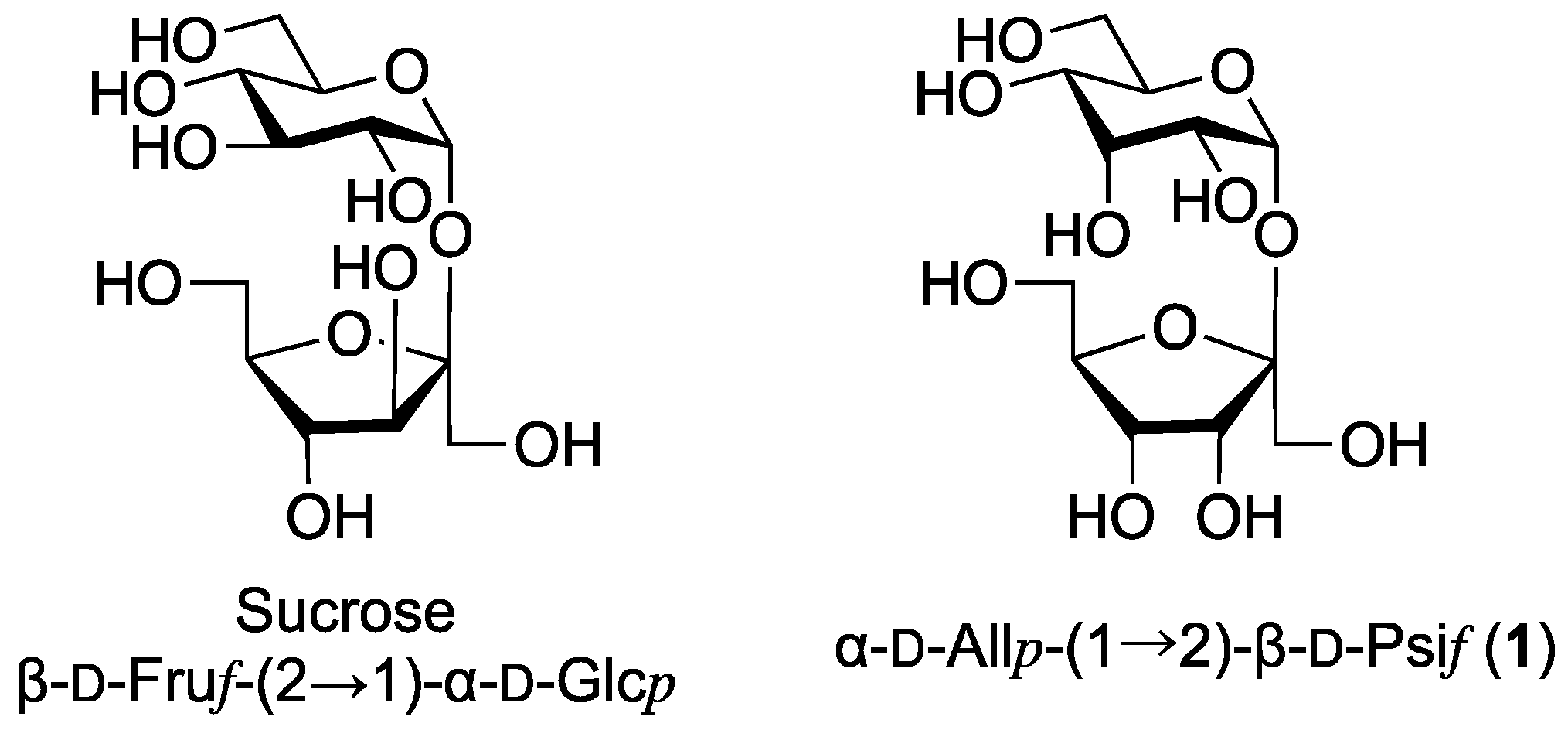
2. Results and Discussion
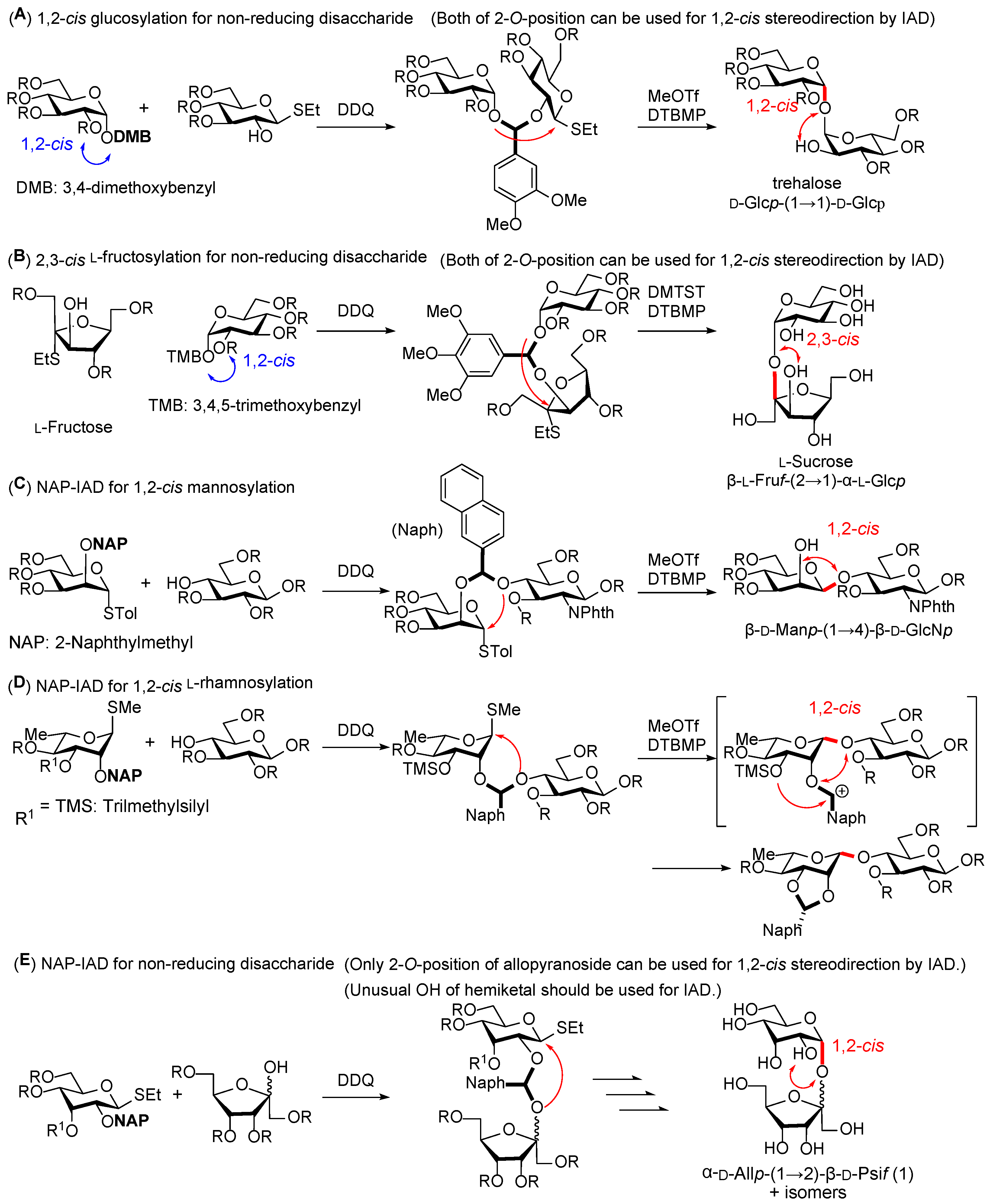
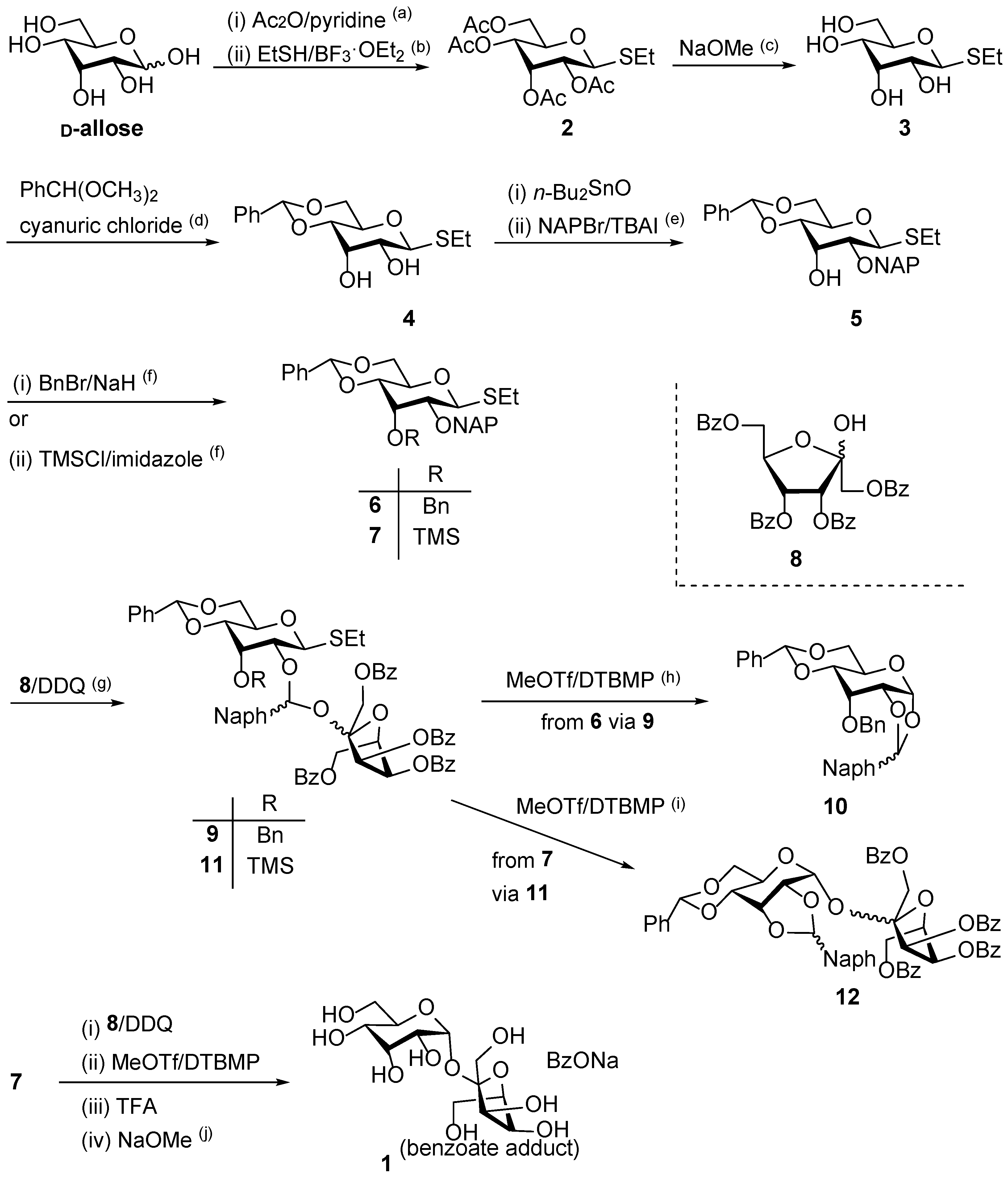
We first attempted the stereoselective α-D-allosylation using the NAP-IAD glycosylation method. The D-allosyl donor with an NAP group at the C-2 position was synthesized from D-allose in six steps. As shown in
Scheme 1, the first step involved the acetylation of the hydroxy group of D-allose with Ac
2O in pyridine. The peracetate was then converted to the thioglycoside as an α/β glycoside mixture (α/β ratio of 23:77) at a yield of 66%, resulting in the recovery of the required β-glycoside-rich fraction. Deacetylation of
2, followed by the regioselective formation of 4,6-
O-benzylidene acetal on the resultant tetraol
3, selectively yielded the desired compound
4. In this step, the α-thioglycoside was completely removed as this anomeric functionality was required for the subsequent general activation with MeOTf in the IAD reaction. However, this process resulted in a yield of 29% over two steps, with 2,3-
O-benzylidene acetal and 2,3,4,6-di-
O-benzylidene obtained as byproducts at 6% and 7% yields, respectively. The remaining hydroxy groups of β-thioglycoside
4 at the C-2 and C-3 positions were protected regioselectively. Initially, the hydroxy group at the C-2 position was protected as an NAP ether, affording the resultant compound
5 at a 52% yield. The hydroxy group at the C-3 position was protected as a benzyl ether to afford
6 at a 99% yield. Following this, the D-allosyl donor
6 was oxidized using 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in the presence of 1,3,4,6-tetra-
O-benzoyl-D-psicofuranose (
8) as the acceptor. This reaction produced mixed acetal isomers
8, forming a simple diastereomeric mixture related to naphthylidene acetal. The major acetal isomer was then isolated, and its subsequent intramolecular glycosylation was performed using MeOTf and 2,6-di-
