

Tropospheric Photochemistry of 2-Butenedial: Role of the Triplet States, CO and Acrolein Formation, and the Experimentally Unidentified Carbonyl Compound—Theoretical Study


Abstract
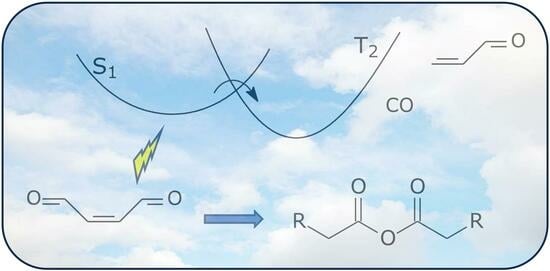
1. Introduction
2. Results
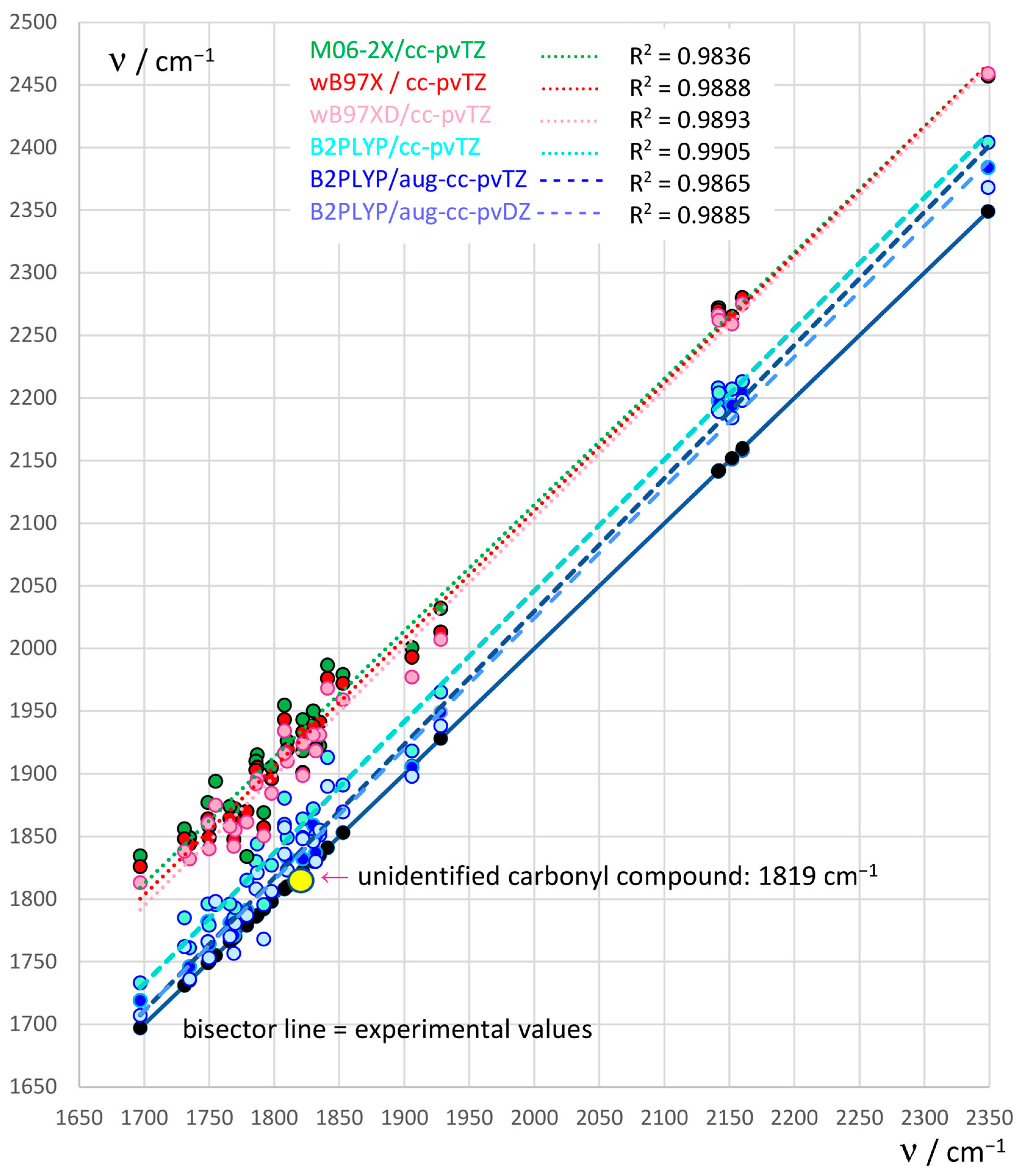
2.1. Assessment of a Suitable Computational Level to Identify the Unknown Carbonyl Product
2.2. Gas-Phase Reaction Pathways Leading to Candidate Products
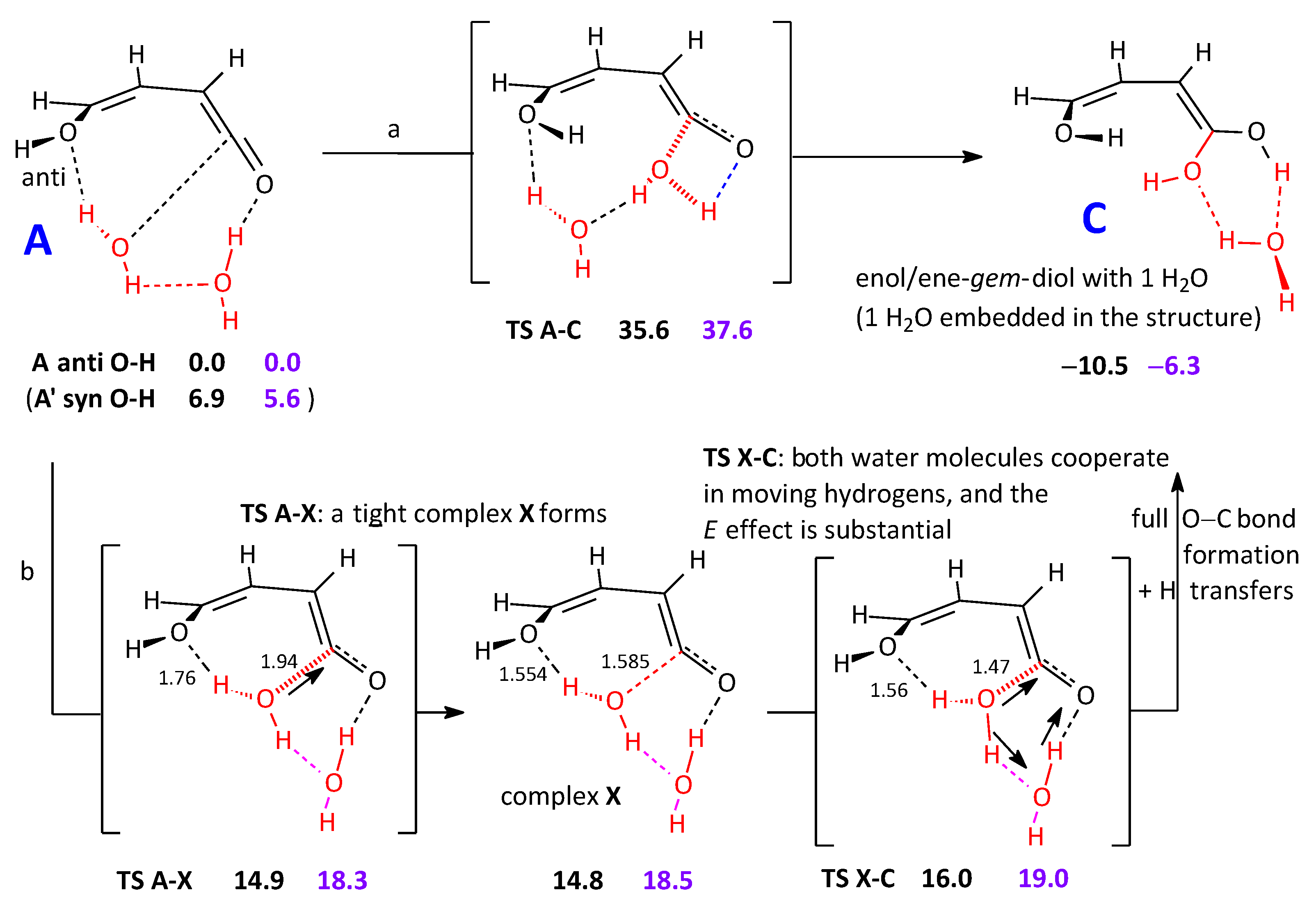
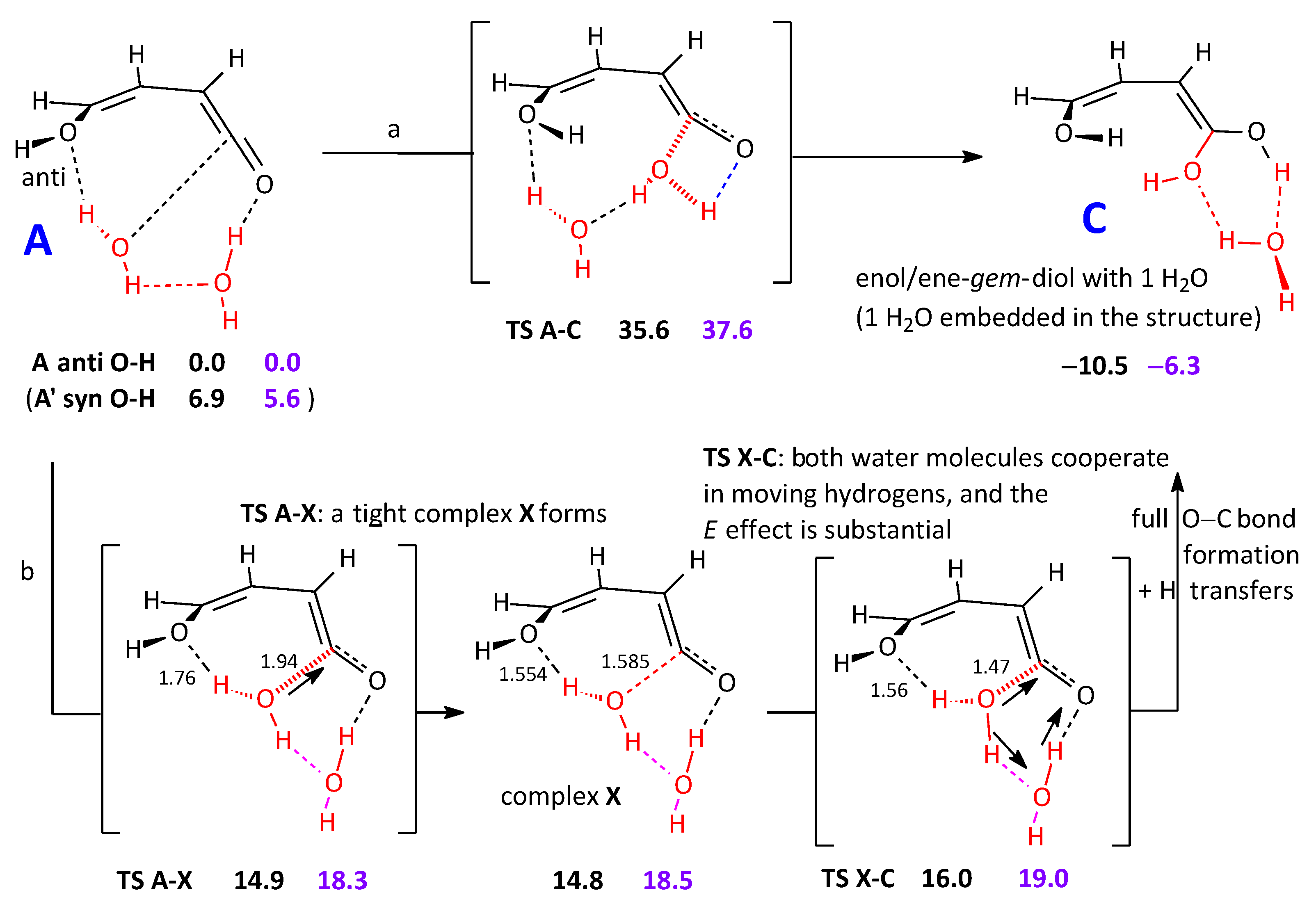
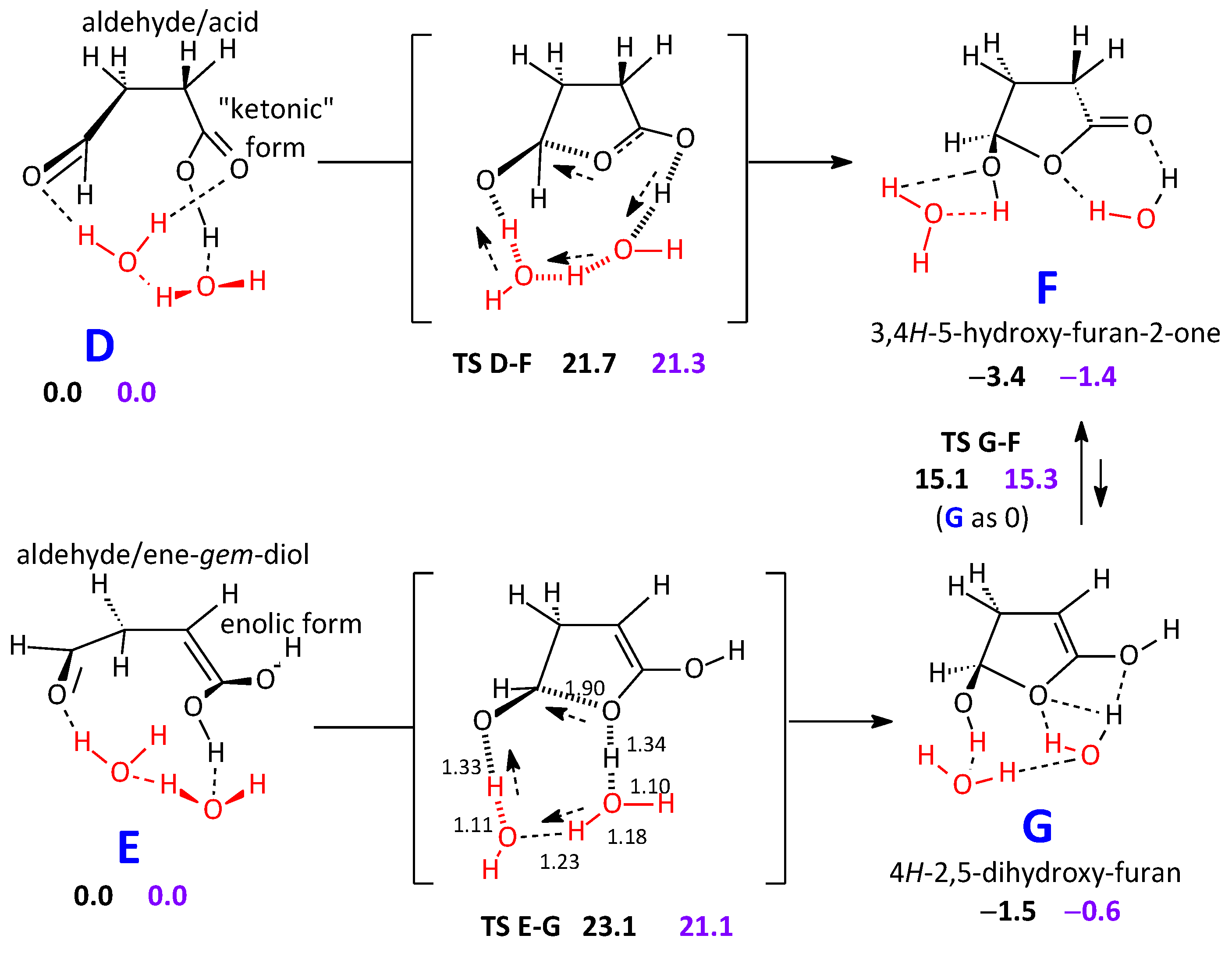
2.2.1. Reaction Pathways to One Cyclic Candidate Product
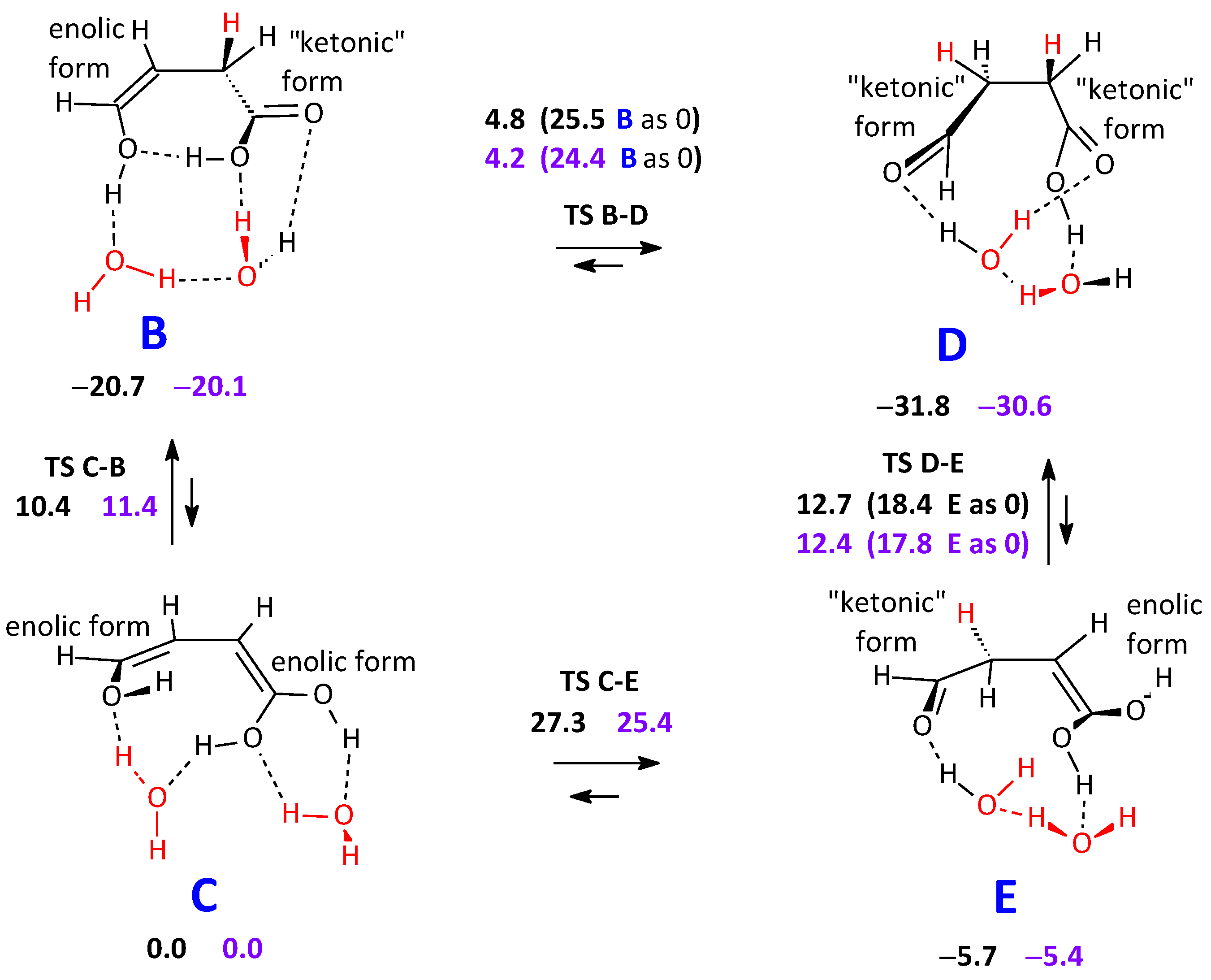
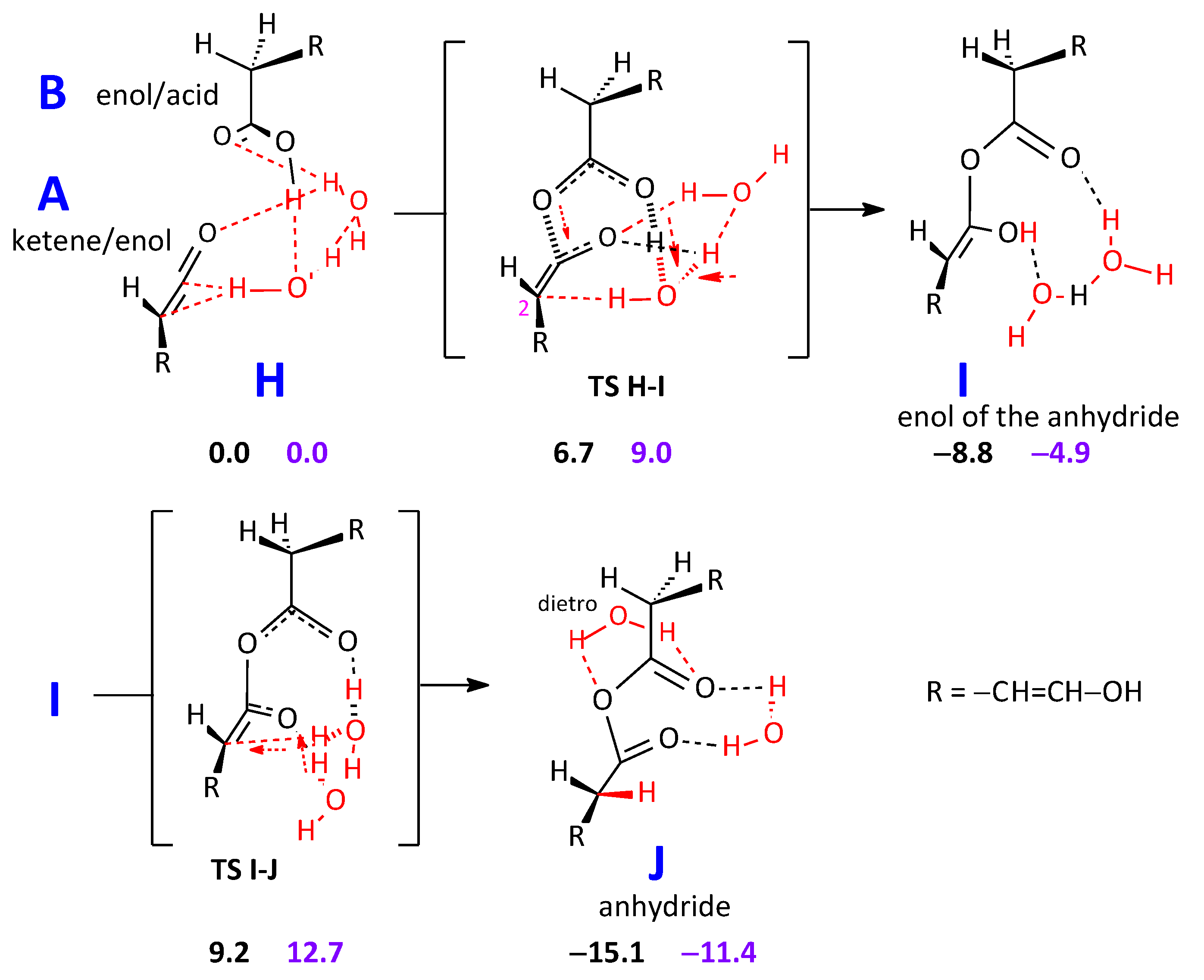
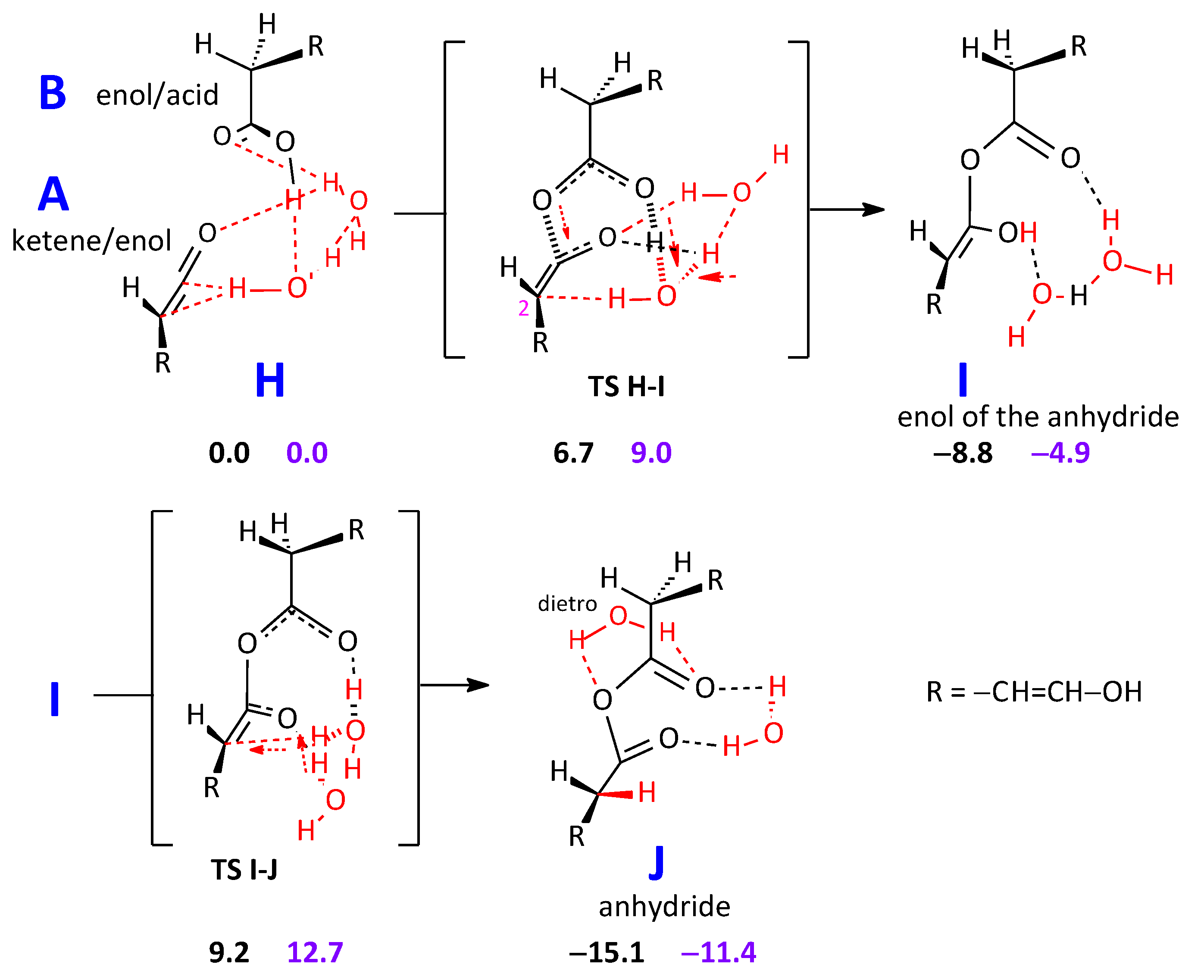
2.2.2. Formation of an Anhydride
2.3. Excited States and CO Formation
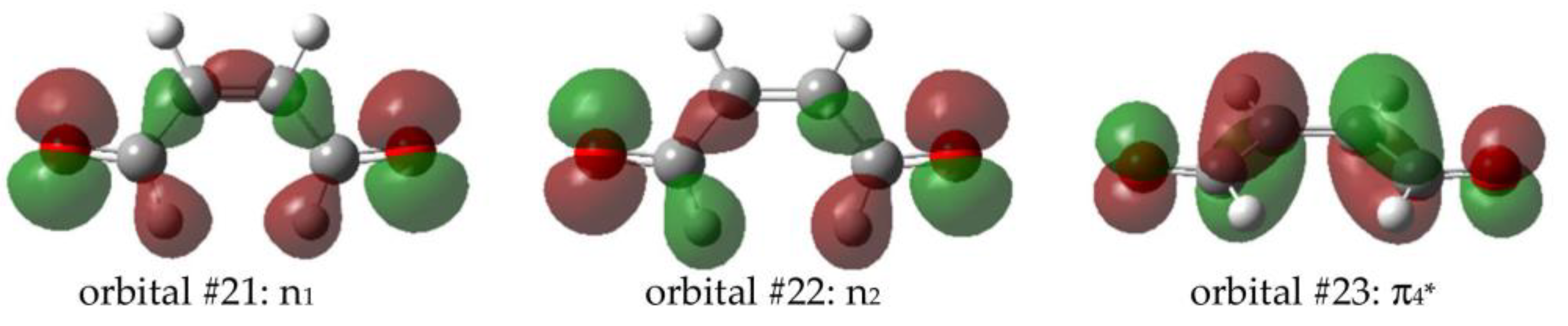
2.3.1. Intersystem Crossing
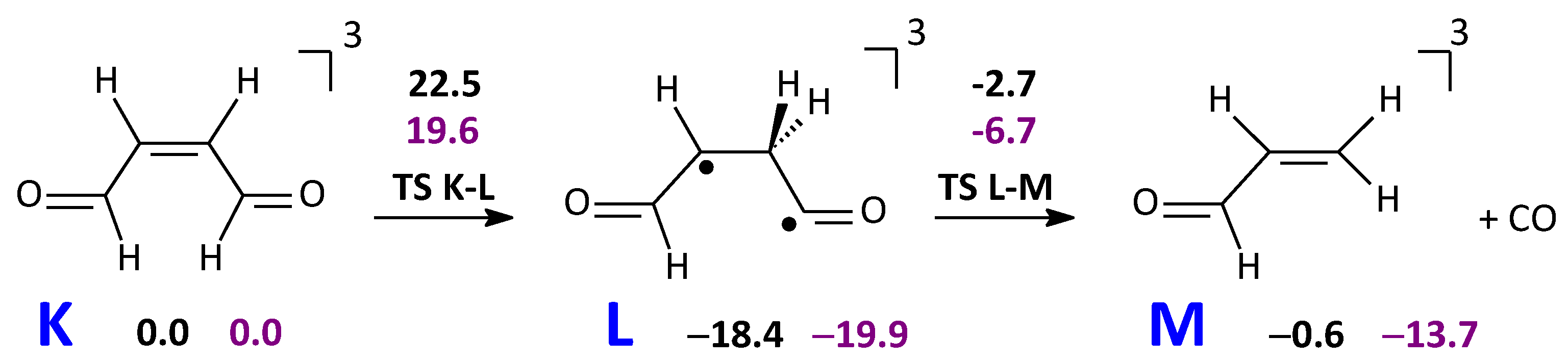
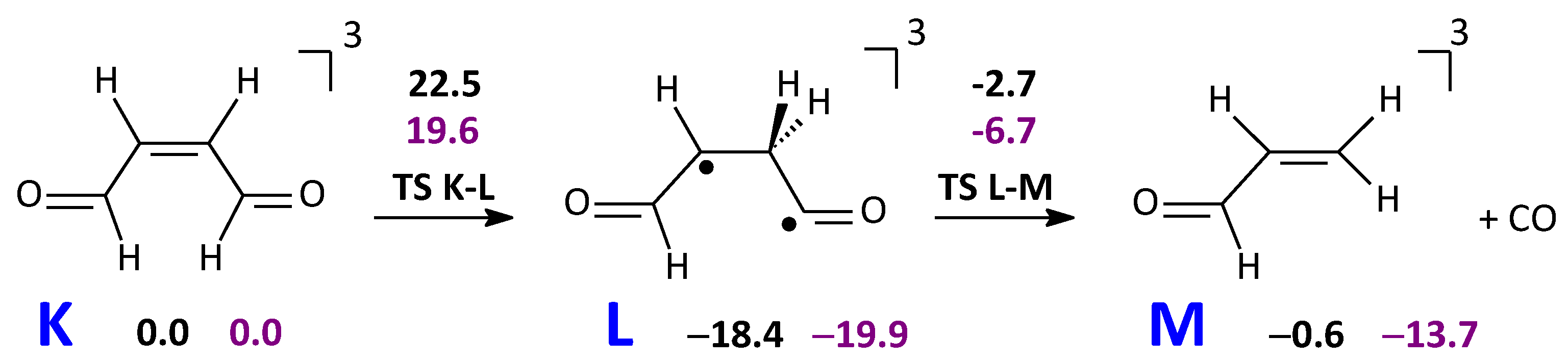
2.3.2. CO and Acrolein Formation in the T1 State
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Obermeyer, G.; Aschmann, S.M.; Atkinson, R.; Arey, J. Carbonyl atmospheric reaction products of aromatic hydrocarbons in ambient air. Atmos. Environ. 2009, 43, 3736–3744. [Google Scholar] [CrossRef]
- Gómez Alvarez, E.; Viidanoja, J.; Muñoz, A.; Wirtz, K.; Hjorth, J. Experimental Confirmation of the Dicarbonyl Route in the Photo-oxidation of Toluene and Benzene. Environ. Sci. Technol. 2007, 41, 8362–8369. [Google Scholar] [CrossRef]
- Yokelson, R.J.; Karl, T.; Artaxo, P.; Blake, D.R.; Christian, T.J.; Griffith, D.W.T.; Guenther, A.; Hao, W.M. The Tropical Forest and Fire Emissions Experiment: Overview and airborne fire emission factor measurements. Atmos. Chem. Phys. 2007, 7, 5175–5196. [Google Scholar] [CrossRef]
