


Screening and Application of DNA Aptamers for Heparin-Binding Protein

 , , , ,
, , , ,  and
and
Abstract
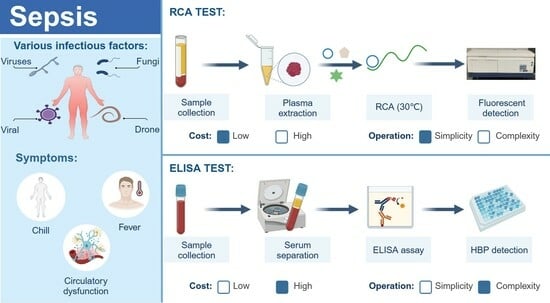
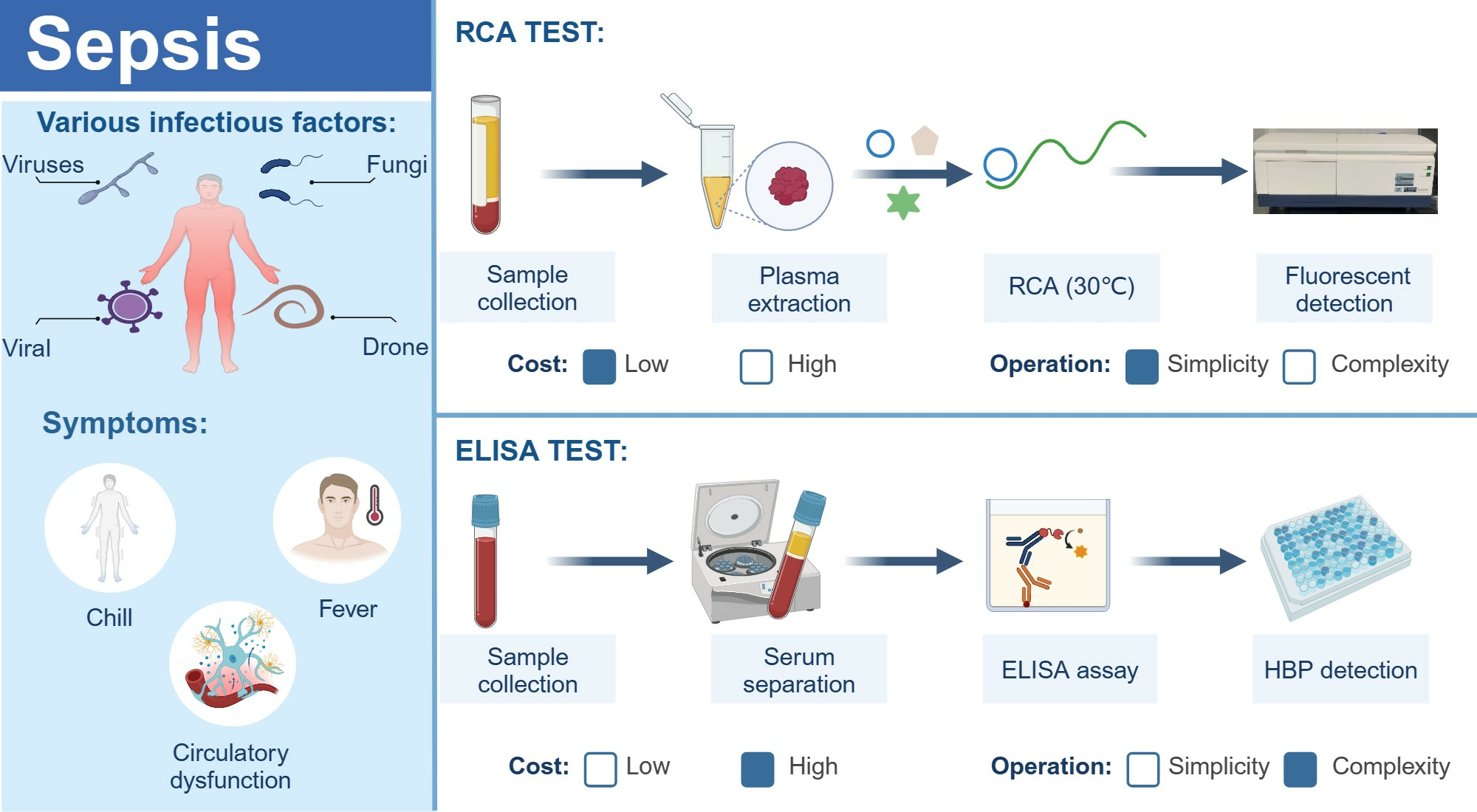
1. Introduction
2. Results and Discussion
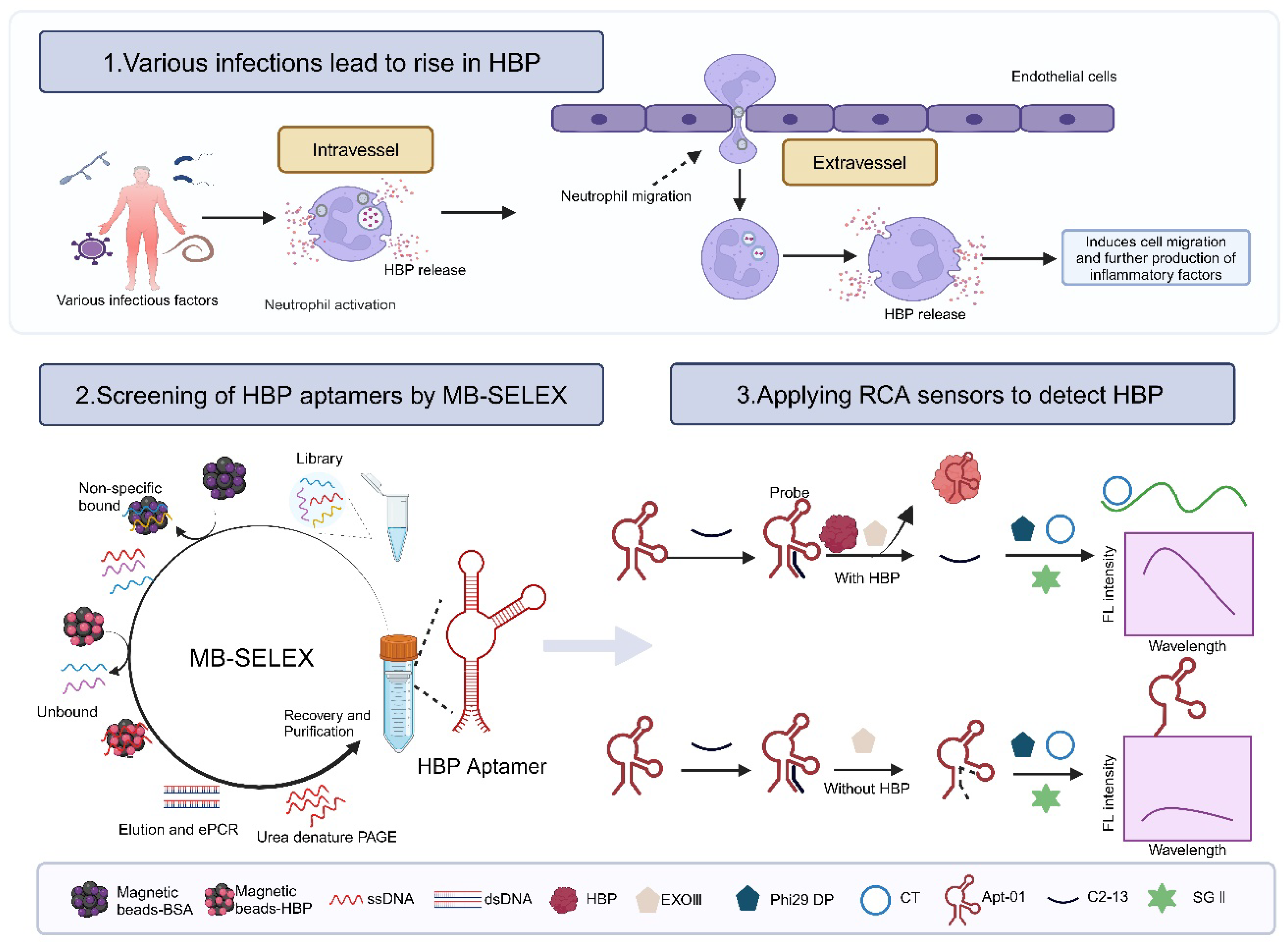
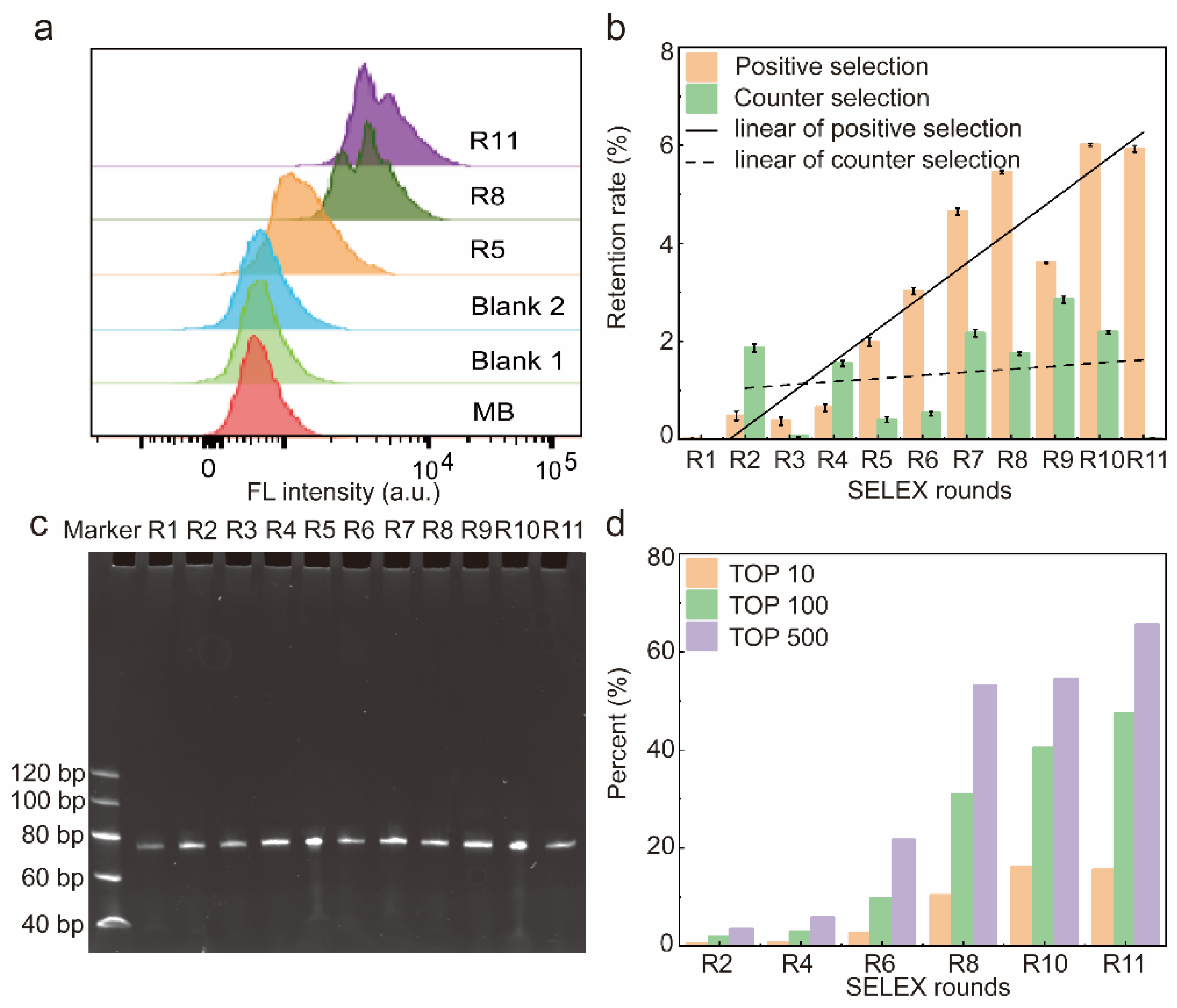
2.1. Selection of Aptamers
2.2. High-Throughput Sequencing
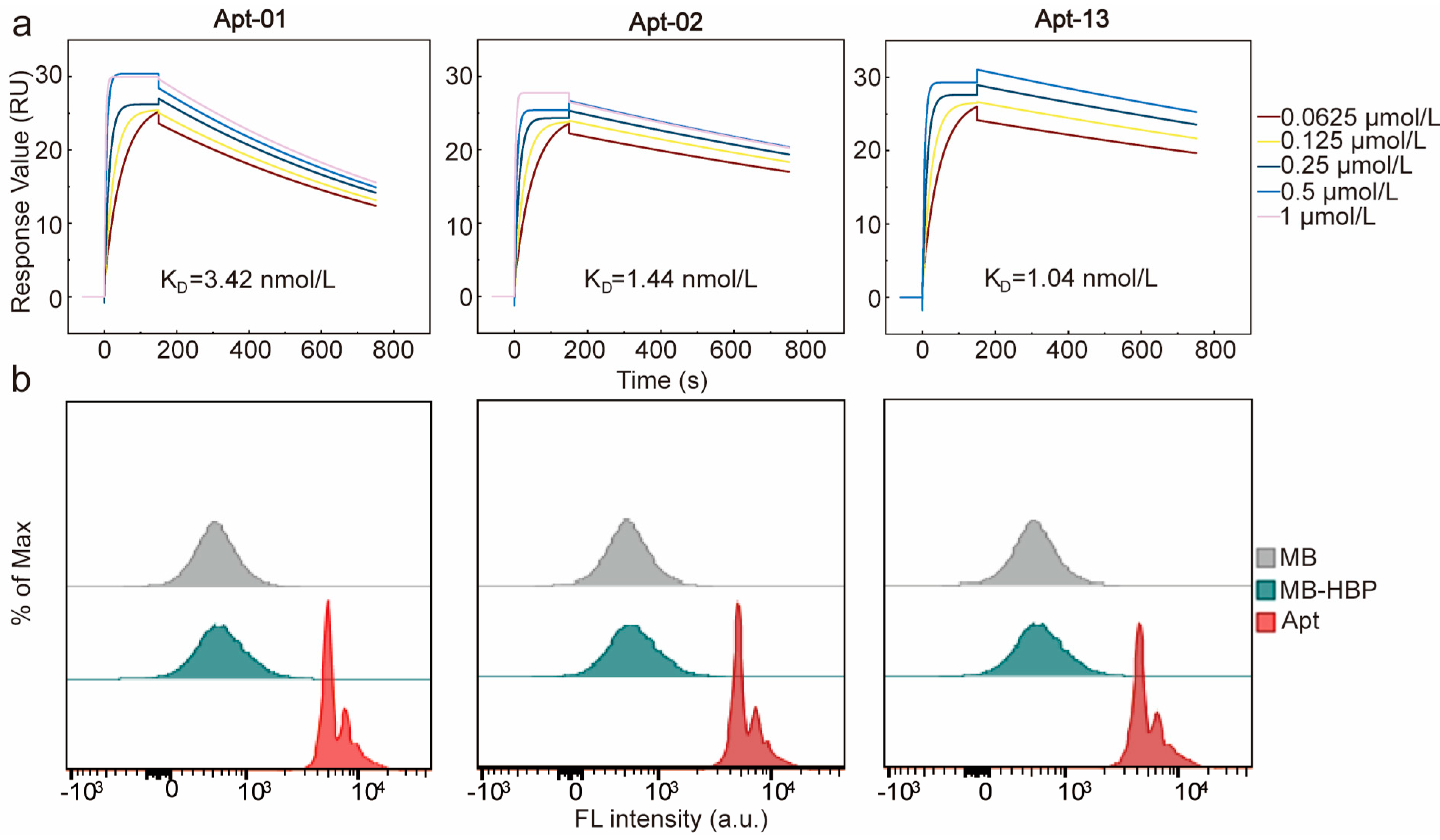
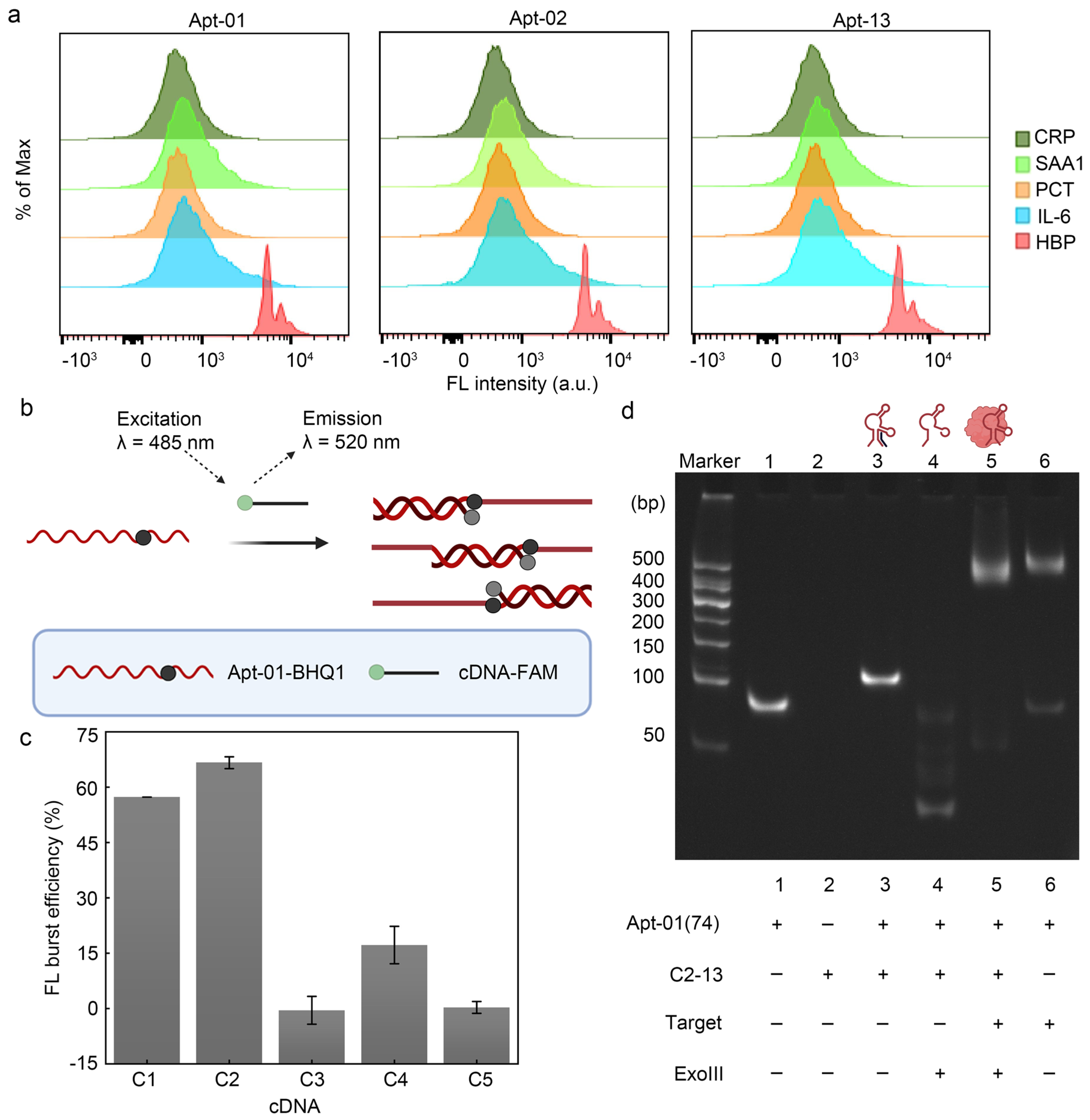
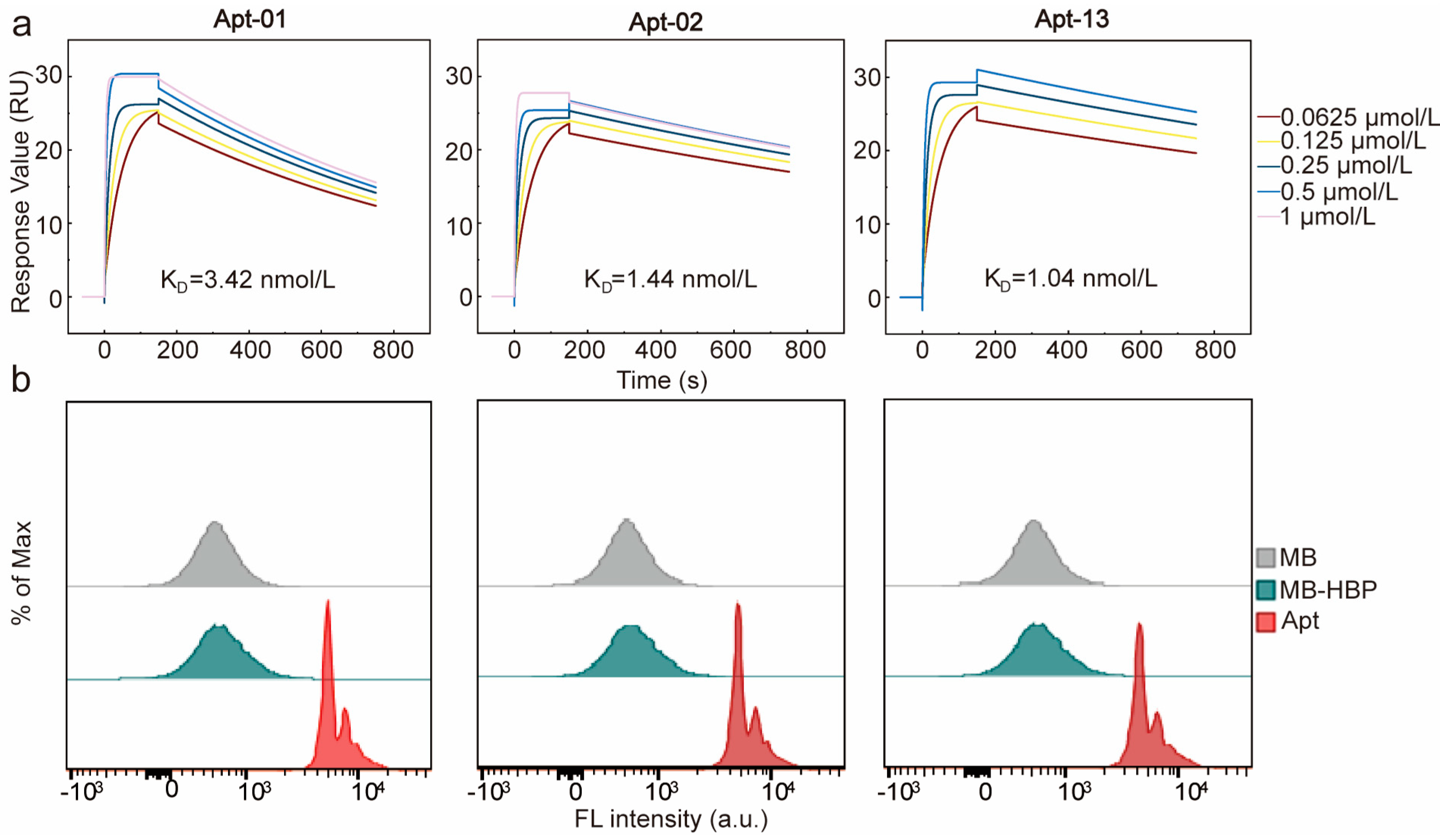
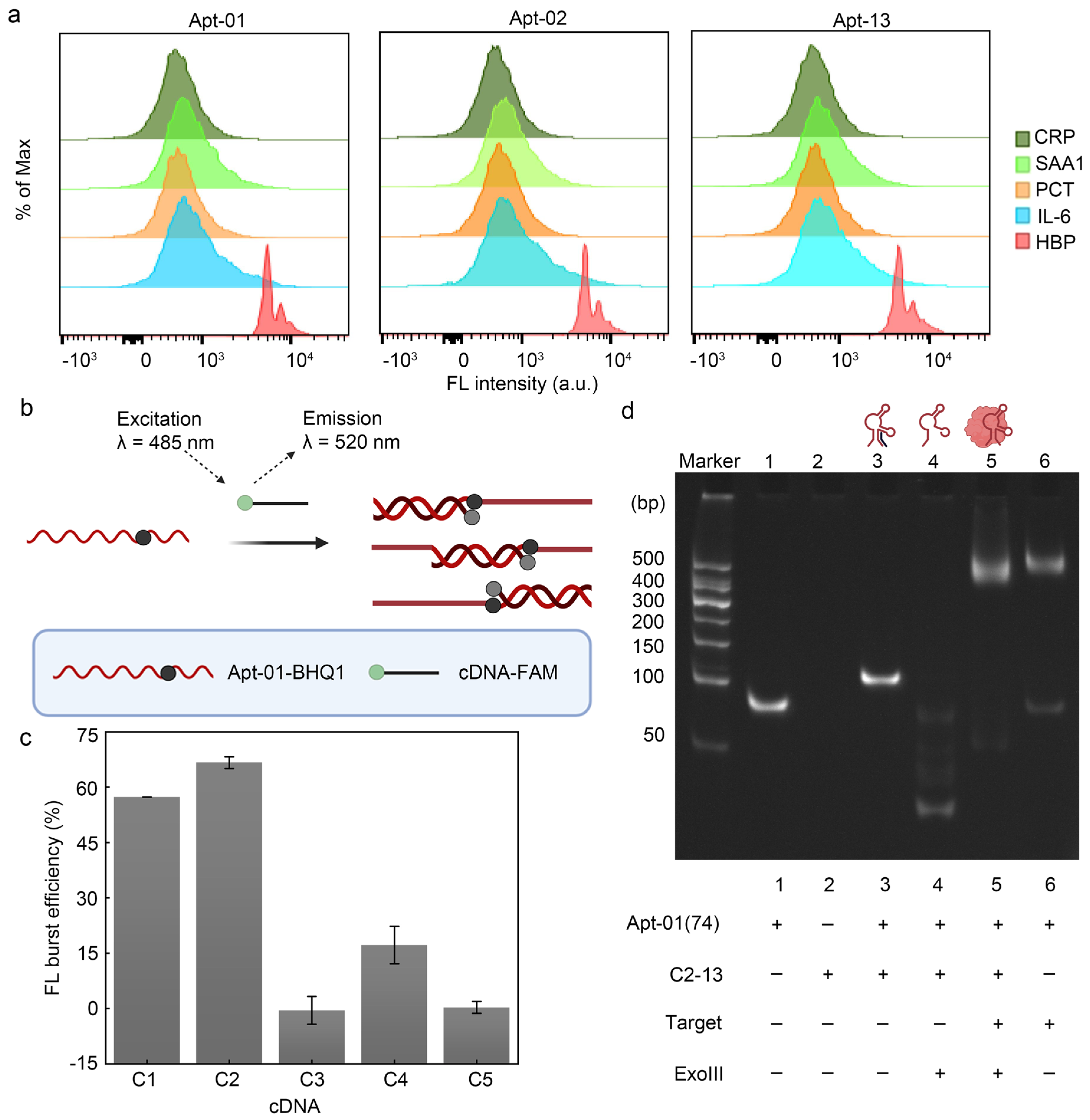
2.3. Affinity and Specificity of Selected Aptamers
2.4. Principle and Feasibility of Fluorescence Biosensor for HBP Based on the Selected Aptamer
2.5. Characterization of Polyacrylamide Gel Electrophoresis
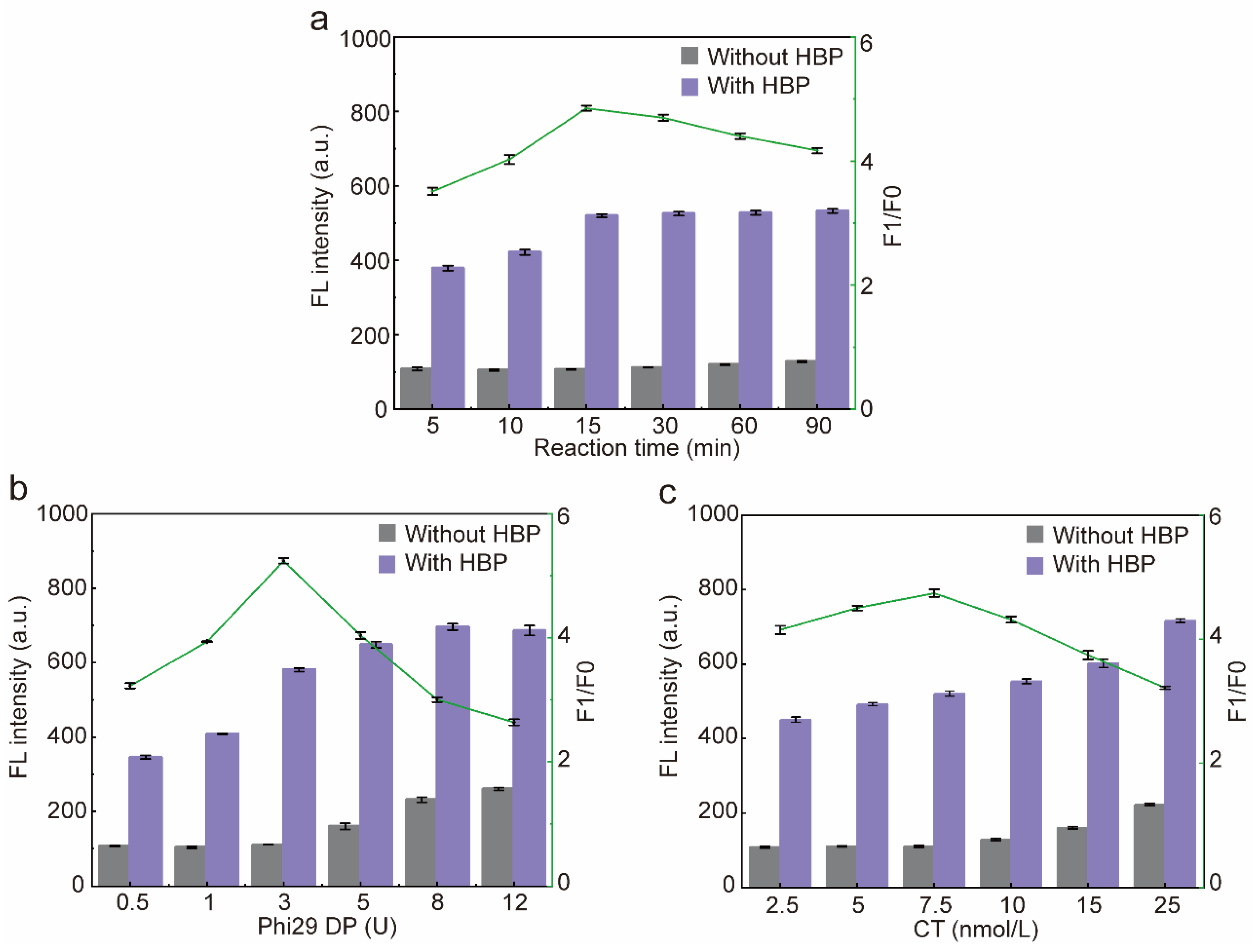
2.6. Optimization of the Reaction Conditions
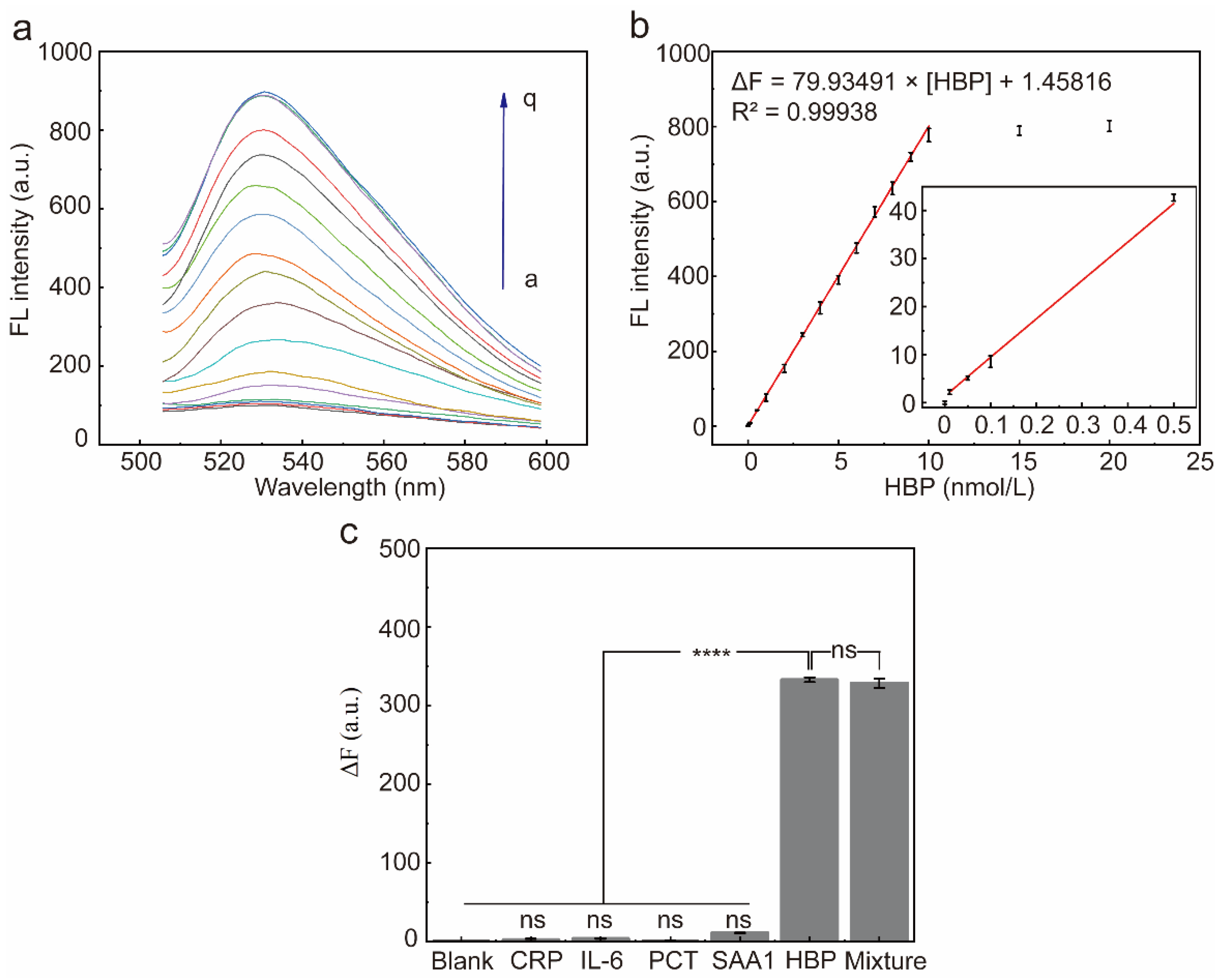
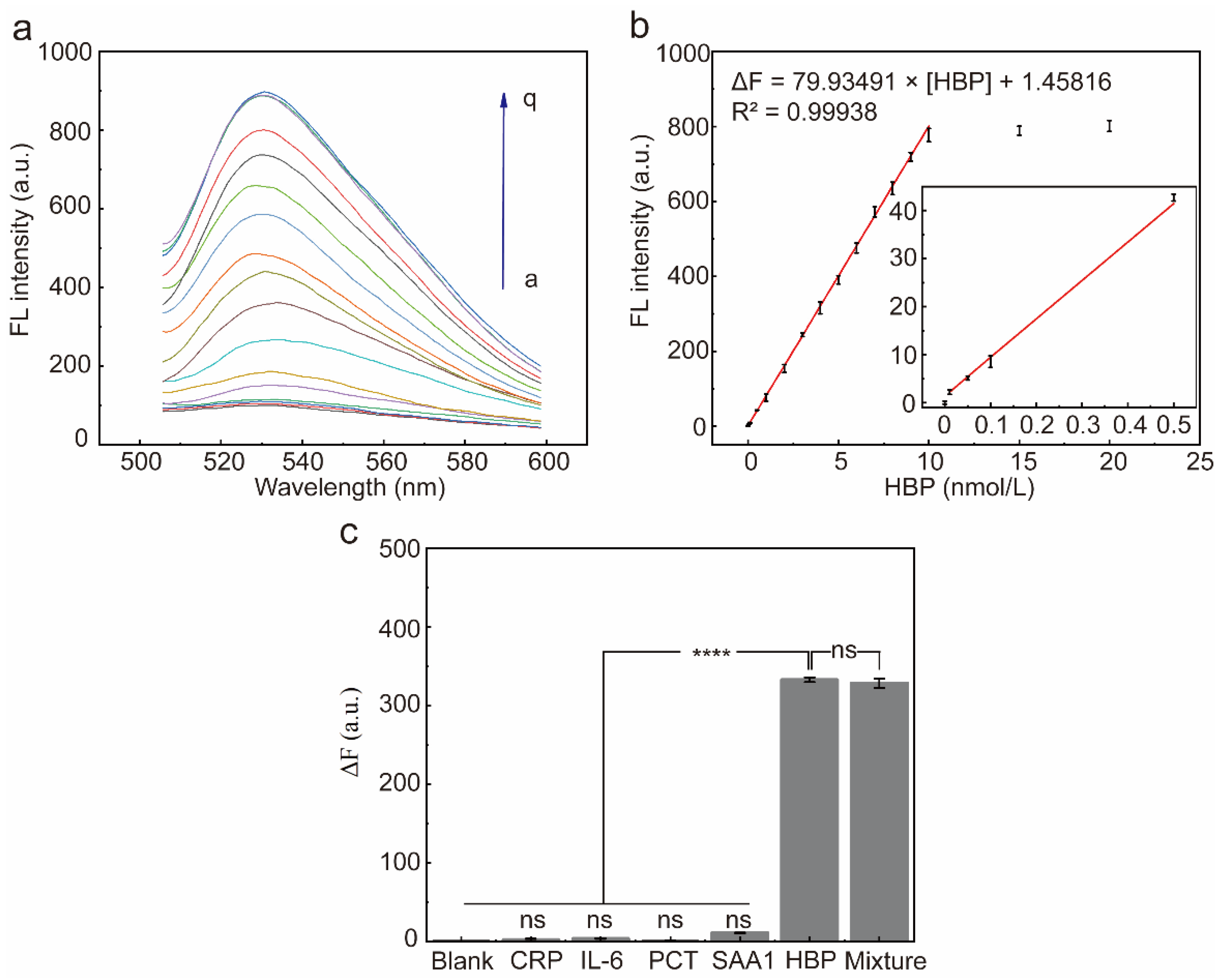
2.7. Performance of the Selected Aptamer for HBP Determination
2.8. Accuracy of the Proposed Fluorescent Sensor in Real Sample Detection
3. Discussion
4. Experimental Section
4.1. Materials and Reagents
4.2. Random Library and Primers
4.3. MB-SELEX Procedure
4.3.1. Preparation of MB-BSA and MB-HBP
4.3.2. MB-SELEX
4.3.3. Flow Cytometry Analysis
4.3.4. Quantitative PCR for Library Enrichment
4.3.5. High-Throughput Sequencing and Sequence Analysis
4.3.6. Affinity Assays by SPR
4.3.7. Specificity of the Aptamers
4.3.8. Circular Dichroism Spectrometer
4.4. Principle and Feasibility of Fluorescence Biosensor for HBP Based on the Selected Aptamer
4.5. Characterization of Polyacrylamide Gel Electrophoresis
4.6. RCA-Based HBP Fluorescent Sensor
4.7. Selectivity of the Sensor
4.8. Analytical Application in Real Samples
