
A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
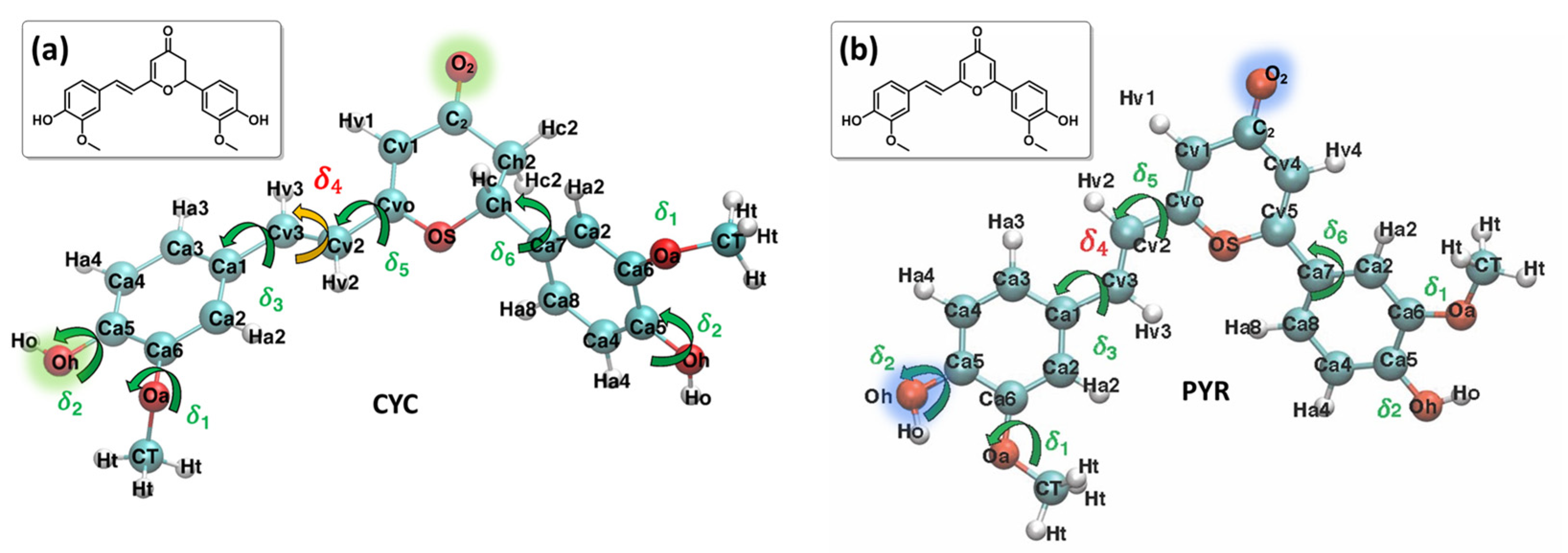
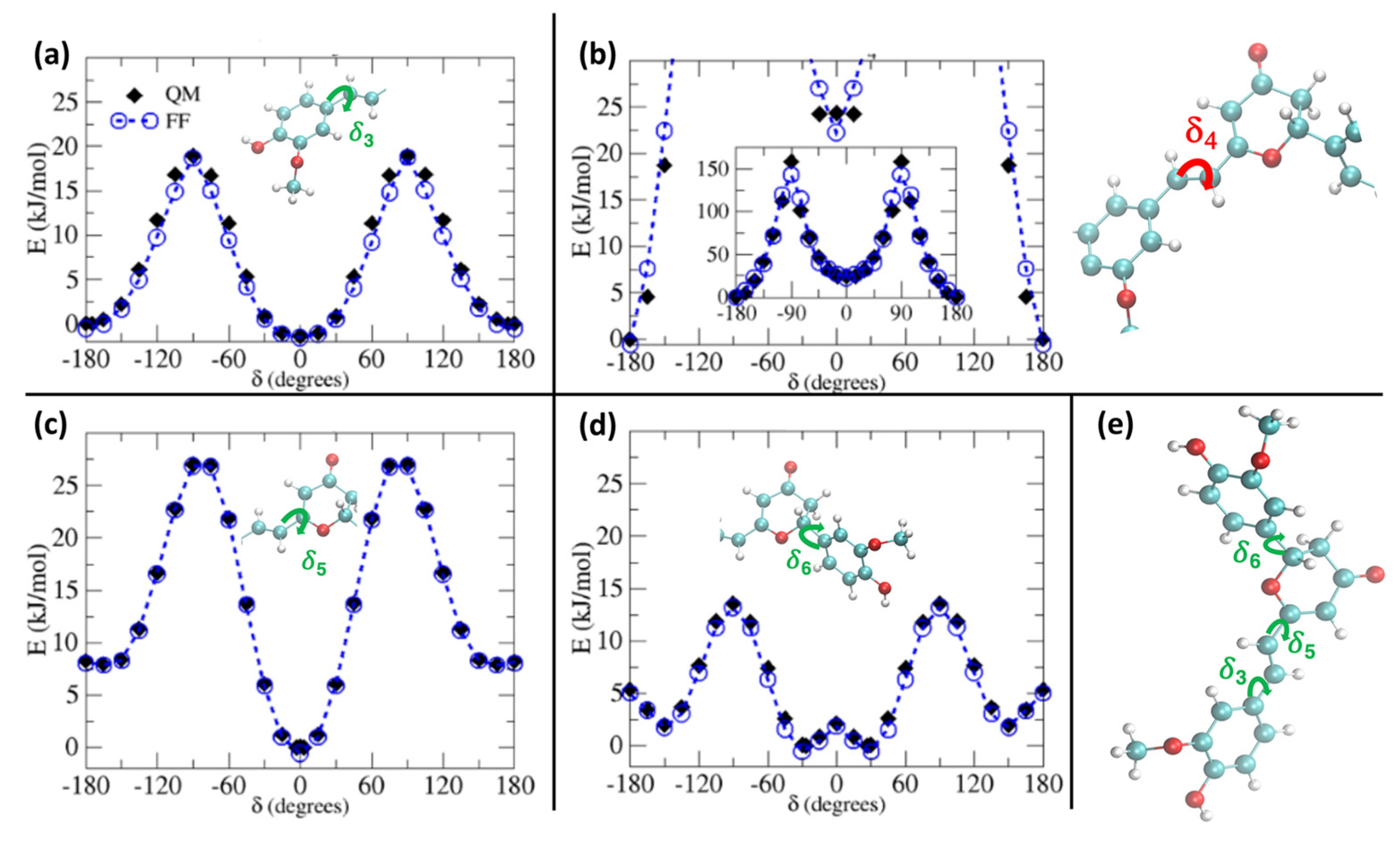
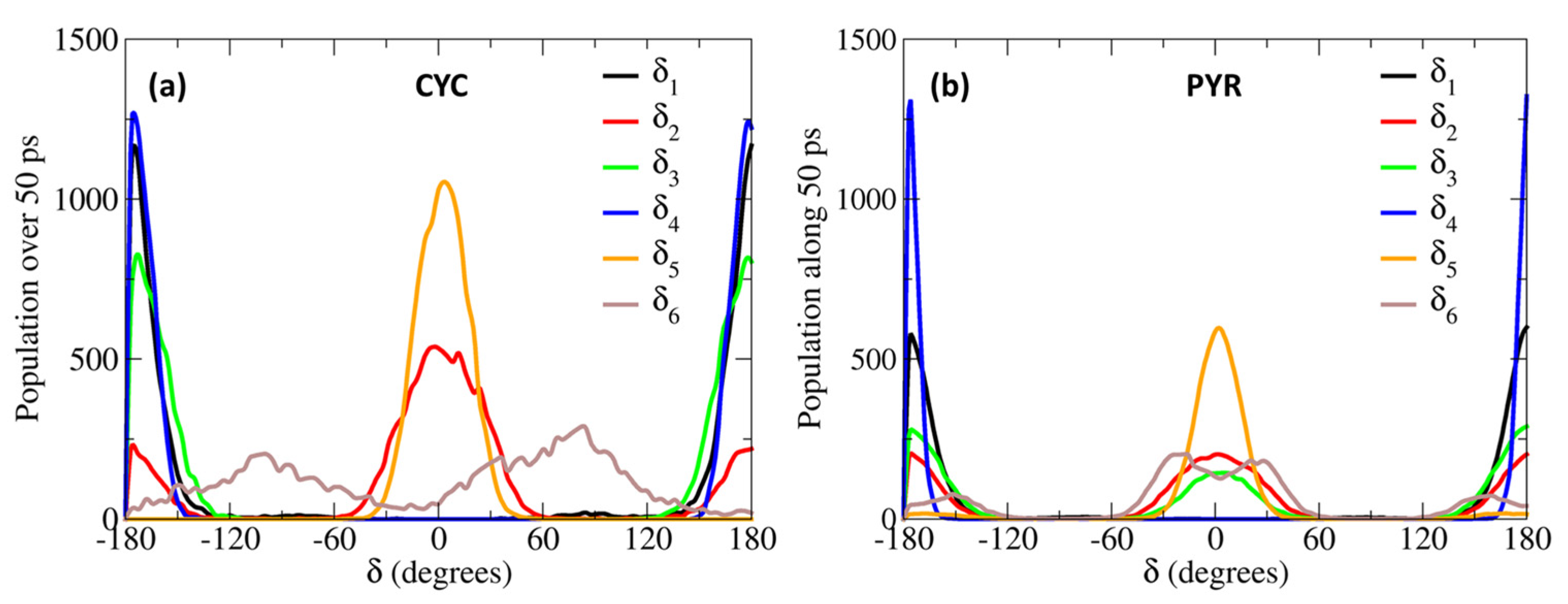
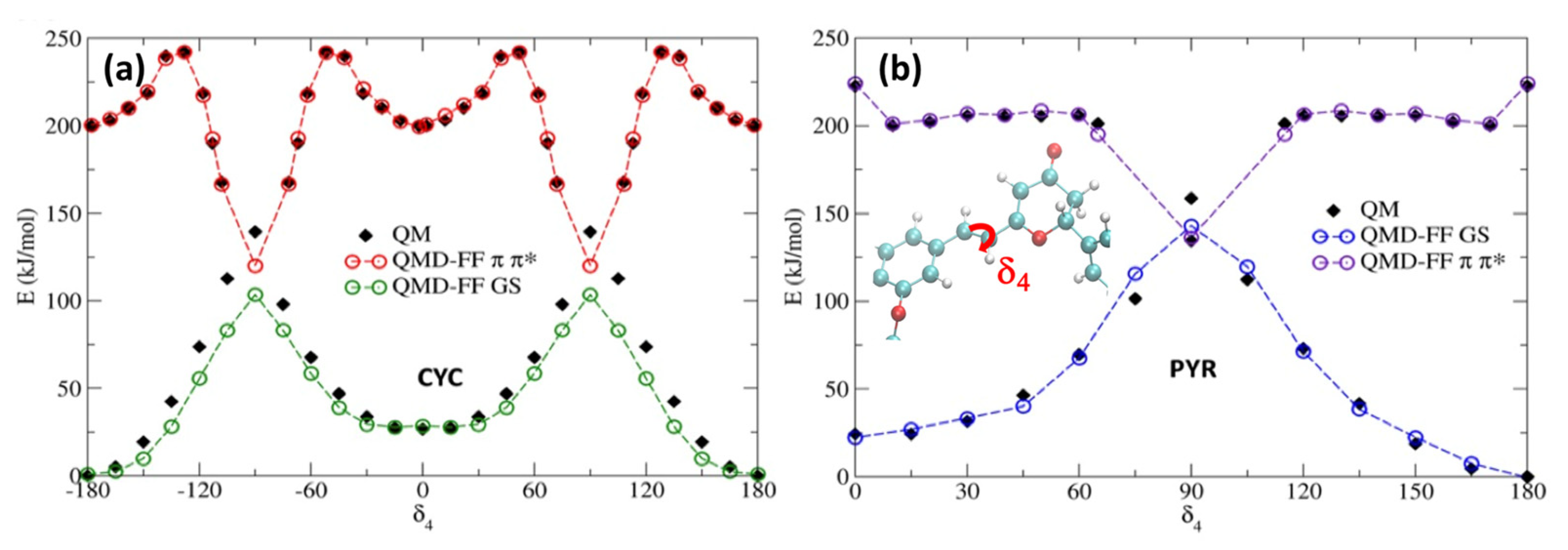
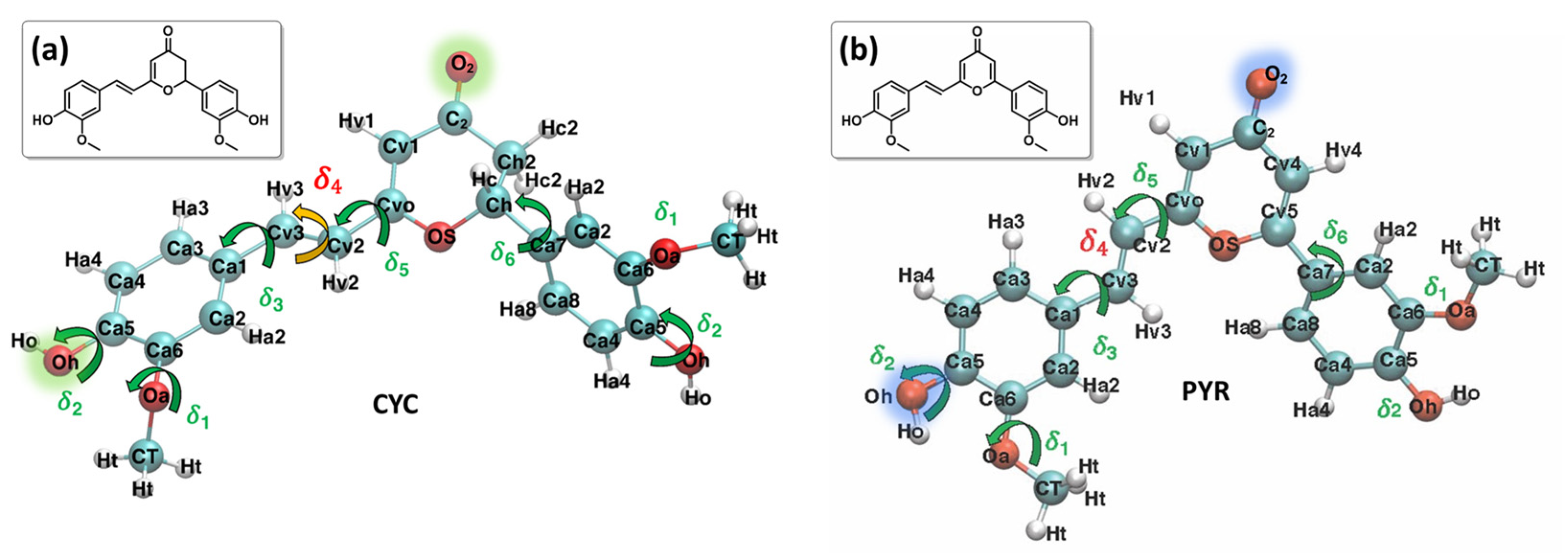
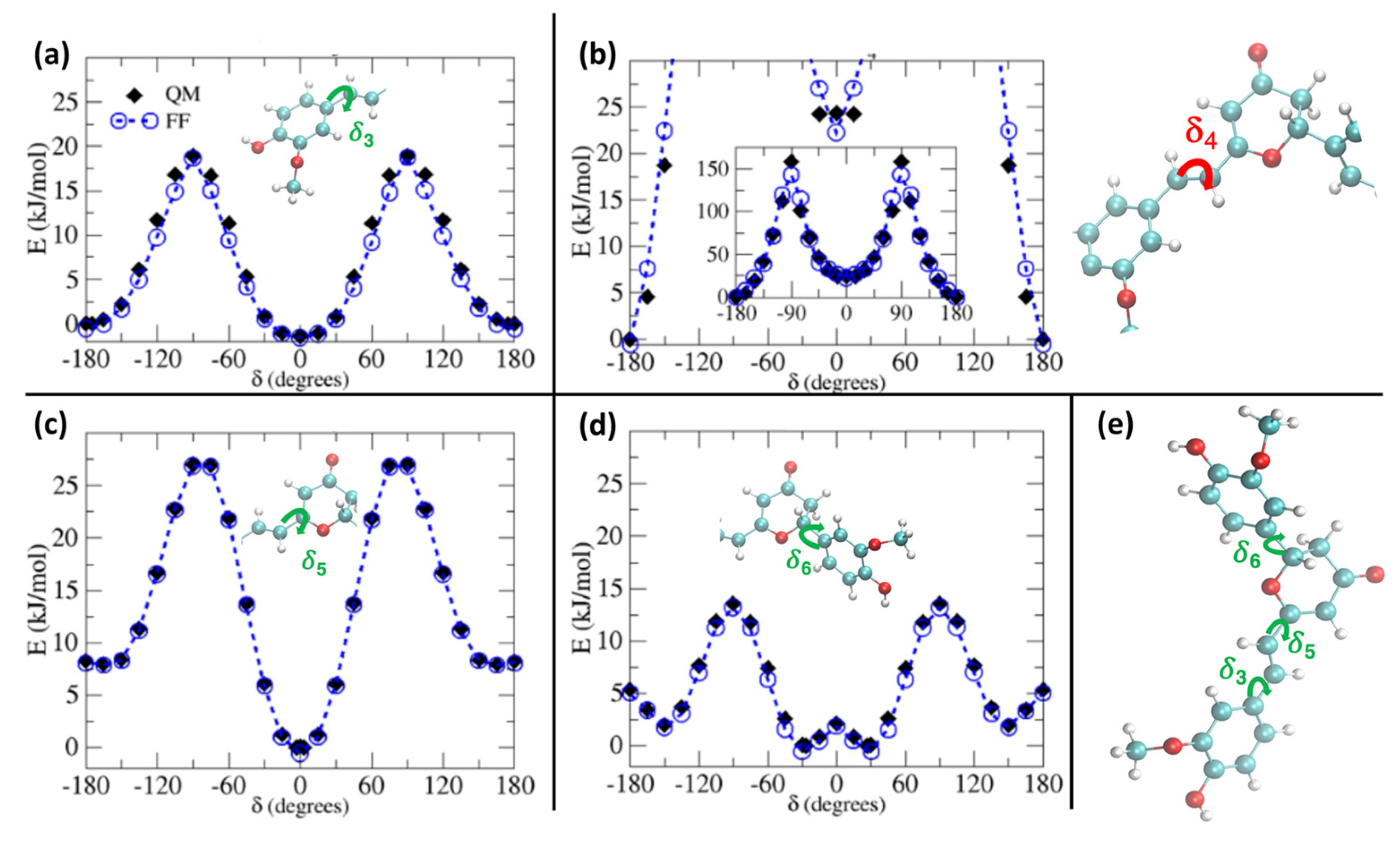
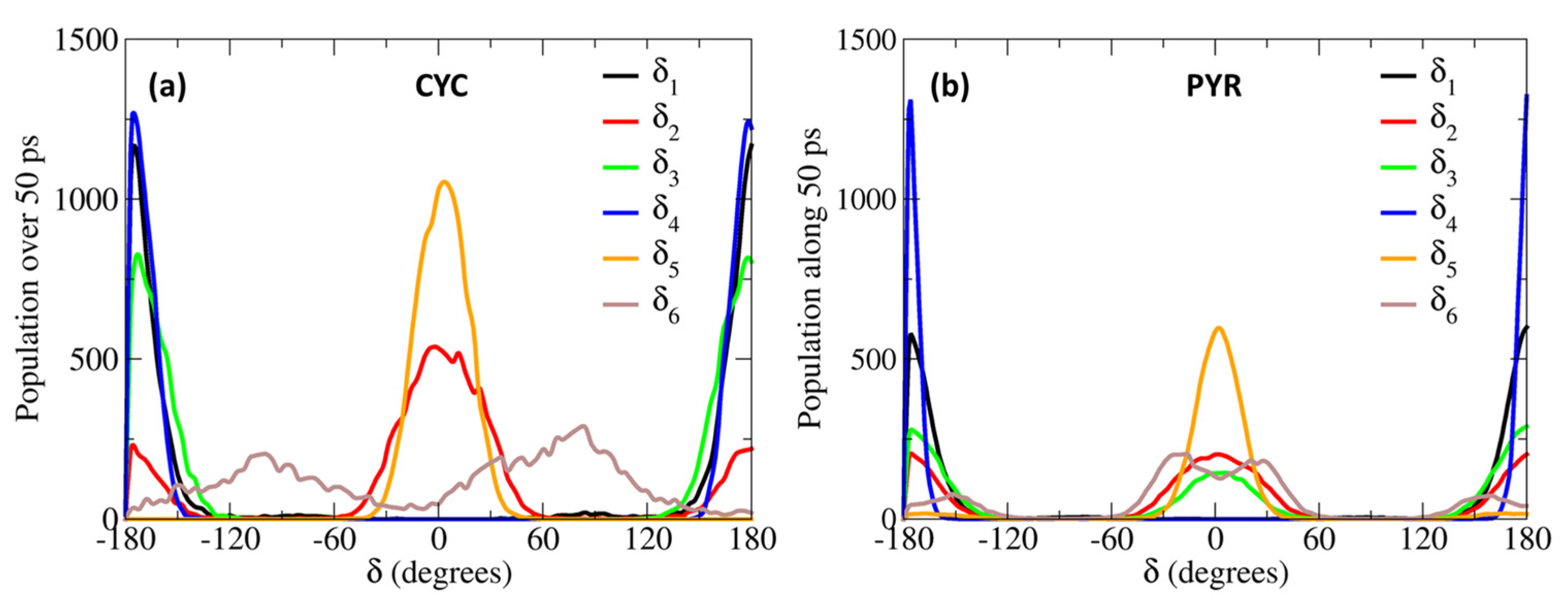
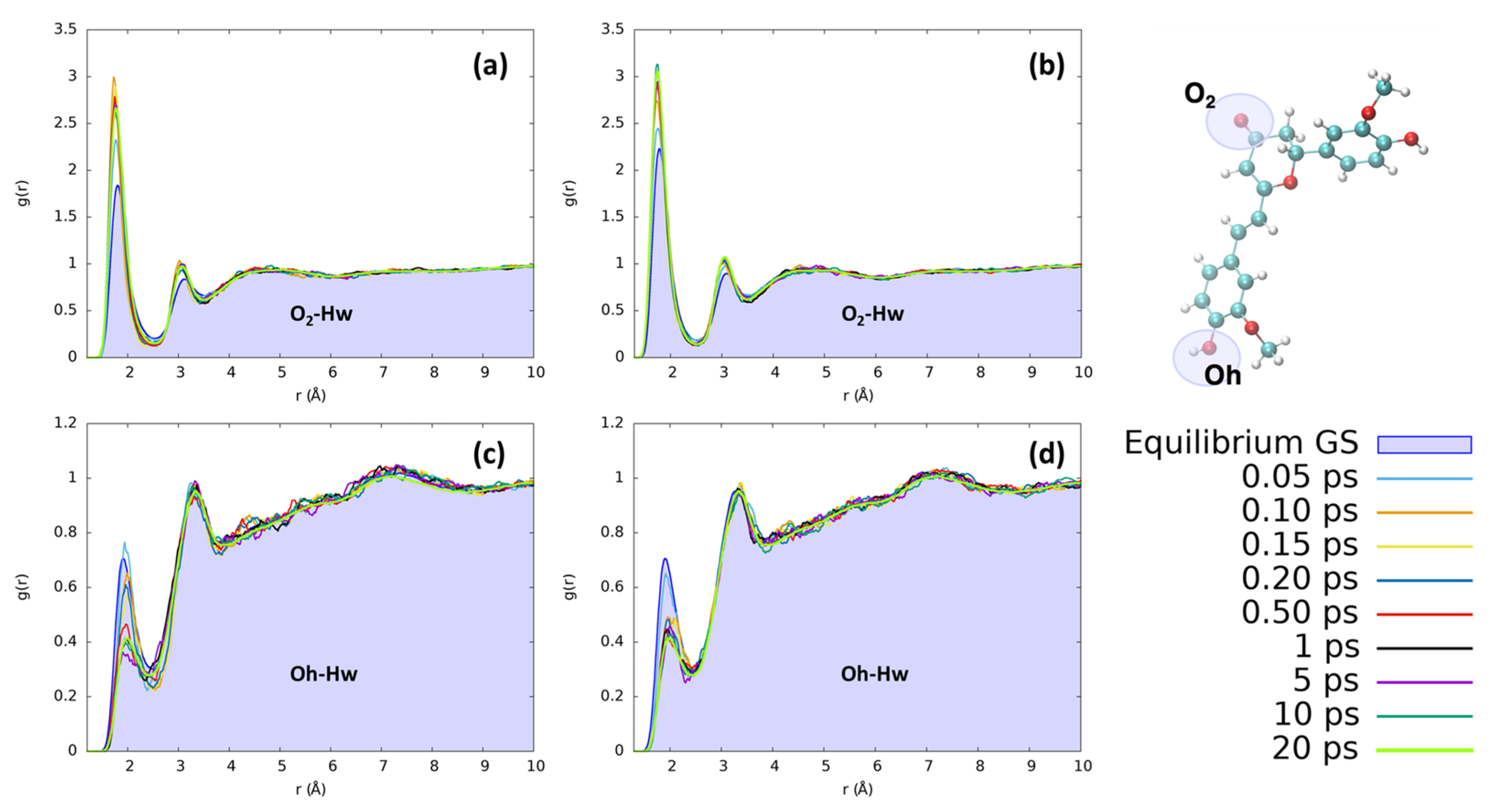
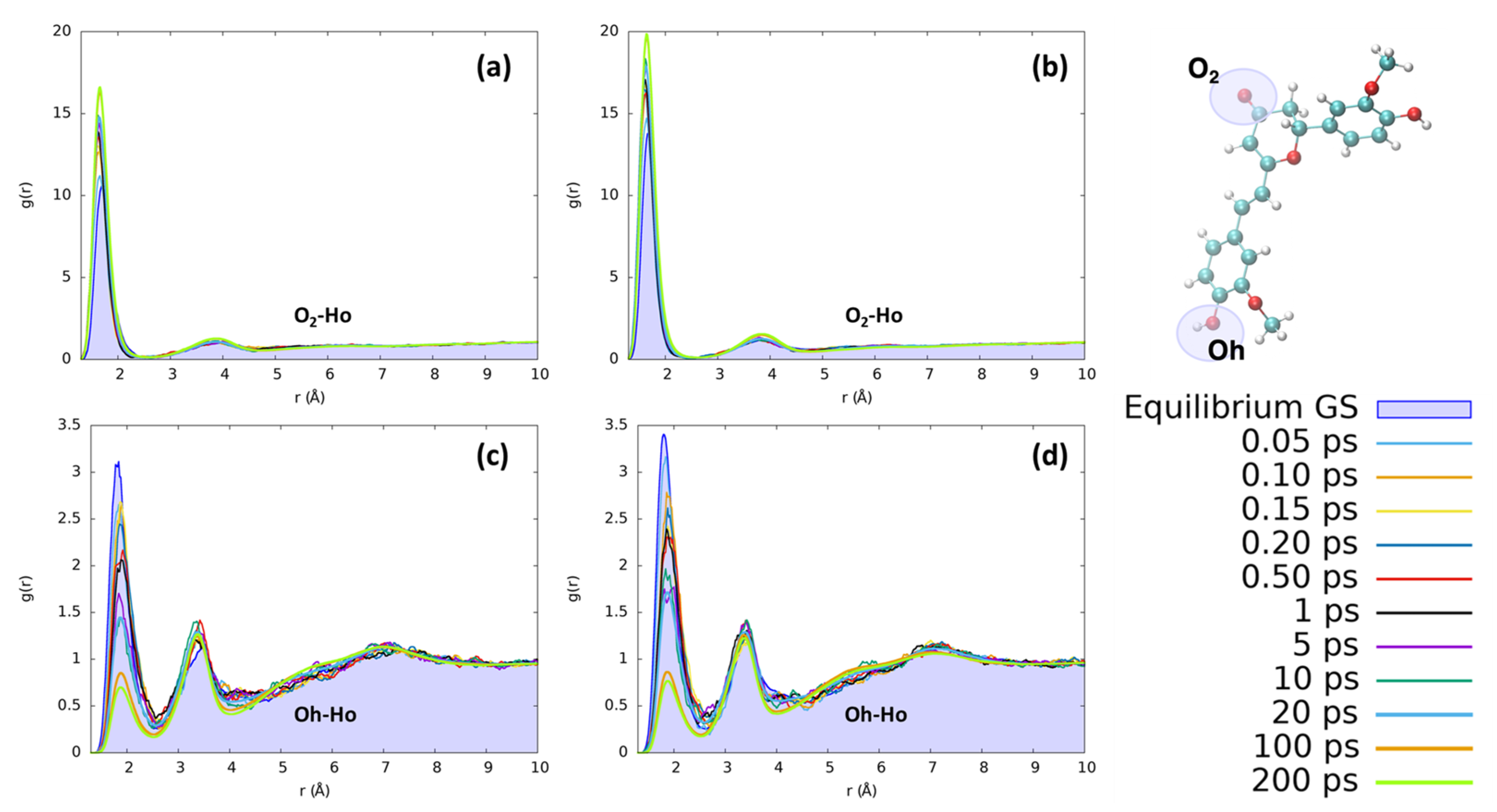
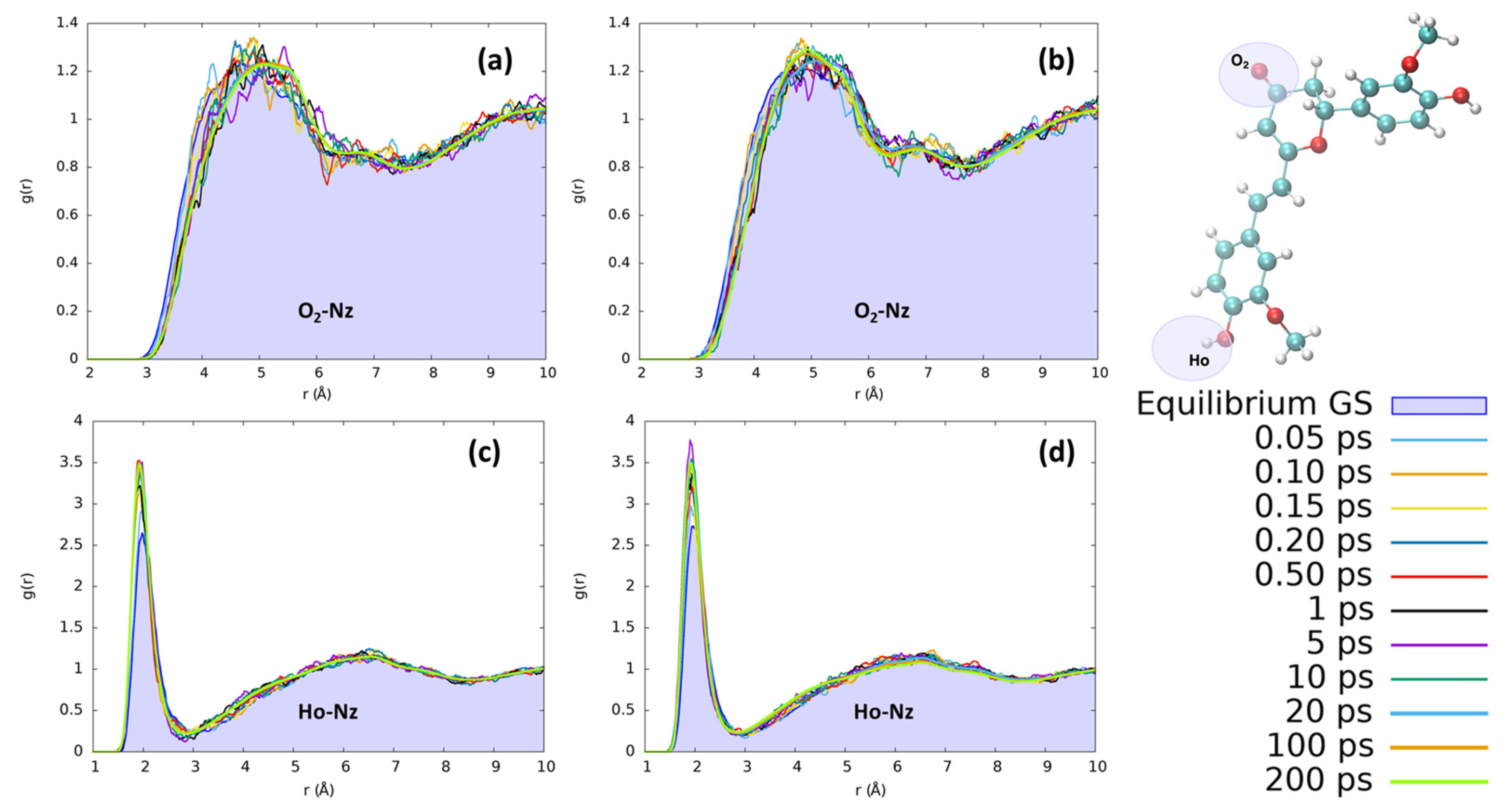
2. Results and Discussion
3. Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broichhagen, J.; Frank, J.A.; Trauner, D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, M.; Garcia-Iriepa, C. Retinal Inspired Photoswitches: From the Isomerization Mechanisms toward Recent Applications. In Photoisomerization: Causes, Behavior and Effects; Sampedro, D., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2019; ISBN 978-1-53615-313-2. [Google Scholar]
- Russew, M.M.; Hecht, S. Photoswitches: From Molecules to Materials. Adv. Mater. 2010, 22, 3348–3360. [Google Scholar] [CrossRef]
- Szymański, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef] [PubMed]
- Wald, G. Molecular Basis of Visual Excitation. Science 1968, 162, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Weingart, O.; Schapiro, I.; Buss, V. Photochemistry of Visual Pigment Chromophore Models by Ab Initio Molecular Dynamics. J. Phys. Chem. B 2007, 111, 3782–3788. [Google Scholar] [CrossRef]