tert-butyl-4-methylpyridine (DTBMP) in 1,2-dichloroethane. The major isolated compound was an unexpected 1,2-
O-naphthylidene acetal of the donor moiety (
10) at a 49% yield. The 1,2-
O-benzylidene-type cyclization from 1,2-
cis glycoside was possible owing to the formation of 1,2-
cis-glycoside in the IAD [
27]. The configuration of allose may support the formation of 1,2-
O-benzylidene acetal when the 3-OH of 1,2-
cis-allopyranoside was protected as a Bn ether, which created a steric hinderance to the naphthylmethyl group at the C-2 position. As previously reported, the activation of ketosidic bonds is largely influenced by the stereochemistry in the sugar ring, which is not clearly understood [
28]. This may have occurred because of the initial nucleophilic attack of the oxygen atom to the resultant naphthylmethyl cation at the 2-position of D-psicofuranoside in the 1,2-
O-naphthylidene acetal formation, leading to the undesired cleavage of the C–O glycosidic bond of 1,2-
cis-D-psicofuranoside. This cleavage likely occurred concomitantly with the enhancement of neighboring group participation by one of the carbonyl oxygens of the benzoyl groups within the D-psicofuranoside moiety. To produce the desired disaccharide derivative, it is necessary to trap the cation species initially on the naphthylmethyl group after glycosidic bond formation. The enhancement effect on the cleavage of the glycosidic bond has been also found in the case of the super arming effect of 2-
O-benzoylated glycosyl donor reported by Demchemko [
29,
30], although we speculate that the electron withdrawing group as the protective group on the psicose stabilizes the psicosidic linkage simply by the disarming effect. Previous studies utilized (TMS)
3SiH for the in situ reductive trapping of the benzylic cation of the NAP ether [
20,