- Atkinson, R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. [Google Scholar] [CrossRef]
- Klotz, B.; Barnes, I.; Becker, K.-H. Kinetic study of the gas-phase photolysis and OH radical reaction of E,Z- and E,E-2,4-Hexadienedial. Int. J. Chem. Kinet. 1999, 31, 689–697. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, J.; Tang, Y.; Zhang, Y.; Wu, W.; Sun, J. Theoretical study on the atmospheric degradation mechanism and subsequent products of E,E-2,4-hexadienal with hydroxyl radical. Int. J. Quantum Chem. 2020, 121, e26563. [Google Scholar] [CrossRef]
- Martín, P.; Cabañas, B.; Colmenar, I.; Salgado, M.S.; Villanueva, F.; Tapia, A. Reactivity of E-butenedial with the major atmospheric oxidants. Atmos. Environ. 2013, 70, 351–360. [Google Scholar] [CrossRef]
- Newland, M.J.; Rea, G.J.; Thuner, L.P.; Henderson, A.P.; Golding, B.T.; Rickard, A.R.; Barnes, I.; Wenger, J. Photochemistry of 2-butenedial and 4-oxo-2-pentenal under atmospheric boundary layer conditions. Phys. Chem. Chem. Phys. 2019, 21, 1160–1171. [Google Scholar] [CrossRef]
- Marshall, P.; Papadimitriou, V.C.; Papanastasiou, D.K.; Roberts, J.M.; Burkholder, J.B. UV and infrared absorption spectra and 248 nm photolysis of maleic anhydride (C4H2O3). J. Photochem. Photobiol. A Chem. 2019, 382, 111953. [Google Scholar] [CrossRef]
- Back, R.A.; Parsons, J.M. The thermal and photochemical decomposition of maleic anhydride in the gas phase. Can. J. Chem. 1981, 59, 1342–1346. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, L. Photolysis of butenedial at 193, 248, 280, 308, 351, 400, and 450 nm. Chem. Phys. Lett. 2005, 409, 151–156. [Google Scholar] [CrossRef]
