.; Hamilton, T.; et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med. 2007, 204, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Winkler, A.; Sun, W.; De, S.; Jiao, A.; Sharif, M.N.; Symanowicz, P.T.; Athale, S.; Shin, J.H.; Wang, J.; Jacobson, B.A.; et al. The Interleukin-1 Receptor-Associated Kinase 4 Inhibitor PF-06650833 Blocks Inflammation in Preclinical Models of Rheumatic Disease and in Humans Enrolled in a Randomized Clinical Trial. Arthritis Rheumatol. 2021, 73, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Lavazais, S.; Jargosch, M.; Dupont, S.; Labéguère, F.; Menet, C.; Jagerschmidt, C.; Ohm, F.; Kupcsik, L.; Parent, I.; Cottereaux, C.; et al. IRAK4 inhibition dampens pathogenic processes driving inflammatory skin diseases. Sci. Transl. Med. 2023, 15, eabj3289. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.A.; Patra, D.; Sinha, A.; Mazumder, S.; Pant, R.; Chouhan, R.; Jha, A.N.; Prusty, B.M.; Manna, D.; Das, S.K.; et al. A small molecule potent IRAK4 inhibitor abrogates lipopolysaccharide-induced macrophage inflammation in-vitro and in-vivo. Eur. J. Pharmacol. 2023, 944, 175593. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ning, Y.; Chen, Z.; Xue, Y.; Wu, Q.; Duan, W.; Ding, J.; Zhou, J.; Xie, H.; Zhang, H. Design, synthesis and pharmacological evaluation of 2,3-dihydrobenzofuran IRAK4 inhibitors for the treatment of diffuse large B-cell lymphoma. Eur. J. Med. Chem. 2023, 256, 115453. [Google Scholar] [CrossRef]
- Ajuebor, M.N.; Das, A.M.; Virág, L.; Flower, R.J.; Szabó, C.; Perretti, M. Role of resident peritoneal macrophages and mast cells in chemokine production and neutrophil migration in acute inflammation: Evidence for an inhibitory loop involving endogenous IL-10. J. Immunol. 1999, 162, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Cao, Z.; Song, X.; Zhang, X.; Mai, B.; Wen, T.; Lin, J.; Chen, J.; Chi, Y.; Su, T.; et al. Rhoifolin Alleviates Inflammation of Acute Inflammation Animal Models and LPS-Induced RAW264.7 Cells via IKKβ/NF-κB Signaling Pathway. Inflammation 2020, 43, 2191–2201. [Google Scholar] [CrossRef]
- Hamesch, K.; Borkham-Kamphorst, E.; Strnad, P.; Weiskirchen, R. Lipopolysaccharide-induced inflammatory liver injury in mice. Lab. Anim. 2015, 49 (Suppl. 1), 37–46. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, R.; Yin, H.; Wang, S.; Liu, B.; Li, J.; Zhou, M.; Yan, Q.; Lu, L. Oral IRAK4 inhibitor BAY-1834845 prevents acute respiratory distress syndrome. Biomed. Pharmacother. 2022, 153, 113459. [Google Scholar] [CrossRef]
- Mizoguchi, A. Animal Models of Inflammatory Bowel Disease. In Animal Models of Molecular Pathology; Progress in Molecular Biology and Translational Science; Academic Press: Cambridge, MA, USA, 2012; pp. 263–320. [Google Scholar]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.11–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kolachala, V.; Dalmasso, G.; Nguyen, H.; Laroui, H.; Sitaraman, S.V.; Merlin, D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS ONE 2009, 4, e6073. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Wells, J.; Cani, P.D.; García-Ródenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human Intestinal Barrier Function in Health and Disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef] [PubMed]
- Förster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xiao, Q.; Li, S.; Chen, L.; Long, J.; Fang, W.; Yu, F.; Huang, J.; Zhao, H.; Liu, D. Bupi Yichang Pill alleviates dextran sulfate sodium-induced ulcerative colitis in mice by regulating the homeostasis of follicular helper T cells. Phytomedicine 2022, 100, 154091. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.I.; Rios-Santos, F.; Balogun, S.O.; Martins, D.T. Vitexin reduces neutrophil migration to inflammatory focus by down-regulating pro-inflammatory mediators via inhibition of p38, ERK1/2 and JNK pathway. Phytomedicine 2016, 23, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Santucci, L.; Fiorucci, S.; Rubinstein, N.; Mencarelli, A.; Palazzetti, B.; Federici, B.; Rabinovich, G.A.; Morelli, A. Galectin-1 suppresses experimental colitis in mice. Gastroenterology 2003, 124, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Jess, T. Implications of the changing epidemiology of inflammatory bowel disease in a changing world. United Eur. Gastroenterol. J. 2022, 10, 1113–1120. [Google Scholar] [CrossRef]