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, L.; Dou, J.; Lin, Q.; He, L.; Zeng, Q.; Song, J. Tissue-Type Plasminogen Activator-Inhibitor Complex as an Early Predictor of Septic Shock: A Retrospective, Single-Center Study. Dis. Markers 2022, 2022, 9364037. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, H.; Kang, Y.; Zhou, L.; Liu, Z.; Qin, B.; Ma, X.; Cao, X.; Chen, D.; Lu, W.; et al. The Epidemiology of Sepsis in Chinese ICUs: A National Cross-Sectional Survey. Crit. Care Med. 2020, 3, e209–e218. [Google Scholar] [CrossRef]
- Lambden, S.; Laterre, P.F.; Levy, M.M.; Francois, B. The SOFA Score-Development, Utility and Challenges of Accurate Assessment in Clinical Trials. Crit. Care 2019, 23, 374. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, M.; Zhu, B.; Zhu, Y.; Xi, X. Prediction of Median Survival Time in Sepsis Patients by the SOFA Score Combined with Different Predictors. Burn. Trauma 2020, 8, tkz006. [Google Scholar] [CrossRef] [PubMed]
- Li, A.T.; Moussa, A.; Gus, E.; Paul, E.; Yii, E.; Romero, L.; Lin, Z.C.; Padiglione, A.; Lo, C.H.; Cleland, H.; et al. Biomarkers for the Early Diagnosis of Sepsis in Burns: Systematic Review and Meta-Analysis. Ann. Surg. 2022, 4, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Tocu, G.; Mihailov, R.; Serban, C.; Stefanescu, B.I.; Tutunaru, D.; Firescu, D. The Contribution of Procalcitonin, C-Reactive Protein and Interleukin-6 in the Diagnosis and Prognosis of Surgical Sepsis: An Observational and Statistical Study. J. Multidiscip. Healthc. 2023, 16, 2351–2359. [Google Scholar] [CrossRef]
- Ruan, L.; Chen, G.Y.; Liu, Z.; Zhao, Y.; Xu, G.Y.; Li, S.F.; Li, C.N.; Chen, L.S.; Tao, Z. The Combination of Procalcitonin and C-reactive Protein or Presepsin Alone Improves the Accuracy of Diagnosis of Neonatal Sepsis: A Meta-Analysis and Systematic Review. Crit. Care 2018, 22, 316. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Linder, A. Heparin-Binding Protein: A Key Player in the Pathophysiology of Organ Dysfunction in Sepsis. J. Intern. Med. 2017, 6, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Russell, J.A.; Bentzer, P.; Parsons, D.; Secchia, S.; Mörgelin, M.; Walley, K.R.; Boyd, J.H.; Linder, A. Heparin-Binding Protein (HBP): A Causative Marker and Potential Target for Heparin Treatment of Human Sepsis-Induced Acute Kidney Injury. Shock 2017, 3, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, J.; Mu, G.; Lu, Z.; Li, W.; Deng, Y. Heparin-Binding Protein (HBP) Worsens the Severity of Pancreatic Necrosis Via Up-Regulated M1 Macrophages Activation in Acute Pancreatitis Mouse Models. Bioengineered 2021, 2, 11978–11986. [Google Scholar] [CrossRef]
- Katsaros, K.; Renieris, G.; Safarika, A.; Adami, E.M.; Gkavogianni, T.; Giannikopoulos, G.; Solomonidi, N.; Halvatzis, S.; Koutelidakis, I.M.; Tsokos, N.; et al. Heparin Binding Protein for the Early Diagnosis and Prognosis of Sepsis in the Emergency Department: The Prompt Multicenter Study. Shock 2022, 4, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, D.; Lou, J.; Lin, J.; Huang, C.; Zou, Y.; Wong, C.; Wu, H.; Yan, G.; Liu, J.; et al. Heparin-Binding Protein as a Biomarker of Severe Sepsis in the Pediatric Intensive Care Unit: A Multicenter, Prospective Study. Clin. Chim. Acta 2022, 539, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Yo, C.H.; Hsu, W.T.; Qian, F.; Wu, B.S.; Dou, Q.L.; Lee, C.C. Accuracy of Heparin-Binding Protein in Diagnosing Sepsis: A Systematic Review and Meta-Analysis. Crit. Care Med. 2020, 1, e80–e90. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Liu, Y.; Zhu, F.; Ma, J.; Gao, J. Time-Resolved Fluorescent Immunoassay-Based Combined Detection of Procalcitonin, C-reactive Protein, Heparin Binding Protein, and Serum Amyloid A1 to Improve the Diagnostic Accuracy of Early Infection. J. Clin. Lab. Anal. 2019, 33, e22694. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Hur, M.; Kim, H.; Moon, H.W.; Yun, Y.M. Reference Interval for the Axis-Shield Clinical Chemistry Heparin-Binding Protein Assay. Diagnostics 2022, 8, 1930. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, M.; Thelaus, L.; Riesbeck, K.; Qvarfordt, I.; Smith, M.E.; Lindén, A.; Linder, A. Heparin-Binding Protein in Lower Airway Samples as a Biomarker for Pneumonia. Respir. Res. 2021, 22, 174. [Google Scholar] [CrossRef] [PubMed]
- Mellhammar, L.; Thelaus, L.; Elén, S.; Fisher, J.; Linder, A. Heparin Binding Protein in Severe COVID-19—A Prospective Observational Cohort Study. PLoS ONE 2021, 4, e0249570. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tam, W.W.; Yu, Y.; Zhuo, Z.; Xue, Z.; Tsang, C.; Qiao, X.; Wang, X.; Wang, W.; Li, Y.; et al. The Application of Aptamer in Biomarker Discovery. Biomark. Res. 2023, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Khoshbin, Z.; Verdian, A.; Housaindokht, M.R.; Izadyar, M.; Rouhbakhsh, Z. Aptasensors as the Future of Antibiotics Test Kits-A Case Study of the Aptamer Application in the Chloramphenicol Detection. Biosens. Bioelectron. 2018, 122, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, Y.; Xu, X.; Liu, Y.; Lin, B.; Zhang, M.; Zhang, J.; Wan, S.; Yang, C.; Tan, W. Aptamer-Based Detection of Circulating Targets for Precision Medicine. Chem. Rev. 2021, 19, 12035–12105. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Z.; Yin, Y.; Jia, F.; Wu, Q.; Tian, P.; Wang, D. Development and Evaluation of a Novel in Situ Target-Capture Approach for Aptamer Selection of Human Noroviruses. Talanta 2019, 193, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Yue, F.; Liu, M.; Huang, J.; Yang, F.; Liu, J.; Li, J.; Li, F.; Sun, X.; Guo, Y.; et al. Non-Immobilized GO-SELEX of Aptamers for Label-Free Detection of Thiamethoxam in Vegetables. Anal. Chim. Acta 2022, 1202, 339677. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, J.; Zhu, L.; Li, C.; Chang, Q.; Xu, W. Advanced Screening and Tailoring Strategies of Pesticide Aptamer for Constructing Biosensor. Crit. Rev. Food Sci. Nutr. 2023, 63, 10974–10994. [Google Scholar] [CrossRef]
- Zhu, C.; Li, L.; Yang, G.; Qu, F. Investigating the Influences of Random-Region Length on Aptamer Selection Efficiency Based On Capillary Electrophoresis-SELEX and High-Throughput Sequencing. Anal. Chem. 2021, 51, 17030–17035. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Agnello, L.; Fedele, M.; Camorani, S.; Cerchia, L. Profiling Cancer Cells by Cell-SELEX: Use of Aptamers for Discovery of Actionable Biomarkers and Therapeutic Applications Thereof. Pharmaceutics 2021, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wan, J.; Wen, X.; Guo, Q.; Jiang, H.; Wang, J.; Ren, Y.; Wang, K. Identification of a New DNA Aptamer by Tissue-SELEX for Cancer Recognition and Imaging. Anal. Chem. 2021, 19, 7369–7377. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Duan, N.; Khan, I.M.; Dong, X.; Zhang, Y.; Wu, S.; Wang, Z. Strategies to Manipulate the Performance of Aptamers in SELEX, post-SELEX and Microenvironment. Biotechnol. Adv. 2022, 55, 107902. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Kalyani, N.; Anand, A.; Khan, E.; Das, S.; Bansal, V.; Kumar, A.; Sharma, T.K. GOLD SELEX: A Novel SELEX Approach for the Development of High-Affinity Aptamers Against Small Molecules without Residual Activity. Microchim. Acta 2020, 187, 618. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Cai, R.; Yue, H.; Tian, Y.; Zhou, N. Screening and Application of a Truncated Aptamer for High-Sensitive Fluorescent Detection of Metronidazole. Anal. Chim. Acta 2020, 1128, 203–210. [Google Scholar] [CrossRef]