- Bierbach, A.; Barnes, I.; Becker, K.H.; Wiesen, E. Atmospheric Chemistry of Unsaturated Carbonyls: Butenedial, 4-Oxo-2-pentenal, 3-Hexene-2,5-dione, Maleic Anhydride, 3H-Furan-2-one, and 5-Methyl-3H-furan-2-one. Env. Sci. Technol. 1994, 28, 715–729. [Google Scholar] [CrossRef]
- Calvert, J.; Mellouki, A.; Orlando, J.; Pilling, M.; Wallington, T. Mechanisms of Atmospheric Oxidation of the Oxygenates; Oxford Academic: New York, NY, USA, 2011. [Google Scholar]
- Tadic, J.M.; Moortgat, G.K.; Bera, P.P.; Loewenstein, M.; Yates, E.L.; Lee, T.J. Photochemistry and photophysics of n-butanal, 3-methylbutanal, and 3,3-dimethylbutanal: Experimental and theoretical study. J. Phys. Chem. A 2012, 116, 5830–5839. [Google Scholar] [CrossRef]
- Shemesh, D.; Nizkorodov, S.A.; Gerber, R.B. Photochemical Reactions of Cyclohexanone: Mechanisms and Dynamics. J. Phys. Chem. A 2016, 120, 7112–7120. [Google Scholar] [CrossRef]
- Scaiano, J.C.; Encinas, M.V.; George, M.V. Photochemistry of o-phthalaldehyde. J. Chem. Soc. Perkin Trans. 2 1980, 7, 724–730. [Google Scholar] [CrossRef]
- Li, Q.; Migani, A.; Blancafort, L. Irreversible phototautomerization of o-phthalaldehyde through electronic relocation. Phys. Chem. Chem. Phys. 2012, 14, 6561–6568. [Google Scholar] [CrossRef]
- Fröbel, S.; Buschhaus, L.; Villnow, T.; Weingart, O.; Gilch, P. The photoformation of a phthalide: A ketene intermediate traced by FSRS. Phys. Chem. Chem. Phys. 2015, 17, 376–386. [Google Scholar] [CrossRef]
- Gebicki, J.; Kuberski, S.; Kamiński, R. Structure and photochemistry of matrix-isolated o-phthalaldehyde. J. Chem. Soc. Perkin Trans. 2 1990, 765–769. [Google Scholar] [CrossRef]