- Wietek, J.; Wiegert, J.S.; Adeishvili, N.; Schneider, F.; Watanabe, H.; Tsunoda, S.P.; Vogt, A.; Elstner, M.; Oertner, T.G.; Hegemann, P. Conversion of Channelrhodopsin into a Light-Gated Chloride Channel. Science 2014, 344, 409–412. [Google Scholar] [CrossRef]
- Fan, W.; Huang, P.; Chen, X. Overcoming the Achilles’ Heel of Photodynamic Therapy. Chem. Soc. Rev. 2016, 45, 6488–6519. [Google Scholar] [CrossRef]
- Reessing, F.; Szymanski, W. Beyond Photodynamic Therapy: Light-Activated Cancer Chemotherapy. Curr. Med. Chem. 2017, 24, 4905–4950. [Google Scholar] [PubMed]
- Babilas, P.; Landthaler, M.; Szeimies, R.-M. Photodynamic Therapy in Dermatology. Eur. J. Dermatol. 2006, 16, 340–348. [Google Scholar]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The Role of Porphyrin Chemistry in Tumor Imaging and Photodynamic Therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Yanovsky, R.L.; Bartenstein, D.W.; Rogers, G.S.; Isakoff, S.J.; Chen, S.T. Photodynamic Therapy for Solid Tumors: A Review of the Literature. Photodermatol. Photoimmunol. Photomed. 2019, 35, 295–303. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Zhang, L.; Ji, Z.; Zhang, J.; Yang, S. Photodynamic Therapy Enhances Skin Cancer Chemotherapy Effects through Autophagy Regulation. Photodiagn. Photodyn. Ther. 2019, 28, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Simon, C.; Finke, M.; Bleses, K.; Birke, M.; Szentmáry, N.; Hüttenberger, D.; Eppig, T.; Stachon, T.; Langenbucher, A.; et al. Photodynamic Inactivation of Multidrug-Resistant Staphylococcus Aureus by Chlorin E6 and Red Light (λ = 670 nm). J. Photochem. Photobiol. B 2016, 162, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Penha, C.B.; Bonin, E.; da Silva, A.F.; Hioka, N.; Zanqueta, É.B.; Nakamura, T.U.; de Abreu Filho, B.A.; Campanerut-Sá, P.A.Z.; Mikcha, J.M.G. Photodynamic Inactivation of Foodborne and Food Spoilage Bacteria by Curcumin. LWT-Food Sci. Technol. 2017, 76, 198–202. [Google Scholar] [CrossRef]
- Yi, F.; Zheng, X.; Fang, F.; Zhang, J.; Zhou, B.; Chen, X. ALA-PDT Alleviates the Psoriasis by Inhibiting JAK Signalling Pathway. Exp. Dermatol. 2019, 28, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Josefsen, L.B.; Boyle, R.W. Photodynamic Therapy and the Development of Metal-Based Photosensitisers. Met. Based Drugs 2008, 2008, 276109. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.C.; Nguyen, V.N.; Choi, Y.; Lee, S.; Yoon, J. Recent Strategies to Develop Innovative Photosensitizers for Enhanced Photodynamic Therapy. Chem. Rev. 2021, 121, 13454–13619. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Photodynamic Therapy for Cancer: What’s Past Is Prologue. Photochem. Photobiol. 2020, 96, 506–516. [Google Scholar] [CrossRef]