- Shi, M.; Liu, R.; Zhang, F.; Chitrakar, B.; Wang, X. Screening of Single-Stranded DNA Aptamer Specific for Florfenicol and Application in Detection of Food Safety. Biosensors 2022, 9, 701. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovska, I.; Renciuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.C.; Patel, K.; Vunikili, R.D.; Johnson, K.W.; Abdu, F.; Belman, S.K.; Glicksberg, B.S.; Tandale, P.; Fontanez, R.; Mathew, O.K.; et al. Sepsis in the era of data-driven medicine: Personalizing risks, diagnoses, treatments and prognoses. Brief. Bioinform. 2020, 4, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Brun-Buisson, C.; Guillemot, D.; Watier, L. Care pathways of sepsis survivors: Sequelae, mortality and use of healthcare services in France, 2015–2018. Crit. Care 2023, 1, 438. [Google Scholar] [CrossRef] [PubMed]
- Mellhammar, L.; Wollter, E.; Dahlberg, J.; Donovan, B.; Olséen, C.J.; Wiking, P.O.; Rose, N.; Schwarzkopf, D.; Friedrich, M.; Fleischmann-Struzek, C.; et al. Estimating Sepsis Incidence Using Administrative Data and Clinical Medical Record Review. JAMA Netw. Open 2023, 8, e2331168. [Google Scholar] [CrossRef]
- Berry, M.; Patel, B.V.; Brett, S.J. New Consensus Definitions for Sepsis and Septic Shock: Implications for Treatment Strategies and Drug Development? Drugs 2017, 4, 353–361. [Google Scholar] [CrossRef]
- Ren, D.; Wu, D.; Liu, F.; Jiao, S.; Wu, Y. Diagnostic value of heparin-binding protein in the cerebrospinal fluid for purulent meningitis in children. Braz. J. Med. Biol. Res. 2021, 11, e11295. [Google Scholar] [CrossRef] [PubMed]
