3.1. Resonance Parameters for Fields along the Vertical -Direction
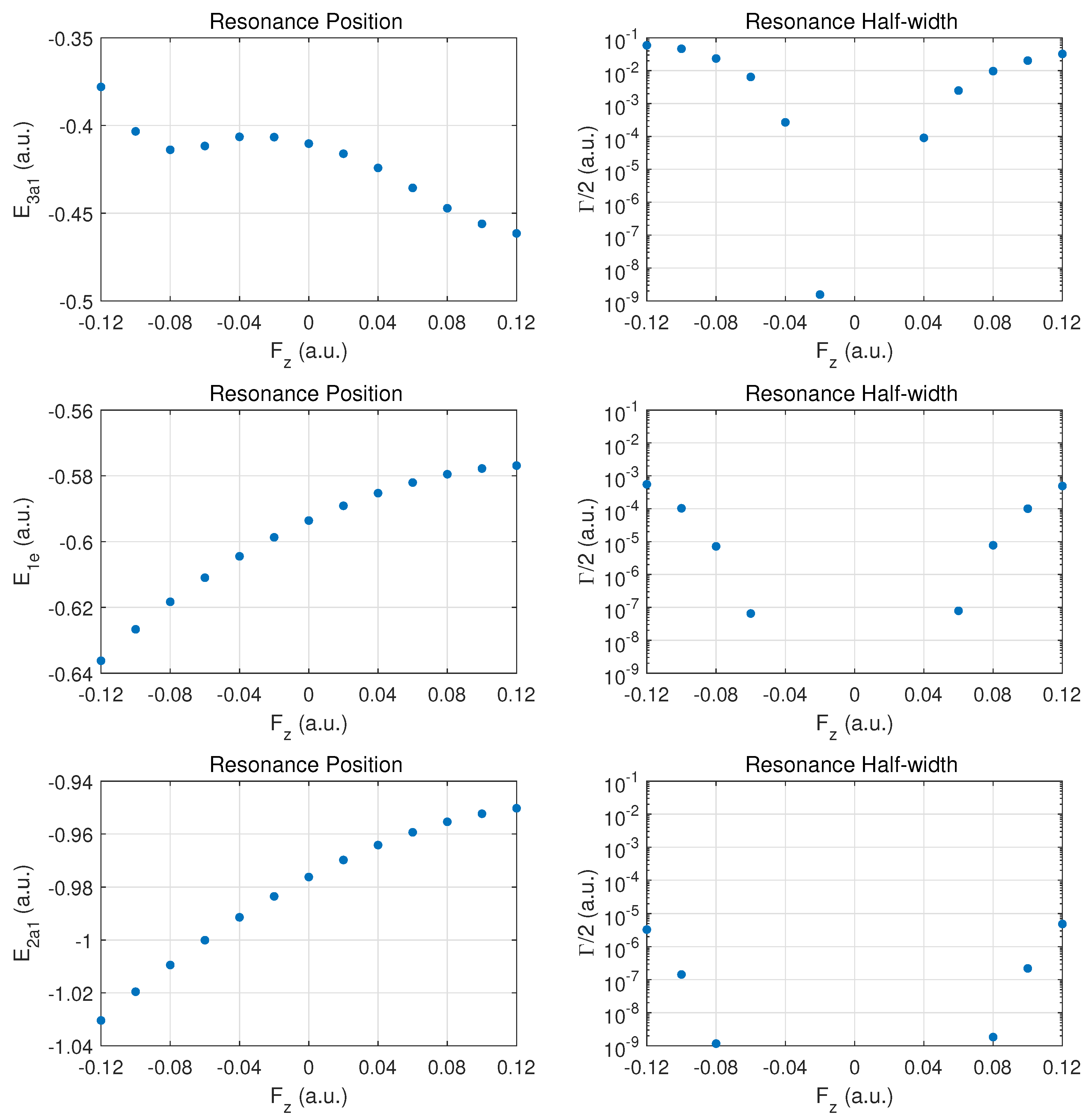
We begin the discussion of resonance parameters with fields along for which the two degenerate MOs and should yield identical results. The dominant contribution to DC field ionization is expected from the weakest bound orbital (). For this orbital the combined molecular and external electric field leads to over-the-barrier ionization at the strongest fields calculated ( of order ).