- Brown, J.M. Tumor Hypoxia in Cancer Therapy. Methods Enzymol. 2007, 435, 297–321. [Google Scholar] [CrossRef]
- Dunkel, P.; Ilaš, J. Targeted Cancer Therapy Using Compounds Activated by Light. Cancers 2021, 13, 3237. [Google Scholar] [CrossRef]
- Askes, S.H.C.; Reddy, G.U.; Wyrwa, R.; Bonnet, S.; Schiller, A. Red Light-Triggered CO Release from Mn2(CO)10 Using Triplet Sensitization in Polymer Nonwoven Fabrics. J. Am. Chem. Soc. 2017, 139, 15292–15295. [Google Scholar] [CrossRef] [PubMed]
- García-López, V.; Chen, F.; Nilewski, L.G.; Duret, G.; Aliyan, A.; Kolomeisky, A.B.; Robinson, J.T.; Wang, G.; Pal, R.; Tour, J.M. Molecular Machines Open Cell Membranes. Nature 2017, 548, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Orozco, C.; Galvez-Aranda, D.; Corona, A.; Seminario, J.M.; Rangel, R.; Myers, J.N.; Tour, J.M. Molecular Jackhammers Eradicate Cancer Cells by Vibronic-Driven Action. Nat. Chem. 2024, 16, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Sinkger, N.K.; Gonzalez, L.; Monari, A. Molecular Photoswitches Regulating the Activity of the Human Serotonin Transporter. J. Phys. Chem. Lett. 2023, 14, 10333–10339. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Morstein, J.; Ladefoged, L.K.; Maesen, J.B.; Schiøtt, B.; Sinning, S.; Trauner, D. A Photoswitchable Inhibitor of the Human Serotonin Transporter. ACS Chem. Neurosci. 2020, 11, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, M.; Xie, Z. Carrier-Free Core–Shell Nanodrugs for Synergistic Two-Photon Photodynamic Therapy of Cervical Cancer. J. Colloid. Interface Sci. 2019, 535, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kuptniratsaikul, V.; Dajpratham, P.; Taechaarpornkul, W.; Buntragulpoontawee, M.; Lukkanapichonchut, P.; Chootip, C.; Saengsuwan, J.; Tantayakom, K.; Laongpech, S. Efficacy and Safety of Curcuma Domestica Extracts Compared with Ibuprofen in Patients with Knee Osteoarthritis: A Multicenter Study. Clin. Interv. Aging 2014, 9, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A Novel Nanoparticle Drug Delivery System: The Anti-Inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Losantos, R.; Pasc, A.; Monari, A. Don’t Help Them to Bury the Light. The Interplay between Intersystem Crossing and Hydrogen Transfer in Photoexcited Curcumin Revealed by Surface-Hopping Dynamics. Phys. Chem. Chem. Phys. 2021, 23, 24757–24764. [Google Scholar] [CrossRef]
- Tao, R.; Zhang, F.; Tang, Q.J.; Xu, C.S.; Ni, Z.J.; Meng, X. Effects of Curcumin-Based Photodynamic Treatment on the Storage Quality of Fresh-Cut Apples. Food Chem. 2019, 274, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, R.; Barnes, C.A.; Trampel, R.L.; Wallace, S.J.; Kee, T.W.; Petrich, J.W. Photoinduced Trans-to-Cis Isomerization of Cyclocurcumin. J. Phys. Chem. B 2011, 115, 10707–10714. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, M.; Francés-Monerris, A.; Mourer, M.; Pasc, A.; Monari, A. Trans -to- Cis Photoisomerization of Cyclocurcumin in Different Environments Rationalized by Computational Photochemistry. Phys. Chem. Chem. Phys. 2020, 22, 4749–4757. [Google Scholar] [CrossRef]
- Pecourneau, J.; Losantos, R.; Monari, A.; Parant, S.; Pasc, A.; Mourer, M. Synthesis and Photoswitching Properties of Bioinspired Dissymmetric γ-Pyrone, an Analogue of Cyclocurcumin. J. Org. Chem. 2021, 86, 8112–8126. [Google Scholar] [CrossRef]
- Losantos, R.; Pecourneau, J.; Mourer, M.; Parant, S.; Pasc, A.; Monari, A. Trans-CisPhotoisomerization of a Biomimetic Cyclocurcumin Analogue Rationalized by Molecular Modelling. Phys. Chem. Chem. Phys. 2021, 23, 12842–12849. [Google Scholar] [CrossRef]
- Delova, A.; Losantos, R.; Pecourneau, J.; Mourer, M.; Pasc, A.; Monari, A. Modelling the Effects of E/Z Photoisomerization of a Cyclocurcumin Analogue on the Properties of Cellular Lipid Membranes. Phys. Chem. Chem. Phys. 2023, 25, 20567–20574. [Google Scholar] [CrossRef]