21]. However, the side reaction could not be suppressed, suggesting that the cation species resulting from the fragmentation of D-psicofuranoside is more stable than the benzylic cation.
To prevent the unfavorable formation of 1,2-
O-naphthylidene acetal, we attempted IAD using the D-allosyl donor
7, which contained a trimethyl silyl (TMS) group at the C-3 position to trap the naphthylmethyl cation in situ by immediate acetal formation [
21]. The trimethylsilylation of derivative
5 proceeded with a yield of 99%. The desired mixed acetal
11 was generated by oxidizing derivative
7 with DDQ, which was confirmed using thin layer chromatography (TLC), MALDI-TOF MS, and NMR spectroscopy. Meanwhile, the actual glycosylation reaction was carried out using MeOTf and DTBMP in 1,2-dichloroethane without purifying the mixed acetals
11. This reaction afforded the 1,2-
cis- D-allosyl-(1→2)-β-D-psicofuranose derivative
12, with a 2,3-
O-naphthylidene acetal group on the D-allosyl moiety (19%). In this reaction, 1,2-
O-naphthylidene formation was not detected by MALDI-TOF MS. This result indicates the importance of introducing the nucleophilic 3-
O-TMS ether to intramolecularly trap benzylic cation species, rather than intermolecularly trapping them using (TMS)
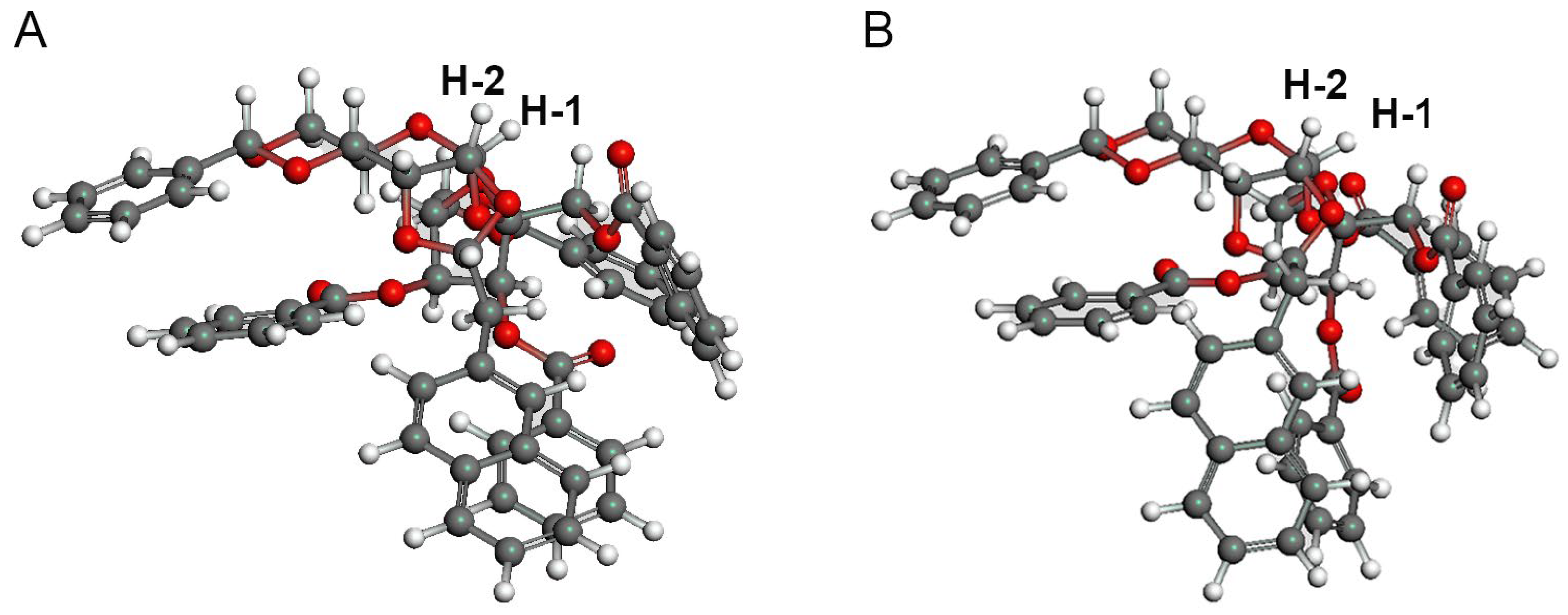
3SiH. In this reaction, 35% of the psicose acceptor was recovered. Based on the TLC analysis, it is possible that the tethered intermediate does not form, that the acetal is cleaved even in the presence of DTBMP as a proton trap, or that the product is cleaved during formation. The coupling constant between the protons at the C-1 and C-2 positions of the allose in derivative
12 is relatively large. However, in the structures obtained using conformational analysis, the dihedral angles of the protons between the C-1 and C-2 protons in the α-alloside were approximately 37°–41°, regardless of the conformers derived from the naphthylidene group, suggesting that the product was an α-anomer (
Figure 3).