- He, H.-Y.; Fang, W.-H.; Phillips, D.L. Photochemistry of Butyrophenone: Combined Complete-Active-Space Self-Consistent Field and Density Functional Theory Study of Norrish Type I and II Reactions. J. Phys. Chem. A 2004, 108, 5386–5392. [Google Scholar] [CrossRef]
- Rowell, K.N.; Kable, S.H.; Jordan, M.J.T. An assessment of the tropospherically accessible photo-initiated ground state chemistry of organic carbonyls. Atmos. Chem. Phys. 2022, 22, 929–949. [Google Scholar] [CrossRef]
- Rowell, K.; Kable, S.; Jordan, M.J.T. Structural Causes of Singlet/triplet Preferences of Norrish Type II Reactions in Carbonyls. ChemRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Rowell, K.N.; Kable, S.H.; Jordan, M.J.T. Structural Effects on the Norrish Type I α-Bond Cleavage of Tropospherically Important Carbonyls. J. Phys. Chem. A 2019, 123, 10381–10396. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Xue, B.C.; Ellison, G.B.; Stanton, J.F. Theoretical Study of Reaction of Ketene with Water in the Gas Phase: Formation of Acetic Acid? J. Phys. Chem. A 2013, 117, 10997–11005. [Google Scholar] [CrossRef]
- Kahan, T.F.; Ormond, T.K.; Ellison, G.B.; Vaida, V. Acetic acid formation via the hydration of gas-phase ketene under ambient conditions. Chem. Phys. Lett. 2013, 565, 1–4. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Raspoet, G. The hydration mechanism of ketene: 15 years later. Can. J. Chem. 1999, 77, 817–829. [Google Scholar] [CrossRef]
- Tidwell, T.T. Ketenes. J. Nat. Prod. 1996, 59, 822. [Google Scholar] [CrossRef]