- Pecourneau, J.; Losantos, R.R.; Delova, A.; Bernhard, Y.; Parant, S.S.; Mourer, M.; Monari, A.; Pasc, A. Biomimetic Photo-Switches Softening Model Lipid Membranes. Langmuir 2022, 38, 15642–15655. [Google Scholar] [CrossRef]
- Delova, A.; Losantos, R.; Pecourneau, J.; Bernhard, Y.; Mourer, M.; Pasc, A.; Monari, A. Perturbation of Lipid Bilayers by Biomimetic Photoswitches Based on Cyclocurcumin. J. Chem. Inf. Model. 2023, 63, 299–307. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B. Polarizable Continuum Model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Barbatti, M. Nonadiabatic Dynamics with Trajectory Surface Hopping Method. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 620–633. [Google Scholar] [CrossRef]
- Crespo-Otero, R.; Barbatti, M. Recent Advances and Perspectives on Nonadiabatic Mixed Quantum-Classical Dynamics. Chem. Rev. 2018, 118, 7026–7068. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Marquetand, P.; González, L. Nonadiabatic Dynamics: The SHARC Approach. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1370. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Avagliano, D.; Heindl, M.; Marquetand, P.; Menger, M.F.S.J.; Oppel, M.; Plasser, F.; Polonius, S.; Ruckenbauer, M.; Shu, Y.; et al. SHARC3.0: Surface Hopping Including Arbitrary Couplings—Program Package for Non-Adiabatic Dynamics. 2023. Available online: https://sharc-md.org/ (accessed on 5 April 2024).