The final target compound 1 was synthesized from compound 7 in four steps without purification. The protecting groups of the coupling products were removed by a short treatment (<1 min) with trifluoroacetic acid (TFA) to prevent glycosidic bond cleavage; nevertheless, the yield of the target compound was very low (4%). Based on the TLC analysis, it is possible that the disaccharide formed is cleaved into monosaccharides. Subsequent removal of benzoyl groups was achieved by treatment with sodium methoxide in methanol, quantitatively affording the target disaccharide as a benzoate adduct. The formation of this adduct was confirmed by 1H and 13C NMR spectroscopy as well as mass spectrometry. High resolution MS indicated the formation of the desired disaccharide (calcd. for C12H22NaO11 [M + Na]+ 365.1054; found 365.1046). It has been suggested that the disaccharide, which has a cis-oriented oxygen functionality around the glycosides, exhibits a strong ionophore-like ability to uptake metal ions such as Na+ ions along with counter anions such as BzO−. However, the NMR spectra of the final compound indicates that it does not release the salt during purification.
The glycosidic linkages of the allosyl residue in the products were determined based on the
1JC–H coupling constant (170 Hz for β or >170 Hz for α) [
31,
32]. The
1JC–H coupling of the deprotected disaccharide was measured at 180 Hz, indicating the formation of an α-allosidic linkage. The anomeric configurations of the psicosyl residue were confirmed with reference to a previous study by Morimoto et al. [
33]. The
13C chemical shift of the C-2 carbon of β-psicoside was recorded as 109.2 ppm (
Table 1). In a previous study, a value of 111.9 Hz was reported when the chemical shift was referenced to the signal corresponding to the methyl group of sodium 3-(trimethylsilyl)[2,2,3,3-
2H
4] propionate (TSP-
d4). The overall chemical shift values shifted downfield by approximately 2 ppm compared to those observed in D
2O. Additionally, the stereoisomer α-D-allosyl-(1→2)-α-D-psicofuranoside was synthesized using the conventional glycosylation method (NIS/TfOH) with ethyl 2,3,4,6-tetra-
O-benzyl-1-thio-β-D-allopyranoside, and the NMR spectra of the two stereoisomers were compared (
Supporting Information Schemes S1 and S2, and Table S1). The
13C chemical shift of the C-2 carbon in the deprotected products obtained via NAP-IAD glycosylation was recorded as approximately 106 ppm, which did not match the reference data. The
13C chemical shifts of the C1 peaks of methyl α-D-glucopyranoside and trehalose (α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside) were recorded as 101 ppm [
34] and 94 ppm [
35], respectively. This difference in the chemical shift value may have been caused by the reduced shielding effect of oxygen between the two sugar moieties in trehalose compared to that of a simple aglycone. Hence, the conformation of the psicofuranoside bond was determined to be β, and the NAP-IAD reaction was confirmed to result in β-psicofuranosylation.
3. Materials and Methods
3.1. General Methods
1H NMR and 13C NMR spectra were recorded on a JEOL (Akishima, Japan) ECS-400 spectrometer at 400 and 100 MHz, respectively. The 1H NMR chemical shifts were referenced to the signals of Me4Si as the internal standard (0.00 ppm in CDCl3 and HDO, and 4.80 ppm in D2O). The 13C NMR chemical shifts were referenced to the signals of the solvent [δC (CDCl3) 77.0] and native scale (D2O). Assignments were aided by COSY, TOCSY, and 1H–13C correlation experiments. All reactions were monitored by TLC using a glass plate coated with silica gel 60F254 (0.2 mm thickness, Merck KGaA, Darmstadt, Germany). Silica gel column chromatography was performed using 60N silica gel (Kanto Chemical, Tokyo, Japan). Anhydrous solvents (superdehydrated grade) were purchased from FUJIFILM Wako Pure Chemical Corp (Osaka, Japan). Optical rotations were measured with a JASCO (Hachioji, Japan) P2200 polarimeter. High resolution mass spectra (HRMS) were recorded on Bruker (Billerica, MA, USA) micrOTOF II and Thermo Scientific (Waltham, MA, USA) Exactive Plus spectrometers using electrospray ionization in acetonitrile or methanol. MALDI-TOF MS was carried out using an Autoflex Speed mass spectrometer (Billerica, MA, USA).