- Newland, M.J.; Nelson, B.S.; Muñoz, A.; Ródenas, M.; Vera, T.; Tárrega, J.; Rickard, A.R. Trends in stabilisation of Criegee intermediates from alkene ozonolysis. Phys. Chem. Chem. Phys. 2020, 22, 13698–13706. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Maranzana, A.; Tonachini, G. Mechanism of the Photochemical Isomerization and Oxidation of 2-Butenedial: A Theoretical Study. Molecules 2023, 28, 4994–5005. [Google Scholar] [CrossRef] [PubMed]
- Houston, P.L.; Kable, S.H. Photodissociation of acetaldehyde as a second example of the roaming mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 16079–16082. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Hoffmann, N. Studies in organic and physical photochemistry—An interdisciplinary approach. Org. Biomol. Chem. 2016, 14, 7392–7442. [Google Scholar] [CrossRef] [PubMed]
- Vörös, D.; Mai, S. Role of Ultrafast Internal Conversion and Intersystem Crossing in the Nonadiabatic Relaxation Dynamics of ortho-Nitrobenzaldehyde. J. Phys. Chem. A 2023, 127, 5872–5886. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Handy, N.C.; Cohen, A.J. Left-right correlation energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Hlavacek, N.C.; McAnally, M.O.; Drucker, S. Lowest triplet (n, π*) electronic state of acrolein: Determination of structural parameters by cavity ringdown spectroscopy and quantum-chemical methods. J. Chem. Phys. 2013, 138, 064303. [Google Scholar] [CrossRef] [PubMed]
- Devaquet, A.; Salem, L. Potential Energy—Sheets for the nπ* and ππ* Triplet States of Acrolein. Can. J. Chem. 1971, 49, 977–979. [Google Scholar] [CrossRef]
- Bokareva, O.S.; Bataev, V.A.; Pupyshev, V.I.; Godunov, I.A. Structure and dynamics of acrolein in lowest excited 1,3(n,π*) electronic states: The quantum-chemical study. Int. J. Quantum Chem. 2008, 108, 2719–2731. [Google Scholar] [CrossRef]