- Polli, D.; Altoè, P.; Weingart, O.; Spillane, K.M.; Manzoni, C.; Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R.A.; et al. Conical Intersection Dynamics of the Primary Photoisomerization Event in Vision. Nature 2010, 467, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Gozem, S.; Schapiro, I.; Ferre, N.; Olivucci, M. The Molecular Mechanism of Thermal Noise in Rod Photoreceptors. Science 2012, 337, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, D.; Bonfanti, M.; Garavelli, M.; González, L. QM/MM Nonadiabatic Dynamics: The SHARC/COBRAMM Approach. J. Chem. Theory Comput. 2021, 17, 4639–4647. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.K.; Kabaciński, P.; Nogueira de Faria, B.E.; Gentile, M.; de Paula, A.M.; Borrego-Varillas, R.; Nenov, A.; Conti, I.; Cerullo, G.; Garavelli, M. Environment-Driven Coherent Population Transfer Governs the Ultrafast Photophysics of Tryptophan. J. Am. Chem. Soc. 2022, 144, 12884–12892. [Google Scholar] [CrossRef] [PubMed]
- Bondanza, M.; Demoulin, B.; Lipparini, F.; Barbatti, M.; Mennucci, B. Trajectory Surface Hopping for a Polarizable Embedding QM/MM Formulation. J. Phys. Chem. A 2022, 126, 6780–6789. [Google Scholar] [CrossRef]
- Stojanović, L.; Aziz, S.G.; Hilal, R.H.; Plasser, F.; Niehaus, T.A.; Barbatti, M. Nonadiabatic Dynamics of Cycloparaphenylenes with TD-DFTB Surface Hopping. J. Chem. Theory Comput. 2017, 13, 5846–5860. [Google Scholar] [CrossRef]
- Dral, P.O.; Barbatti, M.; Thiel, W. Nonadiabatic Excited-State Dynamics with Machine Learning. J. Phys. Chem. Lett. 2018, 9, 5660–5663. [Google Scholar] [CrossRef]
- Zobel, J.P.; Heindl, M.; Plasser, F.; Mai, S.; González, L. Surface Hopping Dynamics on Vibronic Coupling Models. Acc. Chem. Res. 2021, 54, 3760–3771. [Google Scholar] [CrossRef]
- Plasser, F.; Gómez, S.; Menger, M.F.S.J.; Mai, S.; González, L. Highly Efficient Surface Hopping Dynamics Using a Linear Vibronic Coupling Model. Phys. Chem. Chem. Phys. 2019, 21, 57–69. [Google Scholar] [CrossRef]
- Mai, S.; Menger, M.F.S.J.; Marazzi, M.; Stolba, D.L.; Monari, A.; González, L. Competing Ultrafast Photoinduced Electron Transfer and Intersystem Crossing of [Re(CO)3(Dmp)(His124)(Trp122)]+ in Pseudomonas Aeruginosa Azurin: A Nonadiabatic Dynamics Study. Theor. Chem. Acc. 2020, 139, 65. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; González, L. Unconventional Two-Step Spin Relaxation Dynamics of [Re(CO)3(Im)(Phen)]+ in Aqueous Solution. Chem. Sci. 2019, 10, 10405–10411. [Google Scholar] [CrossRef]
- Vilhena, J.G.; Greff Da Silveira, L.; Livotto, P.R.; Cacelli, I.; Prampolini, G. Automated Parameterization of Quantum Mechanically Derived Force Fields for Soft Materials and Complex Fluids: Development and Validation. J. Chem. Theory Comput. 2021, 17, 4449–4464. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; De Mitri, N.; Licari, D.; Monti, S.; Prampolini, G. Joyce and Ulysses: Integrated and User-Friendly Tools for the Parameterization of Intramolecular Force Fields from Quantum Mechanical Data. Phys. Chem. Chem. Phys. 2013, 15, 3736–3751. [Google Scholar] [CrossRef]
- Segalina, A.; Aranda, D.; Green, J.A.; Cristino, V.; Caramori, S.; Prampolini, G.; Pastore, M.; Santoro, F. How the Interplay among Conformational Disorder, Solvation, Local, and Charge-Transfer Excitations Affects the Absorption Spectrum and Photoinduced Dynamics of Perylene Diimide Dimers: A Molecular Dynamics/Quantum Vibronic Approach. J. Chem. Theory Comput. 2022, 18, 3718–3736. [Google Scholar] [CrossRef]
- Prampolini, G.; Ingrosso, F.; Segalina, A.; Caramori, S.; Foggi, P.; Pastore, M. Dynamical and Environmental Effects on the Optical Properties of an Heteroleptic Ru(II)–Polypyridine Complex: A Multilevel Approach Combining Accurate Ground and Excited State QM-Derived Force Fields, MD and TD-DFT. J. Chem. Theory Comput. 2019, 15, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, J.; García-Iriepa, C.; Santoro, F.; Navizet, I.; Prampolini, G. Unraveling the Contributions to the Spectral Shape of Flexible Dyes in Solution: Insights on the Absorption Spectrum of an Oxyluciferin Analogue. Phys. Chem. Chem. Phys. 2023, 25, 5007–5020. [Google Scholar] [CrossRef]
- Cerezo, J.; Prampolini, G.; Cacelli, I. Developing Accurate Intramolecular Force Fields for Conjugated Systems through Explicit Coupling Terms. Theor. Chem. Acc. 2018, 137, 80. [Google Scholar] [CrossRef]
- Cacelli, I.; Prampolini, G. Parametrization and Validation of Intramolecular Force Fields Derived from DFT Calculations. J. Chem. Theory Comput. 2007, 3, 1803–1817. [Google Scholar] [CrossRef] [PubMed]
- Prampolini, G.; Campetella, M.; Ferretti, A. Solvent Effects on Catechol’s Binding Affinity: Investigating the Role of the Intra-Molecular Hydrogen Bond through a Multi-Level Computational Approach. Phys. Chem. Chem. Phys. 2023, 25, 2523–2536. [Google Scholar] [CrossRef]
- Fonseca, T.; Ladanyi, B.M. Breakdown of Linear Response for Solvation Dynamics in Methanol. J. Phys. Chem. 1991, 95, 2116–2119. [Google Scholar] [CrossRef]
- Fonseca, T.; Ladanyi, B.M. Solvation Dynamics in Methanol: Solute and Perturbation Dependence. J. Mol. Liq. 1994, 60, 1–24. [Google Scholar] [CrossRef]
- Cerezo, J.; Gao, S.; Armaroli, N.; Ingrosso, F.; Prampolini, G.; Santoro, F.; Ventura, B.; Pastore, M. Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol. Molecules 2023, 28, 3910. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_C01 2016, Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Mark, J.A.; Teemu, M.; Roland, S.; Szilárd, P.; Jeremy, C.S.; Berk, H.; Erik, L. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losantos, R.; Prampolini, G.; Monari, A. A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives. Molecules 2024, 29, 1752. https://doi.org/10.3390/molecules29081752
Losantos R, Prampolini G, Monari A. A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives. Molecules. 2024; 29(8):1752. https://doi.org/10.3390/molecules29081752
Chicago/Turabian StyleLosantos, Raúl, Giacomo Prampolini, and Antonio Monari. 2024. "A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives" Molecules 29, no. 8: 1752. https://doi.org/10.3390/molecules29081752
APA StyleLosantos, R., Prampolini, G., & Monari, A. (2024). A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives. Molecules, 29(8), 1752. https://doi.org/10.3390/molecules29081752