- Linder, A.; Arnold, R.; Boyd, J.H.; Zindovic, M.; Zindovic, I.; Lange, A.; Paulsson, M.; Nyberg, P.; Russell, J.A.; Pritchard, D.; et al. Heparin-Binding Protein Measurement Improves the Prediction of Severe Infection With Organ Dysfunction in the Emergency Department. Crit. Care Med. 2015, 11, 2378–2386. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 11, 1181–1247. [Google Scholar] [CrossRef] [PubMed]
- Linder, A.; Åkesson, P.; Inghammar, M.; Treutiger, C.J.; Linnér, A.; Sundén-Cullberg, J. Elevated plasma levels of heparin-binding protein in intensive care unit patients with severe sepsis and septic shock. Crit. Care 2012, 3, R90. [Google Scholar] [CrossRef] [PubMed]
- Tverring, J.; Nielsen, N.; Dankiewicz, J.; Linder, A.; Kahn, F.; Åkesson, P. Repeated measures of Heparin-binding protein (HBP) and procalcitonin during septic shock: Biomarker kinetics and association with cardiovascular organ dysfunction. Intensive Care 2020, 1, 51. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, Y.; Wu, Y.; Huang, H.; Sun, C.; Xu, S.; Li, H.; Zhang, X.; Zhao, S.; Huang, L. Heparin-Binding Protein: A Prognostic Biomarker Associated with Severe or Complicated Community-Acquired Pneumonia in Children. J. Inflamm. Res. 2023, 16, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Gu, H.; Duan, N.; Wu, S.; Ma, X.; Xia, Y.; Tao, Z.; Wang, Z. An enhanced chemiluminescence resonance energy transfer aptasensor based on rolling circle amplification and WS2 nanosheet for Staphylococcus aureus detection. Anal. Chim. Acta 2017, 959, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, S.; Ouyang, C.; Wang, Z.; Wu, D.; Liu, Y.; Jiang, Y.; Wu, Z.S. DNA nanostructures from palindromic rolling circle amplification for the fluorescent detection of cancer-related microRNAs. Talanta 2019, 192, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, J.; Zheng, L.; Qian, H.; Xue, F.; Wu, Y.; Pan, D.; Adeloju, S.B.; Chen, W. Rolling chain amplification based signal-enhanced electrochemical aptasensor for ultrasensitive detection of ochratoxin A. Anal. Chem. 2013, 22, 10842–10849. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.Y.; Du, Y.C.; Zhang, L.N.; Liu, K.K.; Zhu, L.N.; Kong, D.M. Sensitive fluorescent detection of DNA methyltransferase using nicking endonuclease-mediated multiple primers-like rolling circle amplification. Biosens. Bioelectron. 2017, 91, 417–423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Detected HBP (nmol/L) | Clinical Diagnosis |
|---|---|---|
| blank | 0 | / |
| 1 | 0.24 | Healthy |
| 2 | 0.22 | Healthy |
| 3 | 2.68 | Sepsis |
| 4 | 6.1 | Sepsis |
| 5 | 2.88 | Sepsis |
| 6 | 6.32 | Sepsis |
| 7 | 3.57 | Sepsis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Cao, Y.; Huang, X.; Qiu, S.; Xiang, X.; Niu, H.; Chen, L.; Wang, S.; Lin, Z.; Zhang, S. Screening and Application of DNA Aptamers for Heparin-Binding Protein. Molecules 2024, 29, 1717. https://doi.org/10.3390/molecules29081717
Zhou X, Cao Y, Huang X, Qiu S, Xiang X, Niu H, Chen L, Wang S, Lin Z, Zhang S. Screening and Application of DNA Aptamers for Heparin-Binding Protein. Molecules. 2024; 29(8):1717. https://doi.org/10.3390/molecules29081717
Chicago/Turabian StyleZhou, Xi, Yingying Cao, Xiaocui Huang, Shuqian Qiu, Xinran Xiang, Huimin Niu, Li Chen, Shuiliang Wang, Zhenyu Lin, and Shenghang Zhang. 2024. "Screening and Application of DNA Aptamers for Heparin-Binding Protein" Molecules 29, no. 8: 1717. https://doi.org/10.3390/molecules29081717
APA StyleZhou, X., Cao, Y., Huang, X., Qiu, S., Xiang, X., Niu, H., Chen, L., Wang, S., Lin, Z., & Zhang, S. (2024). Screening and Application of DNA Aptamers for Heparin-Binding Protein. Molecules, 29(8), 1717. https://doi.org/10.3390/molecules29081717