- Cao, J.; Xie, Z.-Z. Internal conversion and intersystem crossing in α,β-enones: A combination of electronic structure calculations and dynamics simulations. Phys. Chem. Chem. Phys. 2016, 18, 6931–6945. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, K. Theoretical Considerations of Lower Excited States of trans-Polyenecarbaldehydes. II. Radiative and Nonradiative Properties. Bull. Chem. Soc. Jpn. 1975, 48, 779–782. [Google Scholar] [CrossRef]
- Liu, Z.; Nguyen, V.S.; Harvey, J.; Müller, J.-F.; Peeters, J. Theoretically derived mechanisms of HPALD photolysis in isoprene oxidation. Phys. Chem. Chem. Phys. 2017, 19, 9096–9106. [Google Scholar] [CrossRef] [PubMed]
- Laimgruber, S.; Schmierer, T.; Gilch, P.; Kiewisch, K.; Neugebauer, J. The ketene intermediate in the photochemistry of ortho-nitrobenzaldehyde. Phys. Chem. Chem. Phys. 2008, 10, 3872–3882. [Google Scholar] [CrossRef]
- Hansen, D.A.; Lee, E.K.C. Radiative and nonradiative transitions in the first excited singlet state of symmetrical methyl-substituted acetones. J. Chem. Phys. 1975, 62, 183–189. [Google Scholar] [CrossRef]
- Miller, R.G.; Lee, E.K.C. Single vibronic level photochemistry of formaldehydes in the 1A2 state: Radiative and nonradiative processes in H2CO, HDCO, and D2CO. J. Chem. Phys. 1978, 68, 4448–4464. [Google Scholar] [CrossRef]
- Hansen, D.A.; Lee, E.K.C. Radiative and nonradiative transitions in the first excited singlet state of simple linear aldehydes. J. Chem. Phys. 1975, 63, 3272–3277. [Google Scholar] [CrossRef]
- Shinohara, H.; Nishi, N. Laser photofragmentation dynamics of an acrolein supersonic molecular beam at 193 nm. J. Chem. Phys. 1982, 77, 234–245. [Google Scholar] [CrossRef]