3.2. Synthesis of Compound 2
To a solution of D-allose (10.0 g, 55.5 mmol) in pyridine (60.0 mL), acetic anhydride (52.0 mL, 555 mmol) was added at 0 °C. The reaction mixture was stirred at 40 °C for 10 h and then quenched with MeOH at 0 °C. The reaction mixture was azeotropically dried three times with dry toluene; the resulting residue was diluted with ethyl acetate and washed successively with 1 M HCl, brine, saturated aq. NaHCO3, and brine. The organic layer was dried with Na2SO4, filtered, and evaporated in vacuo. The resulting residue was purified by flash silica gel column chromatography using a hexane–ethyl acetate (3/2, v/v) mixture to afford 1 (22.9 g, quant.). To a solution of 1 (22.9 g) and ethanethiol (5.1 mL, 70.4 mmol) in 1,2-dichloroethane (100 mL), boron trifluoride diethyl ether complex (11.0 mL, 88.1 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 12 h and then quenched with saturated aq. NaHCO3 at 0 °C. The reaction mixture was diluted with chloroform and washed successively with saturated aq. NaHCO3 and brine. The organic layer was dried with MgSO4, filtered, and evaporated in vacuo. The resulting residue was purified by flash silica gel column chromatography using a hexane–ethyl acetate (1/1, v/v) mixture to afford compound 2 (14.5 g, 36.9 mmol, 66%, α/β ratio of 23:77). (2α) Rf = 0.51 (hexane/ethyl acetate = 1/1, v/v); [α]23D +159.36 (c 1.00, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 5.60 (t, 1H, J = 3.2 Hz, H-3), 5.53 (d, 1H, J = 5.6 Hz, H-1), 5.11 (dd, 1H, J = 3.2, 6.4 Hz, H-2), 4.95 (dd, 1H, J = 2.4, 10.4 Hz, H-4), 4.59 (ddd, 1H, J = 1.6, 4.0, 10.4 Hz, H-5), 4.33 (dd, 1H, J = 4.8, 12.4 Hz, H-6), 4.16 (dd, 1H, J = 2.4, 12.4 Hz, H-6), 2.63–2.57 (m, 2H, SCH2CH3), 2.19 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 1.28 (t, 3H, J = 8.4 Hz, SCH2CH3). 13C-NMR (100 MHz, CDCl3) δ 170.7 (COCH3), 170.0 (COCH3), 169.6 (COCH3), 169.3 (COCH3), 82.9 (C-1), 67.7 (C-2), 67.5 (C-3), 65.7 (C-4), 64.3 (C-5), 62.1 (C-6), 26.7 (SCH2CH3), 20.9 (COCH3), 20.7 (COCH3), 20.6 (COCH3), 15.3 (SCH2CH3). (2β) Rf = 0.56 (hexane/ethyl acetate = 1/1, v/v); [α]24D + 0.42 (c 1.00, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 5.67 (t, 1H, J = 2.8 Hz, H-3), 5.01–4.95 (m, 2H, H-2, H-4), 4.83 (d, 1H, J = 10.4 Hz, H-1), 4.21 (m, 2H, H-6), 4.05 (td, 1H, J = 3.6, 10.4 Hz, H-5), 2.98–2.64 (m, 2H, SCH2CH3), 2.18 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.29 (t, 3H, J = 7.2 Hz, SCH2CH3). 13C-NMR (100 MHz, CDCl3) δ 170.8 (COCH3), 169.8 (COCH3), 169.1 (COCH3), 169.1 (COCH3), 80.4 (C-1), 72.5 (C-5), 68.3 (C-3), 67.6 (C-2), 66.3 (C-4), 62.5 (C-6), 23.9 (SCH2CH3), 20.8 (COCH3), 20.7 (COCH3), 20.5 (COCH3), 14.9 (SCH2CH3). HRMS ESI-TOF: calcd. for C16H24NaO9S [M + Na]+ 415.1051; found 415.1049.
3.3. Synthesis of Compound 4
To allose derivative 2 (3.0 g, 7.65 mmol), a 1 M solution of sodium methoxide in methanol (0.77 mmol, 770 μL) in a mixture of tetrahydrofuran (15.0 mL) and methanol (15.0 mL) was added at 0 °C and stirred at room temperature for 10 h. The reaction solution was neutralized using an Amberlyst at 0 °C. After diluting the reaction solution with methanol, filtration with a cotton plug was carried out, and the solvent was removed to afford the white powder 3 (3.2 g), which was used without further purification. Benzaldehyde dimethyl acetal (0.96 mL, 6.43 mmol) and 2,4,6-trichloro [1,3,5]triazine (0.30 g, 1.61 mmol) were added to the obtained powder 3 (1.2 g, 5.36 mmol) in DMF (30.0 mL) at 0 °C and stirred at room temperature for 2.5 h. The reaction solution was diluted with ethyl acetate and washed with saturated aq. NaHCO3 and brine. The organic layer was dried with Na2SO4 and concentrated under reduced pressure. The resulting reaction mixture was purified by silica gel column chromatography using a hexane–ethyl acetate (2/3, v/v) mixture to afford derivative 4 (483 mg, 1.55 mmol, 29%). Rf = 0.49 (hexane/ethyl acetate = 2/3, v/v); [α]24D − 25.35 (c 1.00, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.52–7.34 (m, 5H, aromatic H), 5.57 (s, 1H, PhCH), 4.78 (d, 1H, J = 10.4 Hz, H-1), 4.43–4.37 (m, 2H, H-3, H-6), 4.02 (dt, 1H, J = 5.2, 10.0 Hz, H-5), 3.74 (t, 1H, J = 10.0 Hz, H-6), 3.61 (dd, 1H, J = 2.8, 9.6 Hz, H-4), 3.57–3.53 (m, 1H, H-2), 2.81–2.72 (m, 2H, SCH2CH3), 2.70 (d, 1H, J = 6.0 Hz, OH), 2.59 (s, 1H, OH), 1.33 (t, 3H, J = 8.0 Hz, SCH2CH3). 13C-NMR (100 MHz, CDCl3) δ 136.9–126.2 (aromatic C), 101.9 (PhCH), 83.8 (C-1), 78.7 (C-4), 70.0 (C-2), 69.0 (C-6), 68.2 (C-3), 66.1 (C-5), 24.7 (SCH2CH3), 15.3 (SCH2CH3). HRMS ESI-TOF: calcd. for C15H20NaO5S [M + Na]+ 335.0924; found 335.0928.
