

Novel JAK Inhibitors to Reduce Graft-Versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation in a Preclinical Mouse Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
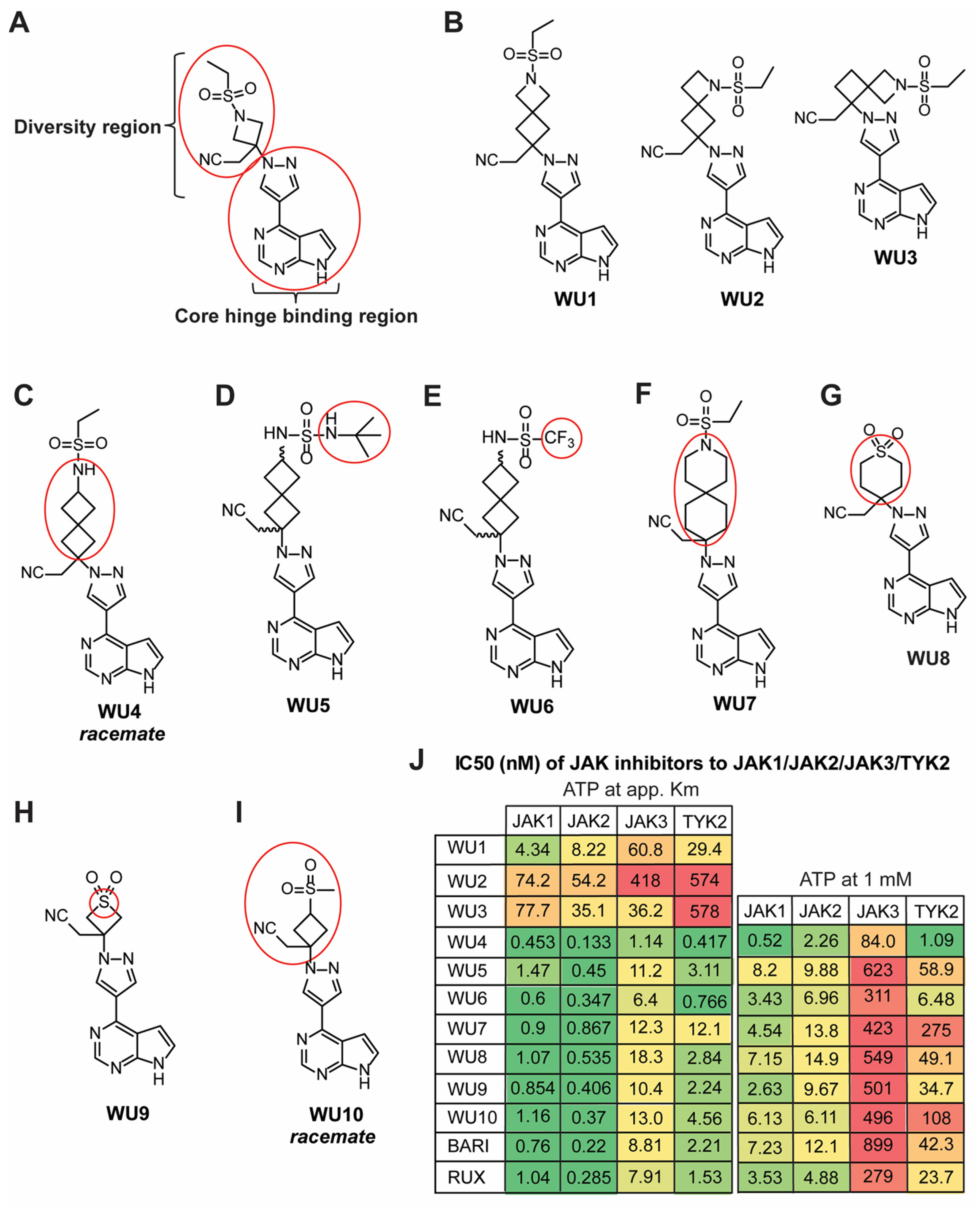
2.1. Design and Synthesis of Novel JAK1/JAK2 Inhibitors: WU Derivatives
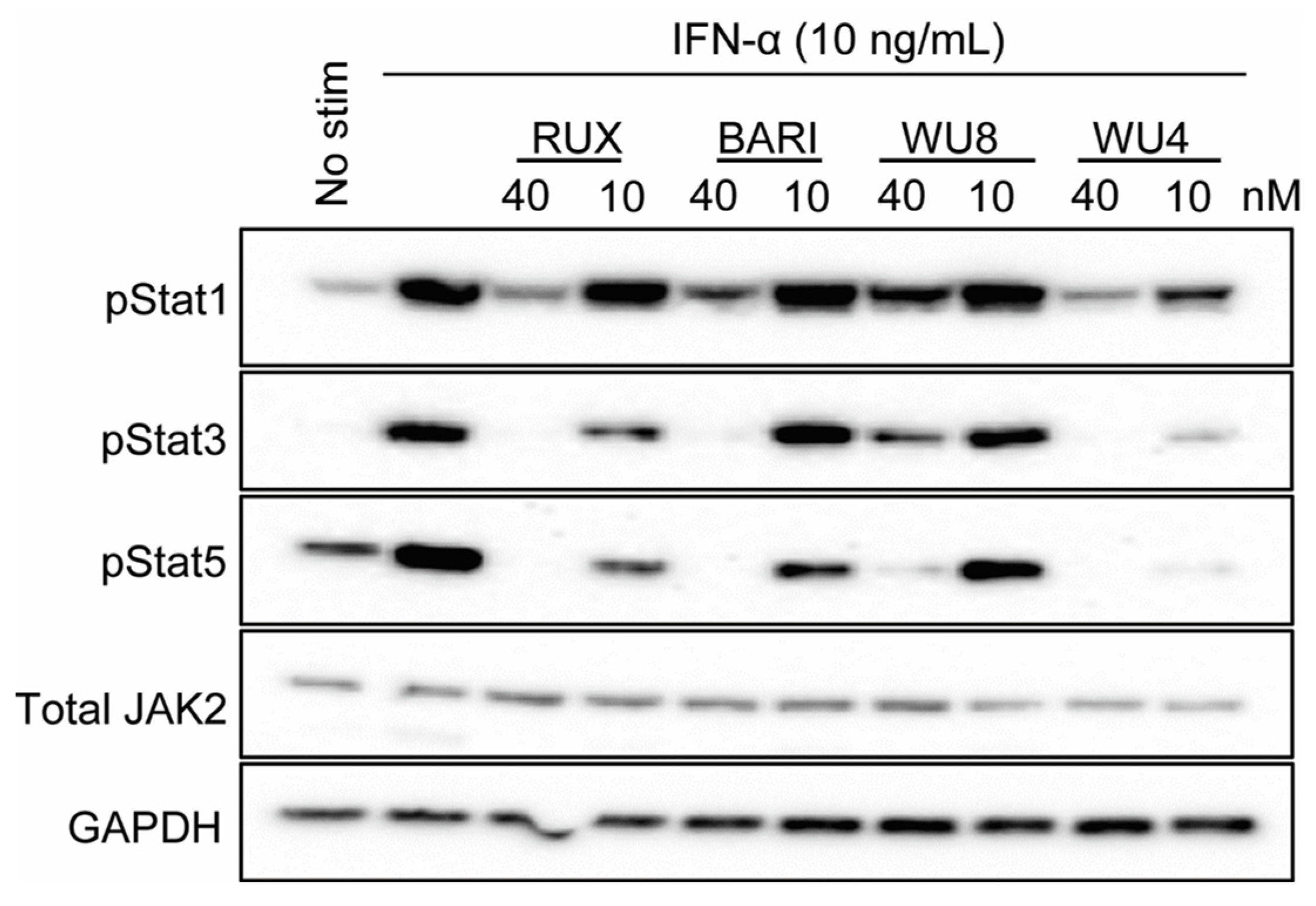
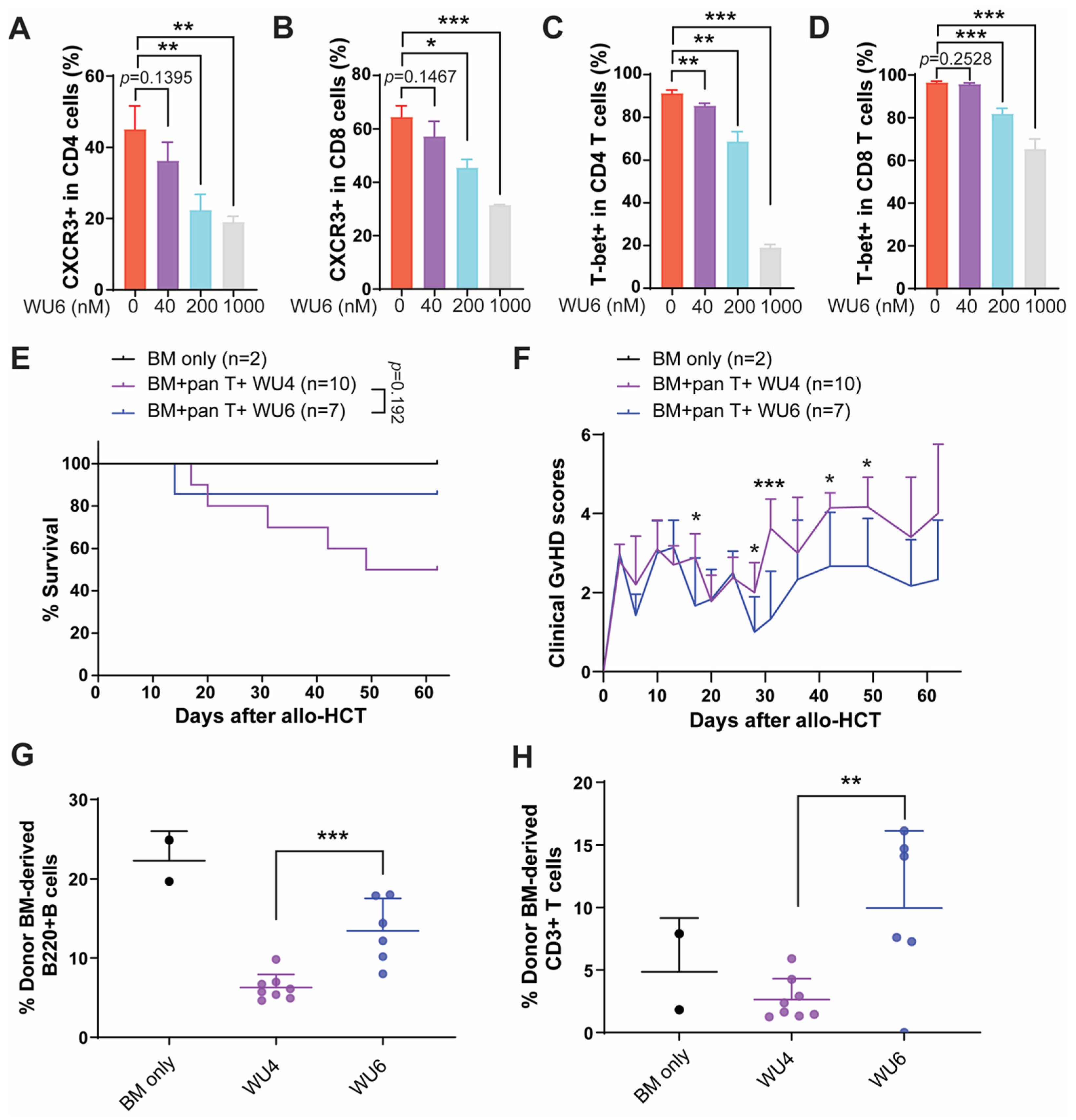
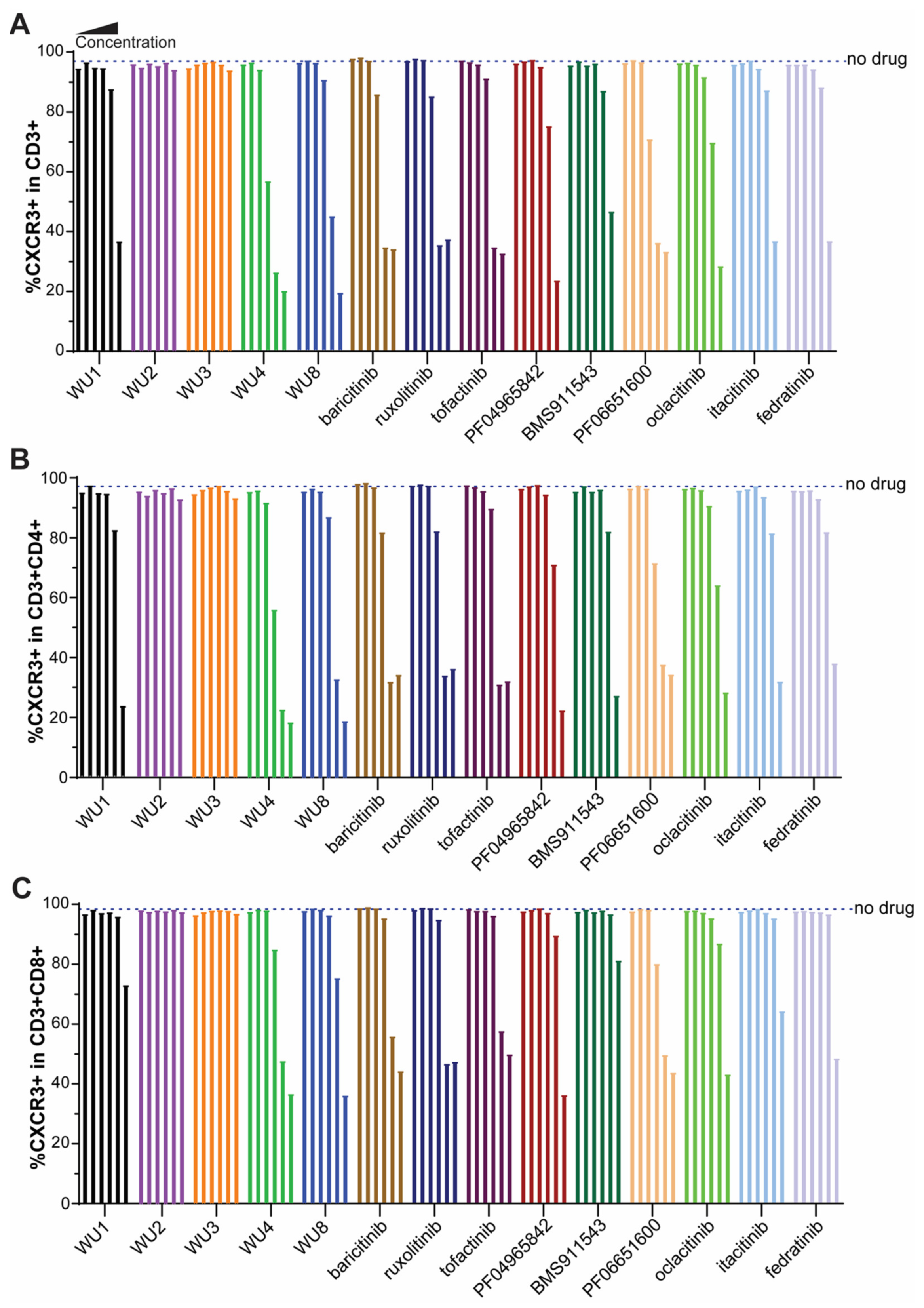
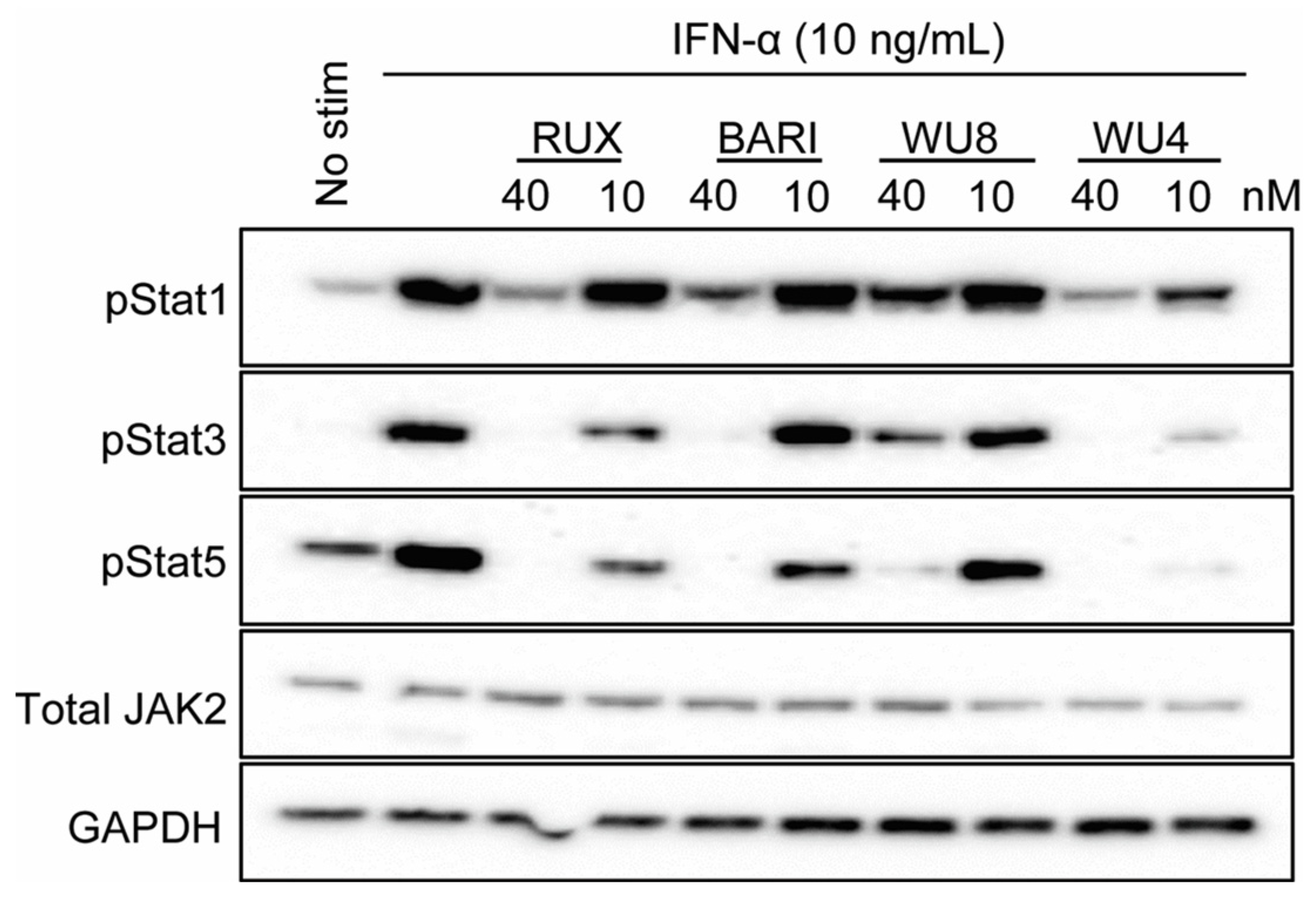
2.2. Inhibitory Effect of WU Derivatives on the Chemokine Receptor CXCR3 and STAT Phosphorylation in Primary T Cells
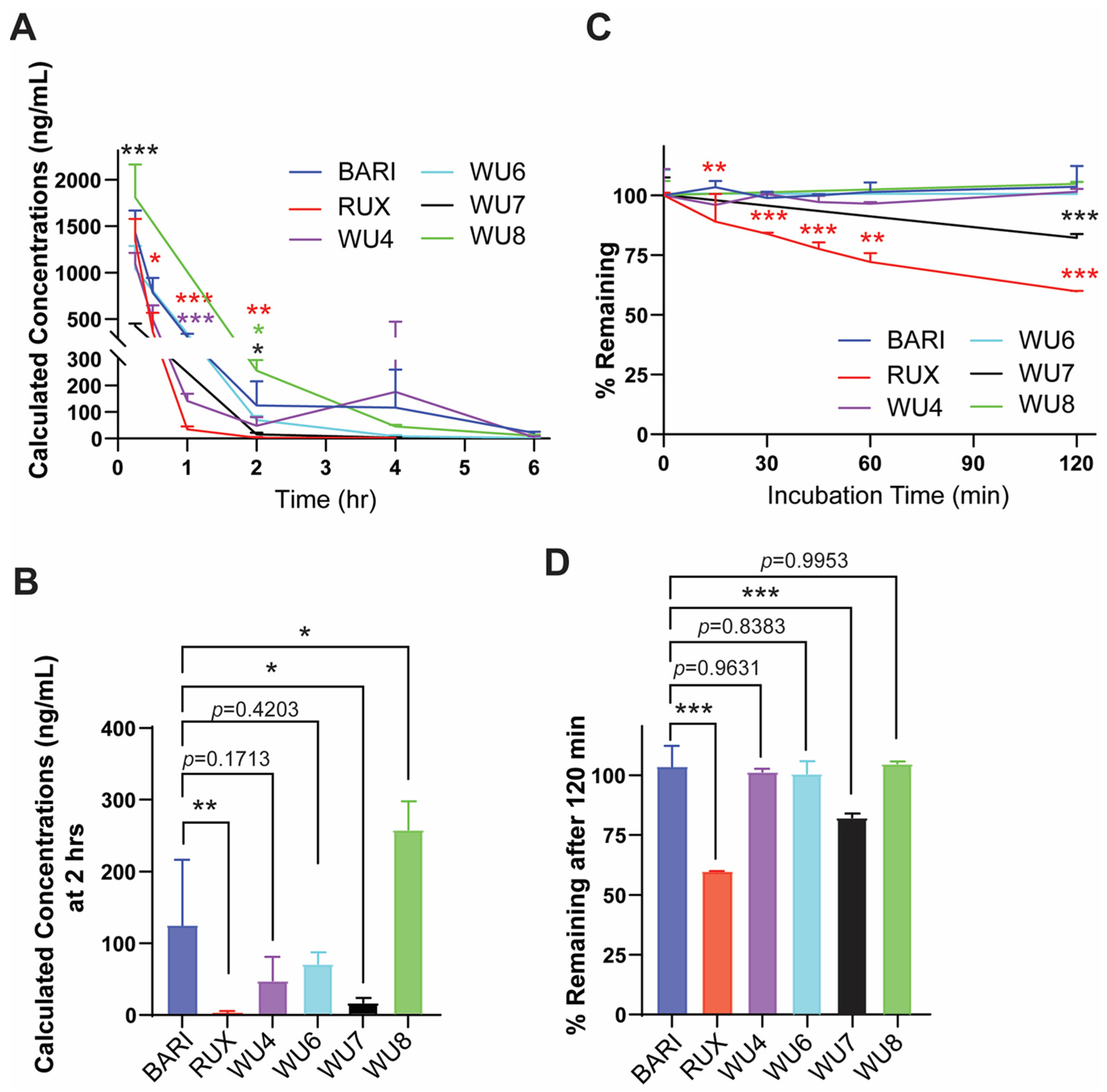
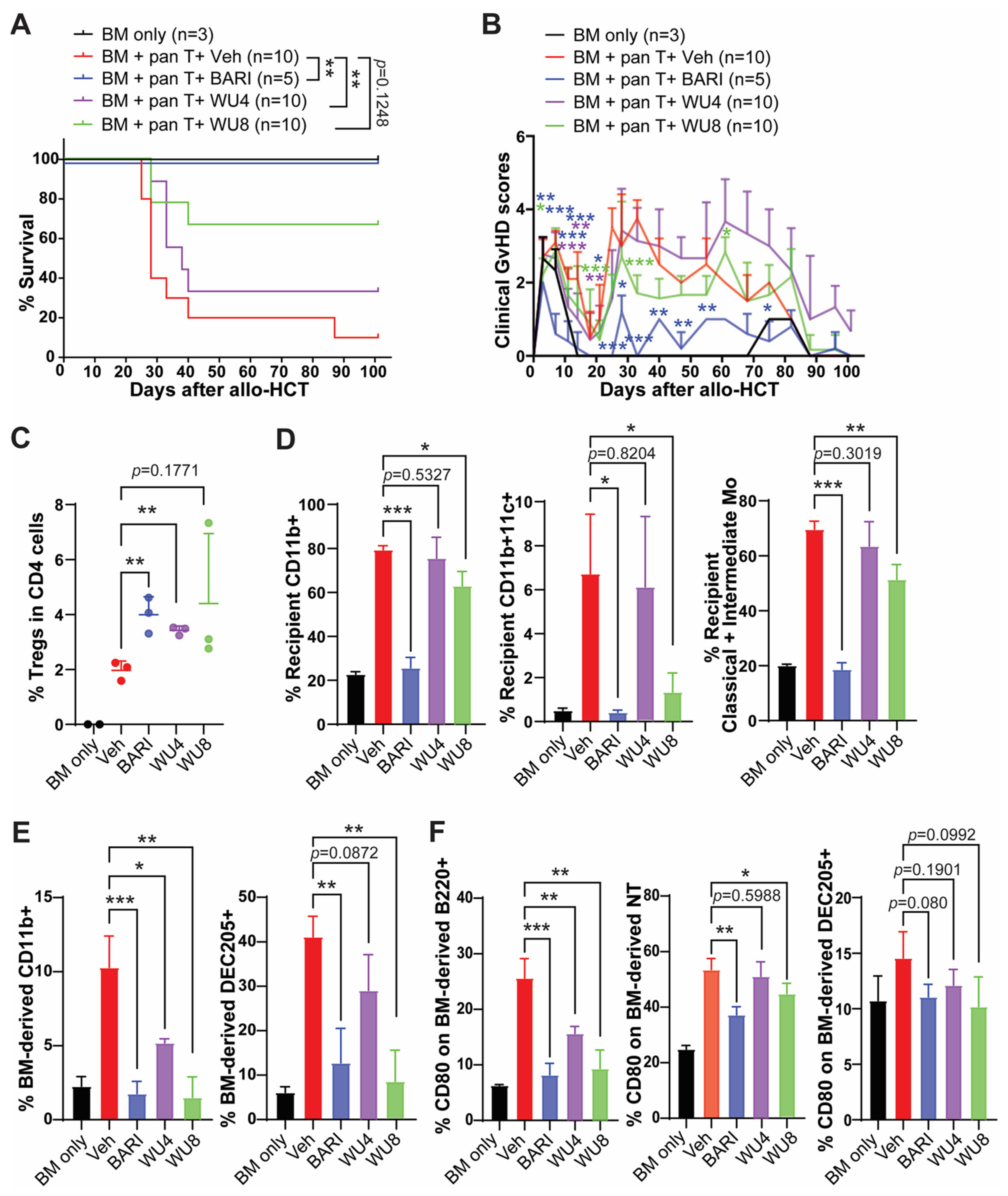
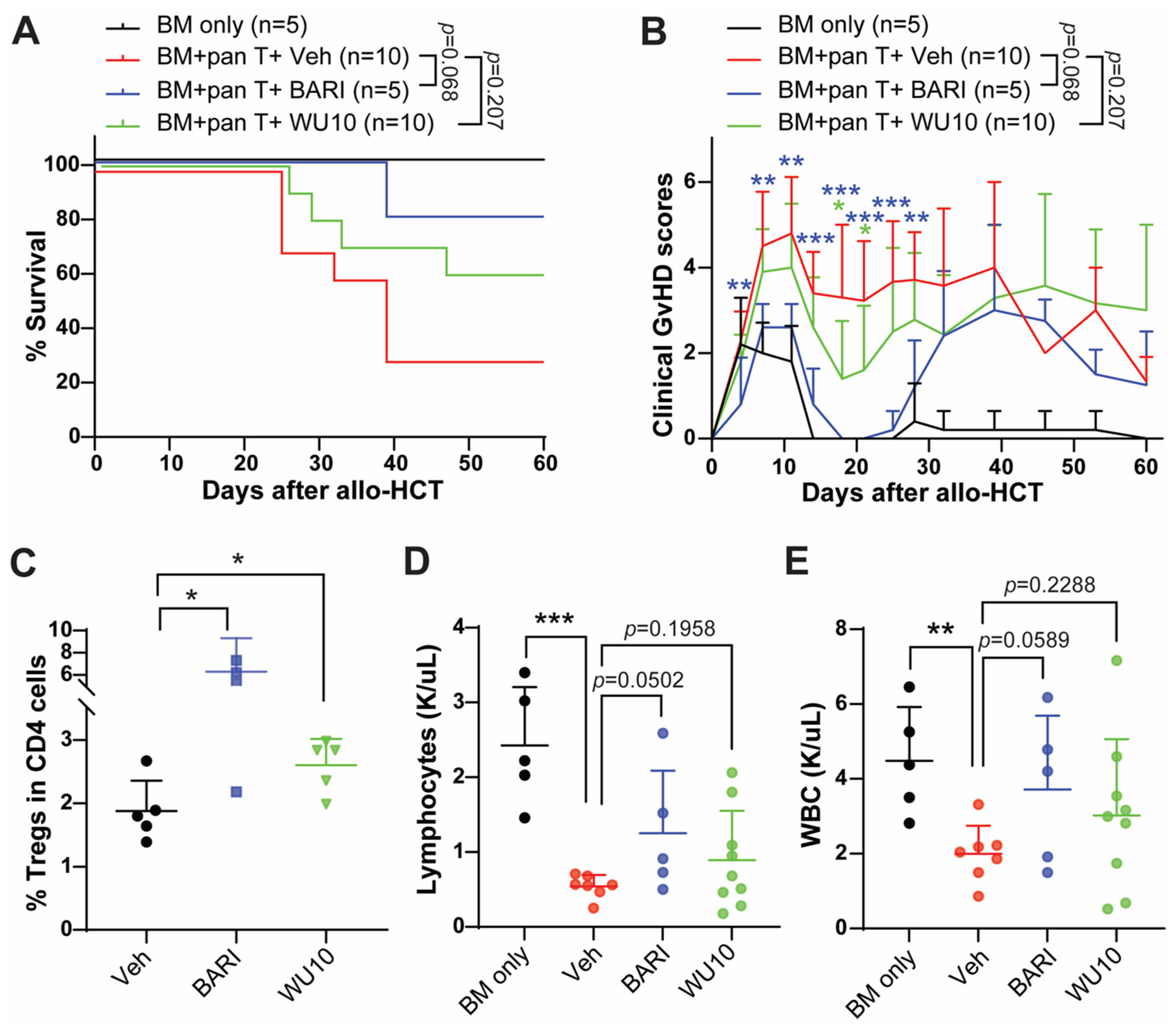
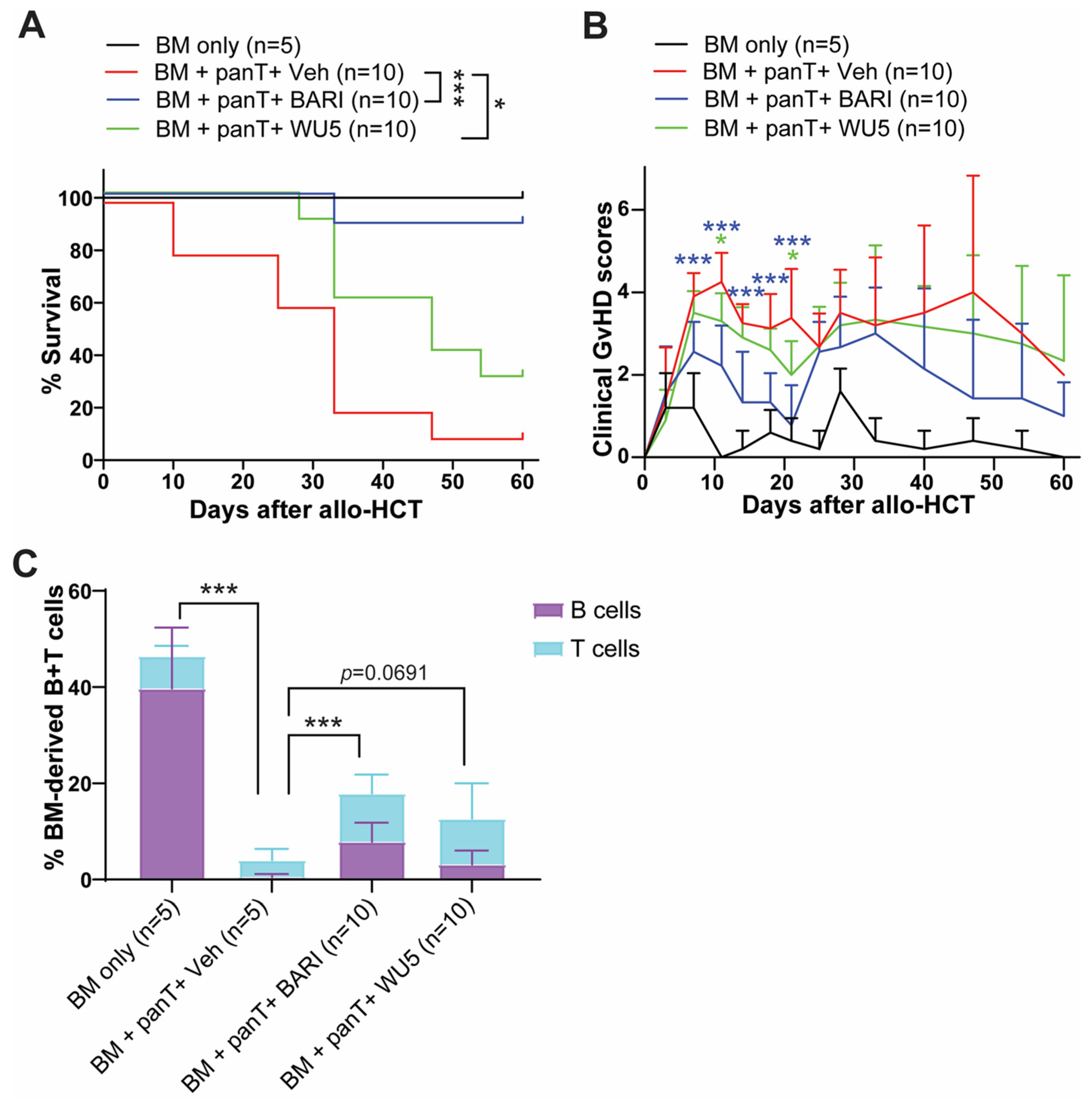
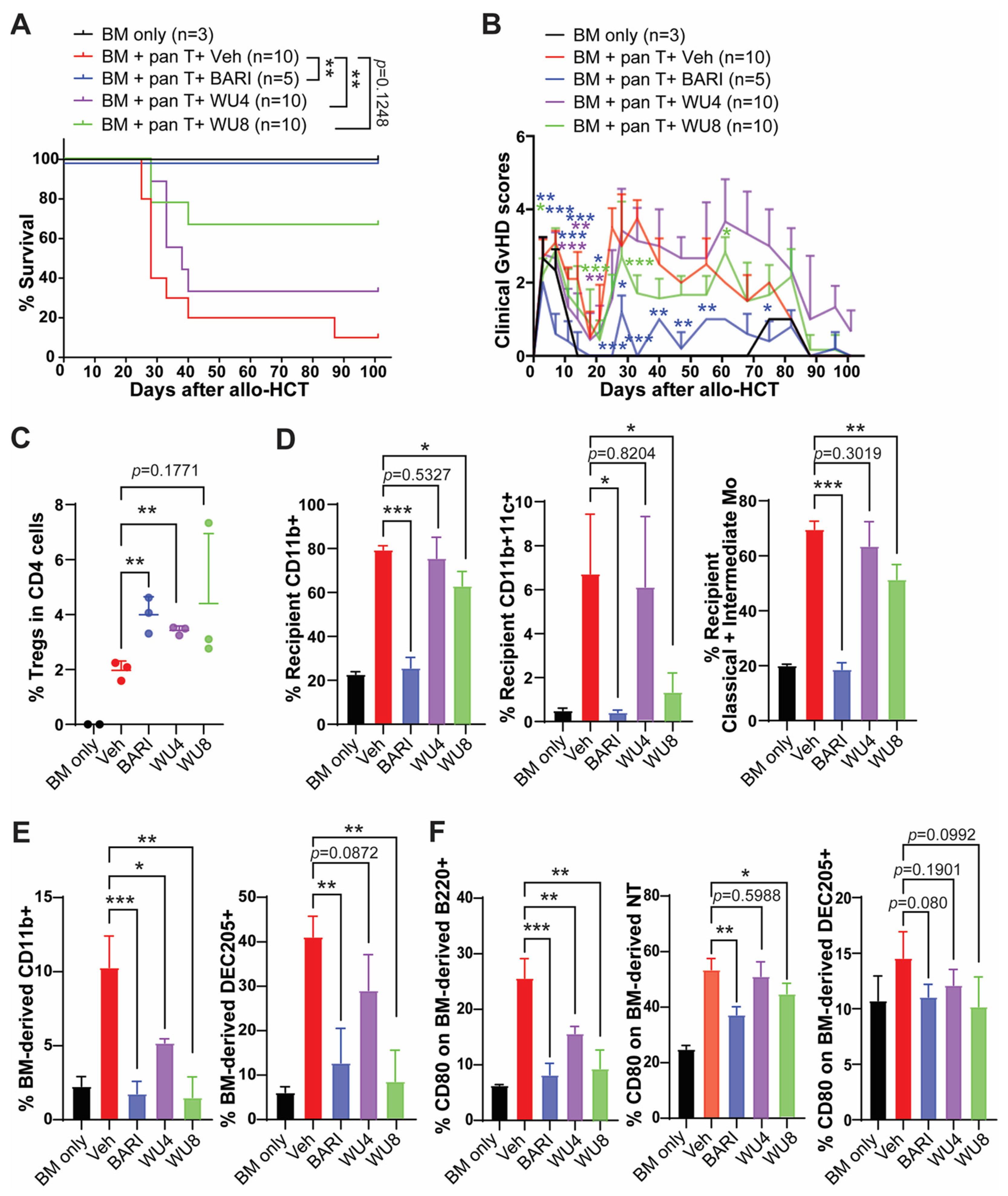
2.3. The Effect of WU Derivatives on GvHD in the Preclinical Mouse Model of Allo-HCT
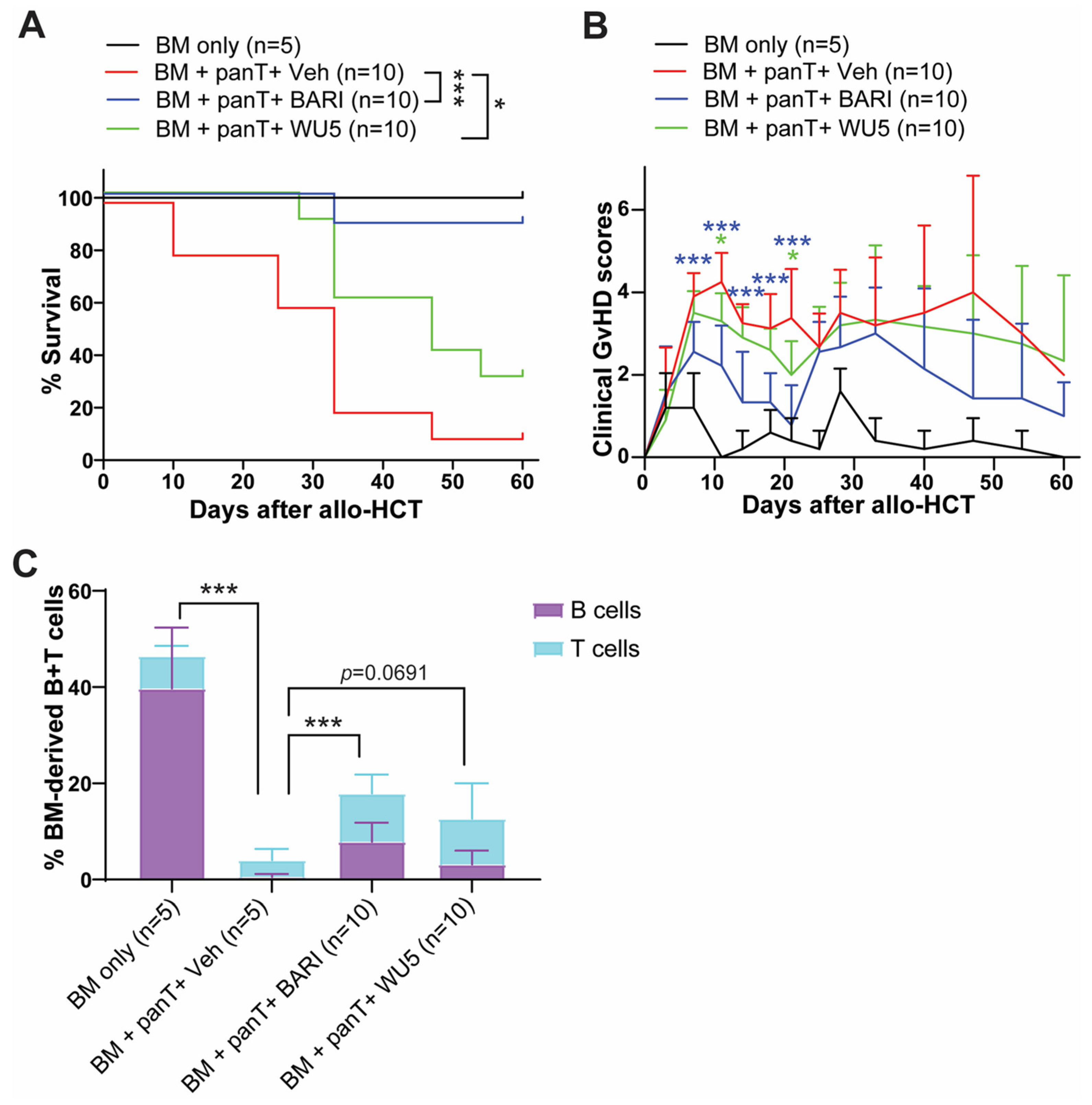
2.4. The Effect of WU6 on the Expression of CXCR3 and T-Bet in Primary Murine T Cells and GvHD in Mouse Model of Allo-HCT
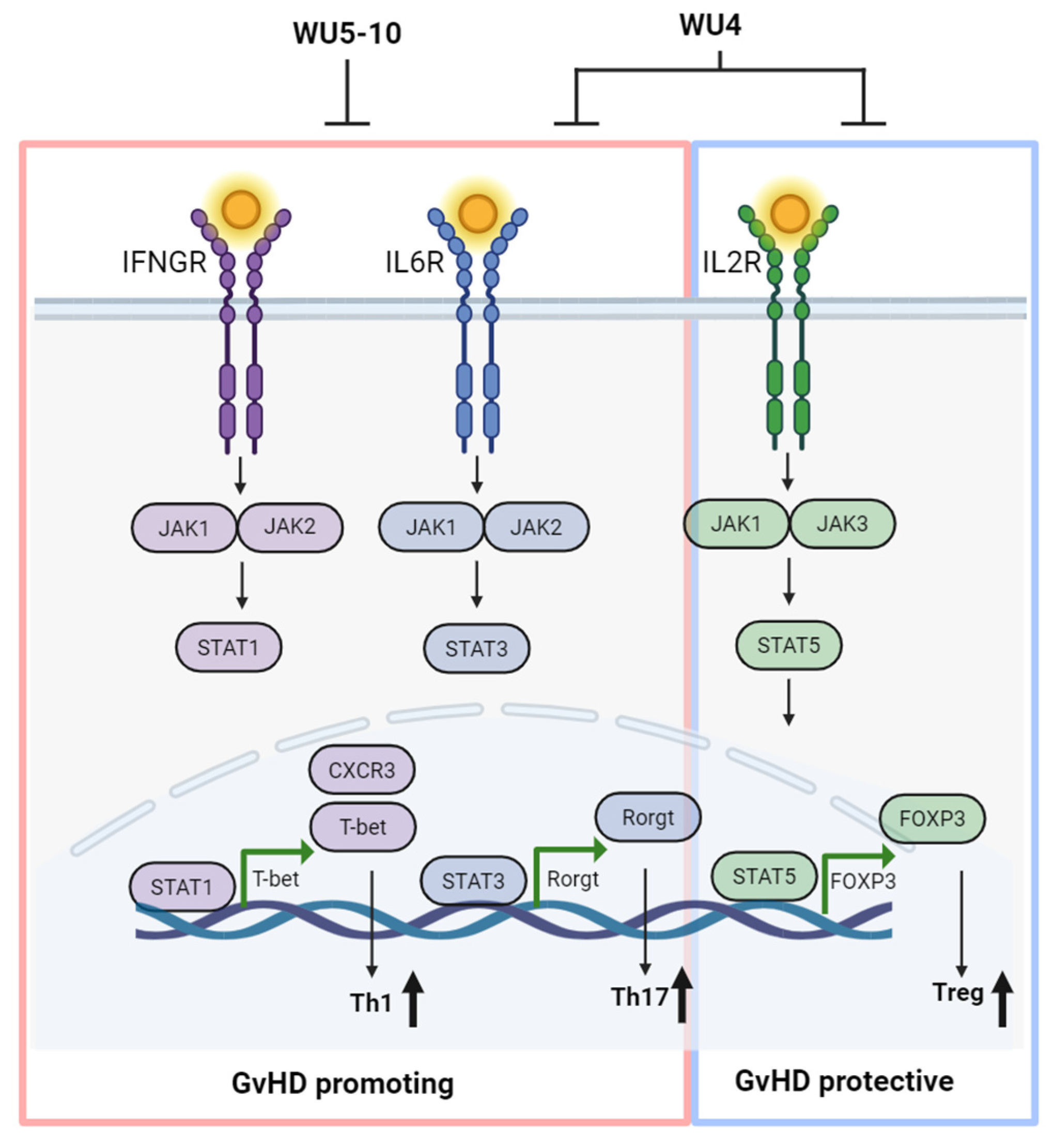
3. Discussion
4. Materials and Methods
4.1. JAK Inhibitor Compounds