- Paulisse, K.W.; Friday, T.O.; Graske, M.L.; Polik, W.F. Vibronic spectroscopy and lifetime of S1 acrolein. J. Chem. Phys. 2000, 113, 184–191. [Google Scholar] [CrossRef]
- Liu, X.; Jeffries, H.E.; Sexton, K.G. Atmospheric Photochemical Degradation of 1,4-Unsaturated Dicarbonyls. Environ. Sci. Technol. 1999, 33, 4212–4220. [Google Scholar] [CrossRef]
- Schuster, D.I.; Dunn, D.A.; Heibel, G.E.; Brown, P.B.; Rao, J.M.; Woning, J.; Bonneau, R. Enone photochemistry. Dynamic properties of triplet excited states of cyclic conjugated enones as revealed by transient absorption spectroscopy. J. Am. Chem. Soc. 2002, 113, 6245–6255. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- McQuarrie, D.A.; Simon, J.D. Molecular Thermodynamics; University Science Books: Sausalito, CA, USA, 1999. [Google Scholar]
- Thermochemistry in Gaussian. Available online: https://gaussian.com/thermo/ (accessed on 21 January 2024).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Roy Dennington, T.K.; Millam, J. GaussView, Version 6.1.1; Semichem Inc.: Shawnee, KS, USA, 2019. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104. [Google Scholar] [CrossRef]
- Risthaus, T.; Hansen, A.; Grimme, S. Excited states using the simplified Tamm–Dancoff-Approach for range-separated hybrid density functionals: Development and application. Phys. Chem. Chem. Phys. 2014, 16, 14408–14419. [Google Scholar] [CrossRef]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- Orca Manual. pp. 405, 1019. Available online: https://orcaforum.kofo.mpg.de/ (accessed on 21 January 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basis Set | cc-pVTZ | aug-cc-pVTZ | ||