3.4. Synthesis of Compound 5
To the allose derivative 4 (483 mg, 1.55 mmol), dibutyltin(IV) oxide (426 mg, 1.71 mmol) in toluene (30.0 mL) was added and stirred at 120 °C for 2 h. The reaction solution was cooled to room temperature and the solvent was removed. To a solution of the resulting reaction mixture in toluene (30.0 mL), 2-bromomethylnaphthalene (483 mg, 2.44 mmol), tetrabutylammonium iodide (860 mg, 2.33 mmol), and cesium fluoride (354 mg, 2.33 mmol) were added at room temperature for 3 h. The reaction solution was cooled to room temperature and diluted with ethyl acetate. The reaction mixture was filtered through Celite and washed with saturated aq. NaHCO3 and brine. The organic layer was dried with MgSO4, filtered, and evaporated in vacuo. The resulting residue was purified by silica gel column chromatography using a toluene–ethyl acetate (3/2, v/v) mixture to afford compound 5 (365 mg, 0.81 mmol, 52%). Rf = 0.54 (hexane/ethyl acetate = 3/2, v/v); [α]24D − 50.21 (c 0.50, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.87–7.32 (m, 12H, aromatic H), 5.49 (s, 1H, PhCH), 4.97–4.94 (m, 2H, H-1, NAPCH2), 4.88 (d, 1H, J = 12.4 Hz, NAPCH2), 4.37 (dd, 1H, J = 5.2, 10.8 Hz, H-6), 4.33 (brdd, 1H, J = 2.4, 5.2 Hz, H-3), 4.04 (dt, 1H, J = 5.6, 10.0 Hz, H-5), 3.69 (t, 1H, J = 10.4 Hz, H-6), 3.49 (dd, 1H, J = 2.0, 9.6 Hz, H-4), 3.43 (dd, 1H, J = 2.8, 9.6 Hz, H-2), 2.85–2.70 (m, 2H, SCH2CH3), 2.50 (s, 1H, OH), 1.34 (t, 3H, J = 8.0 Hz, SCH2CH3). 13C-NMR (100 MHz CDCl3) δ 137.0–126.0 (aromatic C), 101.9 (PhCH), 81.9 (C-1), 78.5 (C-4), 76.8 (C-2), 72.7 (NAPCH2), 69.1 (C-6), 67.1 (C-3), 65.5 (C-5), 25.1 (SCH2CH3), 15.1 (SCH2CH3). HRMS ESI-TOF: calcd. for C26H28NaO5S [M + Na]+ 475.1550; found 475.1560.
3.5. Synthesis of Compound 6
Allose derivative 5 (780 mg, 7.653 mmol) was reacted with benzyl bromide (410 μL, 3.46 mmol) and sodium hydride (60%, 62 mg, 2.60 mmol) in DMF (8.0 mL) at 0 °C and stirred at room temperature for 1.5 h. The reaction was quenched by adding methanol (210 μL, 5.19 mmol) to the reaction solution at 0 °C. After diluting the reaction solution with ethyl acetate, it was washed successively with 1 M HCl, brine, saturated aq. NaHCO3, and brine. The organic layer was dried with Na2SO4 and the solvent was removed. The resulting residue was purified by silica gel column chromatography using a hexane–ethyl acetate (100/0 to 70/30, v/v) solution to afford allose donor 6 (928 mg, 1.71 mmol, 99%). Rf = 0.51 (hexane/ethyl acetate = 3/1, v/v); [α]24D − 82.09 (c 0.50, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.84–7.29 (m, 17H, aromatic H), 5.43 (s, 1H, PhCH), 5.08 (d, 1H, J = 10.0 Hz, H-1), 4.93 (d, 1H, J = 11.6 Hz, PhCH2), 4.84–4.72 (m, 3H, NAPCH2, PhCH2), 4.35 (dd, 1H, J = 5.6, 10.4 Hz, H-6), 4.17 (brt, 1H, J = 2,4, 4.8 Hz, H-3), 4.12 (td, 1H, J = 5.2, 10.0 Hz, H-5), 3.68 (t, 1H, J = 10.4 Hz, H-6), 3.50 (dd, 1H, J = 1.6, 8.8 Hz, H-4), 3.40 (dd, 1H, J = 2.4, 9.6 Hz, H-2), 2.82–2.69 (m, 2H, SCH2CH3), 1.32 (t, 3H, J = 8.0 Hz, SCH2CH3). 13C-NMR (100 MHz, CDCl3) δ 138.6–126.0 (aromatic C), 101.9 (PhCH), 82.5 (C-1), 79.8 (C-4), 77.6 (C-2), 74.0 (PhCH2), 73.7 (C-3), 72.2 (NAPCH2), 69.3 (C-6), 66.1 (C-5), 24.9 (SCH2CH3), 15.0 (SCH2CH3). HRMS ESI-TOF: calcd. for C33H34NaO5S [M + Na]+ 565.2019; found 565.2019.
3.6. Synthesis of Compound 7
Allose derivative 5 (365 mg, 0.81 mmol) was reacted with imidazole (247 mg, 3.63 mmol) and chlorotrimethylsilane (203 μL, 1.61 mmol) in DMF (4.0 mL) at 0 °C and stirred at room temperature for 3 h. The reaction was quenched by adding methanol to the reaction solution at 0 °C. After diluting the reaction solution with ethyl acetate, it was washed successively with 1 M HCl, brine, saturated aq. NaHCO3, and brine. The organic layer was dried with Na2SO4 and the solvent was removed. The resulting residue was purified by silica gel column chromatography using a hexane–ethyl acetate (3/1, v/v) mixture to afford allose donor 7 (420 mg, 0.80 mmol, 99%). Rf = 0.60 (hexane/ethyl acetate = 3/1, v/v); [α]24D − 84.78 (c 1.00, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.84–7.34 (m, 12H, aromatic H), 5.45 (s, 1H, PhCH), 5.02 (d, 1H, J = 9.6 Hz, H-1), 4.88–4.82 (m, 2H, NAPCH2), 4.40 (brt, 1H, J = 2.0 Hz, H-3), 4.32 (dd, 1H, J = 5.2, 10.0 Hz, H-6), 4.01 (dt, 1H, J = 5.2, 10.4 Hz, H-5), 3.67 (t, 1H, J = 10.4 Hz, H-6), 3.39 (dd, 1H, J = 2.0, 9.2 Hz, H-4), 3.34 (dd, 1H, J = 2.8, 10.0 Hz, H-2), 2.82–2.68 (m, 2H, SCH2CH3), 1.32 (t, 3H, J = 8.0 Hz, SCH2CH3), 0.14 (s, 9H, Si(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 137.4–126.0 (aromatic C), 101.7 (PhCH), 82.1 (C-1), 79.2 (C-4), 77.4 (C-2), 72.1 (NAPCH2), 69.2 (C-6), 68.3 (C-3), 65.5 (C-5), 24.9 (SCH2CH3), 15.0 (SCH2CH3), 0.654 (Si(CH3)3). HRMS ESI-TOF: calcd. for C30H40NaO5SSi [M + Na]+ 563.2258; found 563.2257.