4.2. Z’-LYTE Enzyme Activity Assay
4.3. Mice
4.4. PK Studies
4.5. Preclinical Mouse Model of Allo-HCT
4.6. Assessment of Clinical GvHD in Transplanted Mice
4.7. Flow Cytometry Analysis
4.8. Primary Murine T Cell Culture
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Appelbaum, F.R. Hematopoietic-cell transplantation at 50. N. Engl. J. Med. 2007, 357, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Isgro, A.; Sodani, P.; Gaziev, J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb. Perspect. Med. 2012, 2, a011825. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Nasri, U.; Nakamura, R.; Martin, P.J.; Zeng, D. Retention of Donor T Cells in Lymphohematopoietic Tissue and Augmentation of Tissue PD-L1 Protection for Prevention of GVHD While Preserving GVL Activity. Front. Immunol. 2022, 13, 907673. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.K. Current approaches to prevent and treat GVHD after allogeneic stem cell transplantation. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, J.; Wei, W. Advance in Targeted Immunotherapy for Graft-Versus-Host Disease. Front. Immunol. 2018, 9, 1087. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J. Inflamm. Res. 2020, 13, 519–531. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Spinelli, F.R.; Meylan, F.; O’Shea, J.J.; Gadina, M. JAK inhibitors: Ten years after. Eur. J. Immunol. 2021, 51, 1615–1627. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, C.; Deng, J.; Zhou, J. JAK inhibition as a new treatment strategy for patients with COVID-19. Biochem. Pharmacol. 2022, 202, 115162. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cooper, M.L.; Staser, K.; Ashami, K.; Vij, K.R.; Wang, B.; Marsala, L.; Niswonger, J.; Ritchey, J.; Alahmari, B.; et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia 2018, 32, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ziga, E.D.; Ritchey, J.; Collins, L.; Prior, J.L.; Cooper, M.L.; Piwnica-Worms, D.; DiPersio, J.F. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood 2012, 120, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Ashami, K.; DiPersio, J.F.; Choi, J. Targeting IFNGR/IL6R or downstream JAK1/JAK2 to control GvHD. Oncotarget 2018, 9, 35721–35722. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ashami, K.; Lim, S.; Staser, K.; Vij, K.; Santhanam, S.; Ritchey, J.; Peterson, S.; Gao, F.; Ciorba, M.A.; et al. Baricitinib prevents GvHD by increasing Tregs via JAK3 and treats established GvHD by promoting intestinal tissue repair via EGFR. Leukemia 2022, 36, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Richez, C.; Truchetet, M.E.; Kostine, M.; Schaeverbeke, T.; Bannwarth, B. Efficacy of baricitinib in the treatment of rheumatoid arthritis. Expert. Opin. Pharmacother. 2017, 18, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, A.; Vrhovac, R.; Verstovsek, S. Ruxolitinib: A new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol. 2011, 7, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Hoisnard, L.; Lebrun-Vignes, B.; Maury, S.; Mahevas, M.; El Karoui, K.; Roy, L.; Zarour, A.; Michel, M.; Cohen, J.L.; Amiot, A.; et al. Adverse events associated with JAK inhibitors in 126,815 reports from the WHO pharmacovigilance database. Sci. Rep. 2022, 12, 7140. [Google Scholar] [CrossRef]
- Anderson, B.E.; McNiff, J.M.; Jain, D.; Blazar, B.R.; Shlomchik, W.D.; Shlomchik, M.J. Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: Requirements depend on target organ. Blood 2005, 105, 2227–2234. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Jamil, M.O.; Mineishi, S. State-of-the-art acute and chronic GVHD treatment. Int. J. Hematol. 2015, 101, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, K.; Horowitz, M.M.; Gale, R.P.; van Bekkum, D.W.; Gluckman, E.; Good, R.A.; Jacobsen, N.; Kolb, H.J.; Rimm, A.A.; Ringden, O.; et al. Risk factors for chronic graft-versus-host disease after HLA-identical sibling bone marrow transplantation. Blood 1990, 75, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.A.; Khoury, H.J.; Jagasia, M.; Ali, H.; Schiller, G.J.; Staser, K.; Choi, J.; Gehrs, L.; Arbushites, M.C.; Yan, Y.; et al. A phase 1 trial of itacitinib, a selective JAK1 inhibitor, in patients with acute graft-versus-host disease. Blood Adv. 2020, 4, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.; Choi, J.; Ruminski, P.; Schroeder, M.A.; Kim, S.; Abboud, C.N.; DiPersio, J.F. Insights into the role of the JAK/STAT signaling pathway in graft-versus-host disease. Ther. Adv. Hematol. 2020, 11, 2040620720914489. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, S.; Kim, B.; Ritchey, J.; Vij, K.; Prior, J.; Marsala, L.; Stoner, A.; Gao, F.; Achilefu, S.; et al. S100A9 upregulated by IFNGR signaling blockade functions as a novel GVHD suppressor without compromising GVL in mice. Blood 2023, 141, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, S.; Razmkhah, F.; Choi, J. Overexpression of S100A9 in donor T cells is associated with reconstitution of gut microbiota and outcome of allogeneic hematopoietic stem cell transplantation. Blood Res. 2023, 58, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cooper, M.L.; Alahmari, B.; Ritchey, J.; Collins, L.; Holt, M.; DiPersio, J.F. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS ONE 2014, 9, e109799. [Google Scholar] [CrossRef] [PubMed]
- Martini, D.J.; Chen, Y.B.; DeFilipp, Z. Recent FDA Approvals in the Treatment of Graft-Versus-Host Disease. Oncologist 2022, 27, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, S.; Lim, S.; Razmkhah, F. PKN1 is a novel therapeutic target to prevent graft-vs-host disease after allogeneic hematopoietic cell transplantation. J. Immunol. 2023, 210, 173. [Google Scholar] [CrossRef]
- Tall, P.; Qamar, M.A.; Batzu, L.; Leta, V.; Falup-Pecurariu, C.; Ray Chaudhuri, K. Non-oral continuous drug delivery based therapies and sleep dysfunction in Parkinson’s disease. J. Neural Transm. 2023, 130, 1443–1449. [Google Scholar] [CrossRef]
- Sanchez-Mendoza, E.H.; Carballo, J.; Longart, M.; Hermann, D.M.; Doeppner, T.R. Implantation of Miniosmotic Pumps and Delivery of Tract Tracers to Study Brain Reorganization in Pathophysiological Conditions. J. Vis. Exp. 2016, 18, e52932. [Google Scholar]
- Alahmari, B.; Cooper, M.L.; Vij, K.; Ritchey, J.; Ruminski, P.; Gao, F.; Choi, J.; DiPersio, J.F. Selective targeting of alpha4beta1 integrin attenuates murine graft versus host disease. Leukemia 2020, 34, 3100–3104. [Google Scholar] [CrossRef] [PubMed]
- Cooke, K.R.; Kobzik, L.; Martin, T.R.; Brewer, J.; Delmonte, J., Jr.; Crawford, J.M.; Ferrara, J.L. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 1996, 88, 3230–3239. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Ruminski, P.; Singh, M.; Staser, K.; Ashami, K.; Ritchey, J.; Lim, S.; DiPersio, J.F.; Choi, J. Novel JAK Inhibitors to Reduce Graft-Versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation in a Preclinical Mouse Model. Molecules 2024, 29, 1801. https://doi.org/10.3390/molecules29081801
Kim S, Ruminski P, Singh M, Staser K, Ashami K, Ritchey J, Lim S, DiPersio JF, Choi J. Novel JAK Inhibitors to Reduce Graft-Versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation in a Preclinical Mouse Model. Molecules. 2024; 29(8):1801. https://doi.org/10.3390/molecules29081801
Chicago/Turabian StyleKim, Sena, Peter Ruminski, Megh Singh, Karl Staser, Kidist Ashami, Julie Ritchey, Sora Lim, John F. DiPersio, and Jaebok Choi. 2024. "Novel JAK Inhibitors to Reduce Graft-Versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation in a Preclinical Mouse Model" Molecules 29, no. 8: 1801. https://doi.org/10.3390/molecules29081801
APA StyleKim, S., Ruminski, P., Singh, M., Staser, K., Ashami, K., Ritchey, J., Lim, S., DiPersio, J. F., & Choi, J. (2024). Novel JAK Inhibitors to Reduce Graft-Versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation in a Preclinical Mouse Model. Molecules, 29(8), 1801. https://doi.org/10.3390/molecules29081801