|---|---|---|---|---|
| Functional | T1 | T2 | T1 | T2 |
| O3LYP | −8.78 | −0.37 | −8.67 | −0.32 |
| OLYP | −8.18 | −1.95 | −8.05 | −1.90 |
| PBE0 | −10.72 | −2.85 | −10.60 | −2.72 |
| B3LYP | −9.90 | −2.99 | −9.77 | −2.86 |
| Functional 1 | S1/T1 ISC Rate | S1/T2 ISC Rate | T2/T1 MECP Barrier |
|---|---|---|---|
| O3LYP | 8.7 × 107 | 1.0 × 109 | 0.85 |
| OLYP | 1.6 × 106 | 1.1 × 1010 | 1.01 |
| PBE0 | 5.9 × 106 | 1.2 × 1010 | 0.99 |
| B3LYP | 6.9 × 106 | 1.6 × 1010 | 0.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maranzana, A.; Tonachini, G. Tropospheric Photochemistry of 2-Butenedial: Role of the Triplet States, CO and Acrolein Formation, and the Experimentally Unidentified Carbonyl Compound—Theoretical Study. Molecules 2024, 29, 575. https://doi.org/10.3390/molecules29030575
Maranzana A, Tonachini G. Tropospheric Photochemistry of 2-Butenedial: Role of the Triplet States, CO and Acrolein Formation, and the Experimentally Unidentified Carbonyl Compound—Theoretical Study. Molecules. 2024; 29(3):575. https://doi.org/10.3390/molecules29030575
Chicago/Turabian StyleMaranzana, Andrea, and Glauco Tonachini. 2024. "Tropospheric Photochemistry of 2-Butenedial: Role of the Triplet States, CO and Acrolein Formation, and the Experimentally Unidentified Carbonyl Compound—Theoretical Study" Molecules 29, no. 3: 575. https://doi.org/10.3390/molecules29030575
APA StyleMaranzana, A., & Tonachini, G. (2024). Tropospheric Photochemistry of 2-Butenedial: Role of the Triplet States, CO and Acrolein Formation, and the Experimentally Unidentified Carbonyl Compound—Theoretical Study. Molecules, 29(3), 575. https://doi.org/10.3390/molecules29030575