3.7. IAD Reaction between 6 and 8
To a solution of allose derivative 6 (60 mg, 0.11 mmol) and 1,3,4,6-tetra-O-benzoyl-psicofuranoside acceptor 8 (44 mg, 0.07 mmol) in dichloromethane (3.0 mL) in the presence of 4 Å molecular sieves (300 mg), DDQ (30 mg, 0.13 mmol) was added at 0 °C and stirred at room temperature for 1 day. The ascorbic acid/citric acid/sodium hydroxide solution (0.7/1.3/0.9 wt%) was added at 0 °C to quench the reaction. The reaction mixture of tethering intermediate 9 obtained after post-treatment was dissolved in 1,2-dichloroethane (3.0 mL), and in the presence of 4 Å molecular sieves (300 mg), 1 M MeOTf solution (1.33 mL, 1.33 mmol) was added at 0 °C. After starting the reaction for 1 day, triethylamine (24 μL, 0.22 mmol) was added at 0 °C to quench the reaction. The reaction solution was diluted with ethyl acetate, filtered through celite, and washed with saturated aq. NaHCO3 and brine. The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The resulting residue was purified by gel filtration chromatography with chloroform to give allose derivative 10 (27 mg, 49%). Rf = 0.50 (hexane/ethyl acetate = 3/2, v/v); [α]24D 161.59 (c 0.30, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.01–7.03 (m, 15H), 6.11 (s, 1H, PhCH), 5.63 (d, 1H, J = 4.8 Hz, H-1), 5.57 (s, 1H, NAPCH), 4.76 (dt, 1H, J = 5.6, 10.0 Hz, H-5), 4.56 (d, 1H, J = 12.4 Hz, PhCH2), 4.52 (dd, 1H, J = 5.2, 10.8 Hz, H-6), 4.44 (d, 1H, J = 12.4 Hz, PhCH2), 4.26 (t, 1H, J = 5.6 Hz, H-2), 4.07 (dd, 1H, J = 2.0, 4.8 Hz, H-3), 3.76 (t, 1H, J = 10.4 Hz, H-6), 3.66 (dd, J = 2.0, 9.6 Hz, H-4). 13C-NMR (100 MHz, CDCl3) δ 138.3–124.7 (aromatic C), 102.0 (NaphCH), 100.8 (PhCH), 98.5 (C-1), 77.3–77.2 (C-4), 74.6 (PhCH2), 72.5 (C-3), 71.1 (C-2), 69.4 (C-6), 59.4 (C-5). HRMS ESI-TOF: calcd. for C31H28NaO6 [M + Na]+ 519.1778; found 519.1782.
3.8. IAD Reaction between 7 and 8
To a solution of compound 7 (50 mg, 0.095 mmol), 1,3,4,6-tetra-O-benzoyl-psicofuranoside acceptor 8 (38 mg, 0.063 mmol), and dried MS (4 Å, 250 mg) in 1,2-dichloroethane (5.0 mL), DDQ (26 mg, 0.013 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 19 h and then quenched with an ascorbate buffer [L-ascorbic acid (70 mg), citric acid monohydrate (120 mg), and NaOH (92 mg) in H2O (10.0 mL)] at 0 °C. The reaction mixture was diluted with ethyl acetate and filtered with celite. The filtrate was washed successively with saturated aq. NaHCO3 and brine. The organic layer was dried with MgSO4, filtered, and evaporated in vacuo to obtain intermediate 11. To a solution of mixed acetal (11) and dried MS (4 Å, 500 mg) in 1,2-dichloroethane (5.0 mL), a solution of DTBMP (129 mg, 0.63 mmol) in 1,2-dichloroethane (5 mL) and 1 M MeOTf (1.3 mL, 1.33 mmol) was added under an argon atmosphere at 0 °C. The reaction mixture was stirred at 40 °C for 20 h and then quenched with triethylamine (185 μL, 2.00 mmol) at 0 °C. The reaction mixture was diluted with ethyl acetate and filtered through celite. The filtrate was washed with saturated aq. NaHCO3 and brine. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by preparative TLC using a toluene–ethyl acetate (8/1, v/v) mixture and preparative liquid chromatography (hexane/ethyl acetate = 85/15 to 77/23, v/v) to afford 12-1 (6.5 mg, 6.6 μmol, 10%) and 12-2 (5.8 mg, 5.9 μmol, 9%), respectively. Compound 12-1: [α]23D +45.95 (c 0.60, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.03–7.19 (aromatic H), 6.08 (s, 1H, PhCH), 5.87 (dd, 1H, J3,4 = 5.6 Hz, J4,5 = 11.6 Hz, H-4I), 5.85 (d, 1H, J3,4 = 5.2 Hz, H-3I), 5.70 (d, 1H, J1,2 = 5.2 Hz, H-1II), 5.63 (s, 1H, NaphCH), 4.71–4.59 (m, 6H, H-5I, H-6I, H-6I, H-3II, H-5II, H-6II), 4.49 (t, 1H, J1,2 = 4.8 Hz, J2,3 = 5.6 Hz, H-2II), 4.41 (d, 1H, J1,1 = 12.4 Hz, H-1I), 4.11 (d, 1H, J1,1 = 12.4 Hz, H-1I), 3.96 (dd, 1H, J3,4 = J4,5 = 9.2 Hz, H-4II), 3.77 (q, 1H, J5,6 = 4.0 Hz, J6,6 = 12.4 Hz, H-6II). 