- Lewis, J.D.; Parlett, L.E.; Jonsson Funk, M.L.; Brensinger, C.; Pate, V.; Wu, Q.; Dawwas, G.K.; Weiss, A.; Constant, B.D.; McCauley, M.; et al. Incidence, Prevalence, and Racial and Ethnic Distribution of Inflammatory Bowel Disease in the United States. Gastroenterology 2023, 165, 1197–1205.e1192. [Google Scholar] [CrossRef]
- Wu, X.F.; Ouyang, Z.J.; Feng, L.L.; Chen, G.; Guo, W.J.; Shen, Y.; Wu, X.D.; Sun, Y.; Xu, Q. Suppression of NF-κB signaling and NLRP3 inflammasome activation in macrophages is responsible for the amelioration of experimental murine colitis by the natural compound fraxinellone. Toxicol. Appl. Pharmacol. 2014, 281, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Nahidi, L.; Leach, S.T.; Mitchell, H.M.; Kaakoush, N.O.; Lemberg, D.A.; Munday, J.S.; Huinao, K.; Day, A.S. Inflammatory bowel disease therapies and gut function in a colitis mouse model. Biomed. Res. Int. 2013, 2013, 909613. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Scarpignato, C.; Strate, L.L.; Lanas, A.; Kruis, W.; Lahat, A.; Danese, S. Colonic diverticular disease. Nat. Rev. Dis. Primers 2020, 6, 20. [Google Scholar] [CrossRef]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L., 3rd; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Dudhgaonkar, S.; Ranade, S.; Nagar, J.; Subramani, S.; Prasad, D.S.; Karunanithi, P.; Srivastava, R.; Venkatesh, K.; Selvam, S.; Krishnamurthy, P.; et al. Selective IRAK4 Inhibition Attenuates Disease in Murine Lupus Models and Demonstrates Steroid Sparing Activity. J. Immunol. 2017, 198, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Varfolomeev, E.; Setiadi, A.F.; Francis, R.; Klabunde, S.; Senger, K.; Sujatha-Bhaskar, S.; Drobnick, J.; Do, S.; Suto, E.; et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in B cells and plasmacytoid dendritic cells. Sci. Signal 2020, 13, eaaz1053. [Google Scholar] [CrossRef]
- Guidetti, F.; Arribas, A.J.; Sartori, G.; Spriano, F.; Barnabei, L.; Tarantelli, C.; Von Roemeling, R.; Martinez, E.; Zucca, E.; Bertoni, F. Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors. J. Clin. Med. 2023, 12, 399. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | DW18134 | PF-06650833 |
|---|---|---|
| Structure |  | 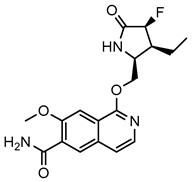 |
| IC50 (nM) | 11.2 ± 2.0 | 0.9 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Ning, Y.; Chen, Z.; Song, P.; Tang, H.; Shi, W.; Wan, Z.; Huang, G.; Liu, Q.; Chen, Y.; et al. A Novel IRAK4 Inhibitor DW18134 Ameliorates Peritonitis and Inflammatory Bowel Disease. Molecules 2024, 29, 1803. https://doi.org/10.3390/molecules29081803
Huang Y, Ning Y, Chen Z, Song P, Tang H, Shi W, Wan Z, Huang G, Liu Q, Chen Y, et al. A Novel IRAK4 Inhibitor DW18134 Ameliorates Peritonitis and Inflammatory Bowel Disease. Molecules. 2024; 29(8):1803. https://doi.org/10.3390/molecules29081803
Chicago/Turabian StyleHuang, Yuqing, Yi Ning, Zhiwei Chen, Peiran Song, Haotian Tang, Wenhao Shi, Zhipeng Wan, Gege Huang, Qiupei Liu, Yun Chen, and et al. 2024. "A Novel IRAK4 Inhibitor DW18134 Ameliorates Peritonitis and Inflammatory Bowel Disease" Molecules 29, no. 8: 1803. https://doi.org/10.3390/molecules29081803
APA StyleHuang, Y., Ning, Y., Chen, Z., Song, P., Tang, H., Shi, W., Wan, Z., Huang, G., Liu, Q., Chen, Y., Zhou, Y., Li, Y., Zhan, Z., Ding, J., Duan, W., & Xie, H. (2024). A Novel IRAK4 Inhibitor DW18134 Ameliorates Peritonitis and Inflammatory Bowel Disease. Molecules, 29(8), 1803. https://doi.org/10.3390/molecules29081803