13C-NMR (100 MHz, CDCl3) δ 166.1 (CO), 165.7 (CO), 165.0 (CO), 164.4 (CO), 137.0–124.2 (aromatic C), 107.1 (C-2I), 106.0 (PhCH), 102.7 (NaphCH), 89.6 (C-1II), 80.0 (C-5I), 76.2 (C-4II), 75.7 (C-3I), 74.1 (C-3II), 73.6 (C-2II), 71.8 (C-4I), 69.3 (C-6II), 64.9 (C-6I), 63.3 (C-1I), 58.7 (C-5II). HRMS ESI-TOF: calcd. for C58H48NaO15 [M + Na]+ 1007.2885; found 1007.2886. Compound 12-2: [α]24D +144.51 (c 0.60, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.25–6.98 (aromatic H), 6.11 (PhCH), 5.95 (dd, 1H, J3,4 = 2.4 Hz, J4,5 = 9.6 Hz, H-4I), 5.60 (s, 1H, PhCH), 5.49 (d, 1H, J3,4 = 2.4 Hz, H-3I), 5.42 (d, 1H, J1,2 = 5.26 Hz, H-1II), 5.03 (d, 1H, J1,1 = 17.6 Hz, H-1I), 4.81 (dd, 1H, J5,6 = 2.8 Hz, J6,6 = 12.4 Hz, H-6I), 4.67–4.65 (m, 2H, H-1I, H-3II), 4.61–4.51 (m, 2H, H-2II, H-5II), 4.44 (dd, 1H, J5,6 = 2.8 Hz, J6,6 = 12.4 Hz, H-6I), 4.41–4.35 (m, 2H, H-5I, H-6II), 3.98 (dd, 1H, J3,4 = 3.6 Hz, J4,5 = 9.6 Hz, H-4II), 3.69 (t, 1H, J5,6 = J6,6 = 10.4 Hz, H-6II). 13C-NMR (100 MHz, CDCl3) δ 166.4 (CO), 165.5 (CO), 164.7 (CO), 164.6 (CO), 136.9–126.4 (aromatic C), 106.7 (PhCH), 102.7 (NaphCH), 98.8 (C-1II), 77.2 (C-5I), 76.2 (C-3I, C-4II), 74.7 (C-3II), 73.1 (C-2II), 70.3 (C-4I), 69.1 (C-6II), 67.7 (C-1I), 63.7 (C-6I), 57.5 (C-5II). HRMS ESI-TOF: calcd. for C58H48NaO15 [M + Na]+ 1007.2885; found 1007.2886.
3.9. Synthesis of Compound 1
To a solution of allose derivative 7 (300 mg, 0.57 mmol) and 1,3,4,6-tetra-O-benzoyl-psicofuranoside acceptor (8) (316 mg, 0.530 mmol) in 1,2-dichloroethane (20.0 mL) in the presence of 4 Å molecular sieves (1.58 g), DDQ (155 mg, 0.684 mmol) was added at 0 °C and stirred at room temperature for one day. The ascorbic acid/citric acid/sodium hydroxide solution (0.7/1.3/0.9 wt%) was added at 0 °C to quench the reaction. The reaction mixture of tethering intermediate 11 obtained after post-treatment was dissolved in 1,2-dichloroethane (30.0 mL), and in the presence of 4 Å molecular sieves (1.58 g), 1 M MeOTf solution (1.33 mL, 1.33 mmol) was added at 0 °C. After the reaction proceeded for one day at 40 °C, triethylamine (185 μL, 2.00 mmol) was added at 0 °C to quench the reaction. The reaction solution was diluted with ethyl acetate, filtered through celite, and washed with saturated aq. NaHCO3 and brine. The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure to obtain the mixture 12 (259 mg). To a solution of the mixture (234 mg) in dry dichloromethane (2.0 mL), TFA (200 μL) was added at 0 °C, and the entire mixture was stirred for 1 min. The reaction mixture was dried with N2 gas, and pyridine (200 μL) was added at 0 °C. The reaction mixture was azeotropically dried with toluene, diluted with chloroform, and washed successively with saturated aq. NaHCO3 and brine. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting residue was purified by gel filtration chromatography using chloroform and by flash silica gel column chromatography using a chloroform–MeOH (15/1, v/v) mixture to give an intermediate compound (18.7 mg, 4%). To a solution of the intermediate compound (18.7 mg, 0.025 mmol) in MeOH (1.5 mL), 1 M NaOMe in MeOH (20 µL) was added at 0 °C. The reaction mixture was stirred at room temperature for 4 h, neutralized with Amberlyst, filtered, and concentrated in vacuo. The resulting residue was purified by reversed-phase column chromatography (H2O) to give compound 1 (8.6 mg, 0.025 mmol, quant.). [α]23D +120.24 (c 0.10, H2O); 1H-NMR (400 MHz, D2O) δ 7.70–7.27 (m, 5H, aromatic H of benzoate adduct), 5.32 (d, 1H, J = 4.0 Hz, H-1II), 4.25 (dd, 1H, J = 4.4, 8.8 Hz), 4.20 (d, 1H, J = 4.8 Hz), 4.09 (t, 1H, J = 4.8 Hz), 4.04 (td, 1H, J = 5.2, 8.4 Hz), 3.87 (m, 1H), 3.81–3.42 (m, 8H). 13C-NMR (100 MHz, D2O) δ 175.8 (C=O of benzoate adduct), 136.1–128.3 (aromatic C of benzoate adduct), 109.2 (C-2I), 91.6 (C-1II), 83.5 (C-5I), 73.5 (C-3I), 71.0 (C-3II), 70.4 (C-4I), 67.5 (C-5II), 66.7 (C-2II), 65.6 (C-4II), 62.4 (C-6I), 60.2 (C-1I), 59.8 (C-6II). HRMS ESI-TOF: calcd. for C12H22NaO11 [M + Na]+ 365.1054; found 365.1046.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}