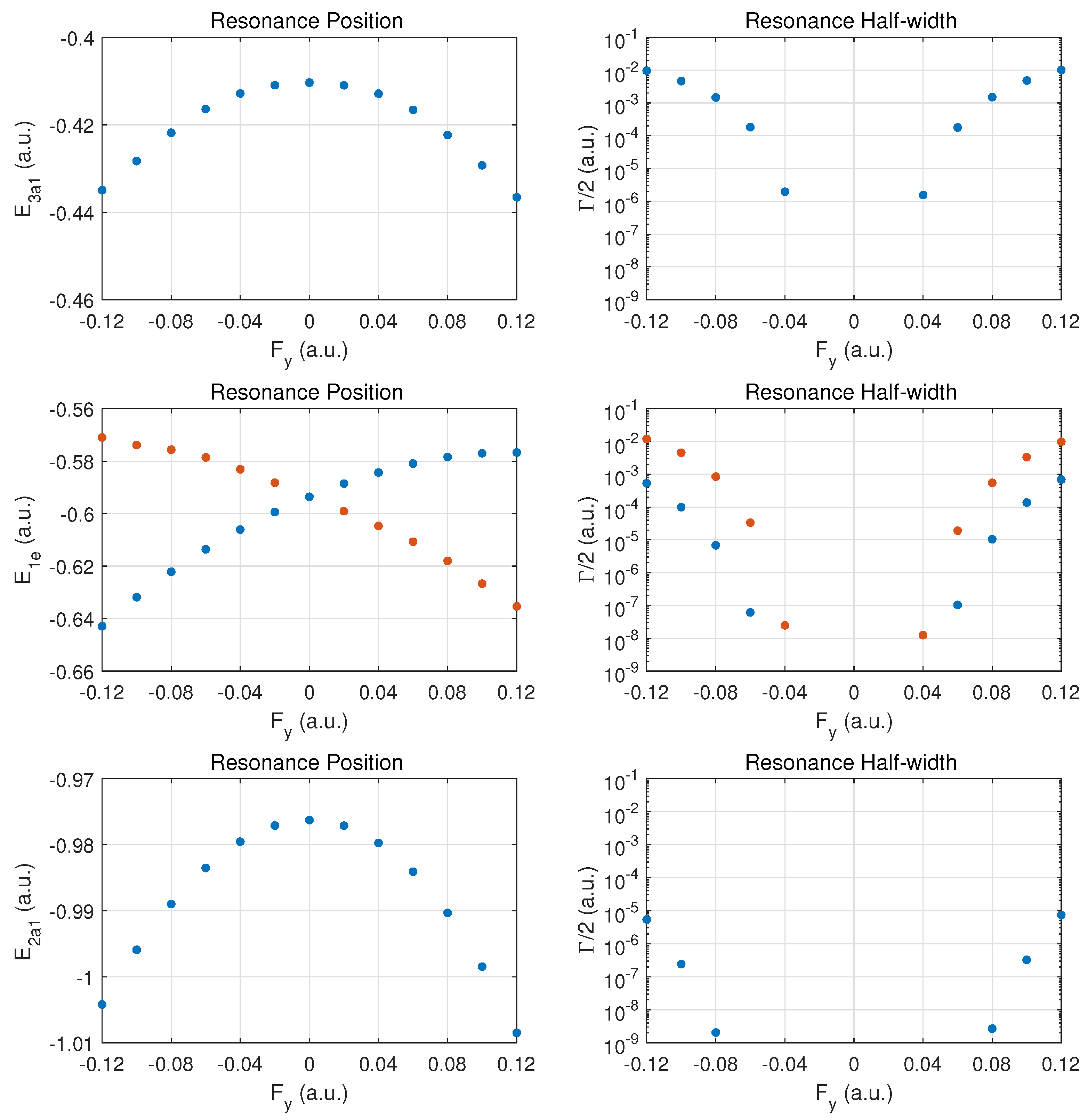
In
Figure 2 the top row shows the resonance position (left column) and resonance half width (right column) for this orbital as a function of field strength
. Positive values
correspond to the field direction pushing electrons out in the direction from the hydrogen atom plane past the nitrogen atom, while negative values
are for electrons ejected from the hydrogen plane (at negative
z) away from the nitrogen atom which is located at
.
The change of the resonance position with field strength can be described as monotonically stronger binding for , since electron density is transferred from the hydrogen atoms in the direction of the central nitrogen atom. For the opposite field direction (), we observe non-monotonic behavior. First, one expects marginally weaker binding when transferring electron density from a nitrogen to a hydrogen atom.
In the molecule, the shift in electron density will be towards regions around the partially shielded protons where the electron binding is weaker. This feature becomes apparent at strong fields (over-the-barrier regime), but there is an intermediate range of field strengths () where there is a non-monotonic variation in the resonance position with field strength.
The resonance widths are obviously small in the tunneling regime. They change by orders of magnitude as the field is increased, and the ionization rate for emission from the hydrogen plane () is stronger than in the opposite direction, by more than a factor of two. For field strengths of the order of , saturation in the ionization rate sets in, which is associated with the over-the-barrier regime. At these field strengths, one may reach the limitations of the exponential decay model, and, thus, results for stronger fields are not reported.
We note that the behavior of the resonance position is consistent with the change in ionization rate (or resonance width) as a function of field direction. In the strong field regime (at about and beyond), the binding energies are quite different and the ionization rates change by a factor of two when the field is reversed. An interesting observation for is the rise in the ionization rate even though the resonance position indicates stronger binding. This phenomenon is associated with density being driven by the field towards the barrier region.
In the middle panel, the results are shown for the two degenerate MOs. The dependence of the resonance position on field strength is monotonic in this case, and varies only at the level in the given field strength range. The corresponding decay rates are weaker by orders of magnitude as compared to the MO, and remain in the tunneling regime. This conclusion will be supported further below by probability density plots for
The bottom panel shows results for the more deeply bound MO. Here, the variation in the resonance position is only at the level of , and the ionization rate is suppressed by two to three additional orders of magnitude. The shape of the DC Stark shift (left panel) as a function of field strength and orientation is similar to what is observed for the pair of MOs. A small asymmetry can be observed in the decay rates, with a small enhancement for vs. .
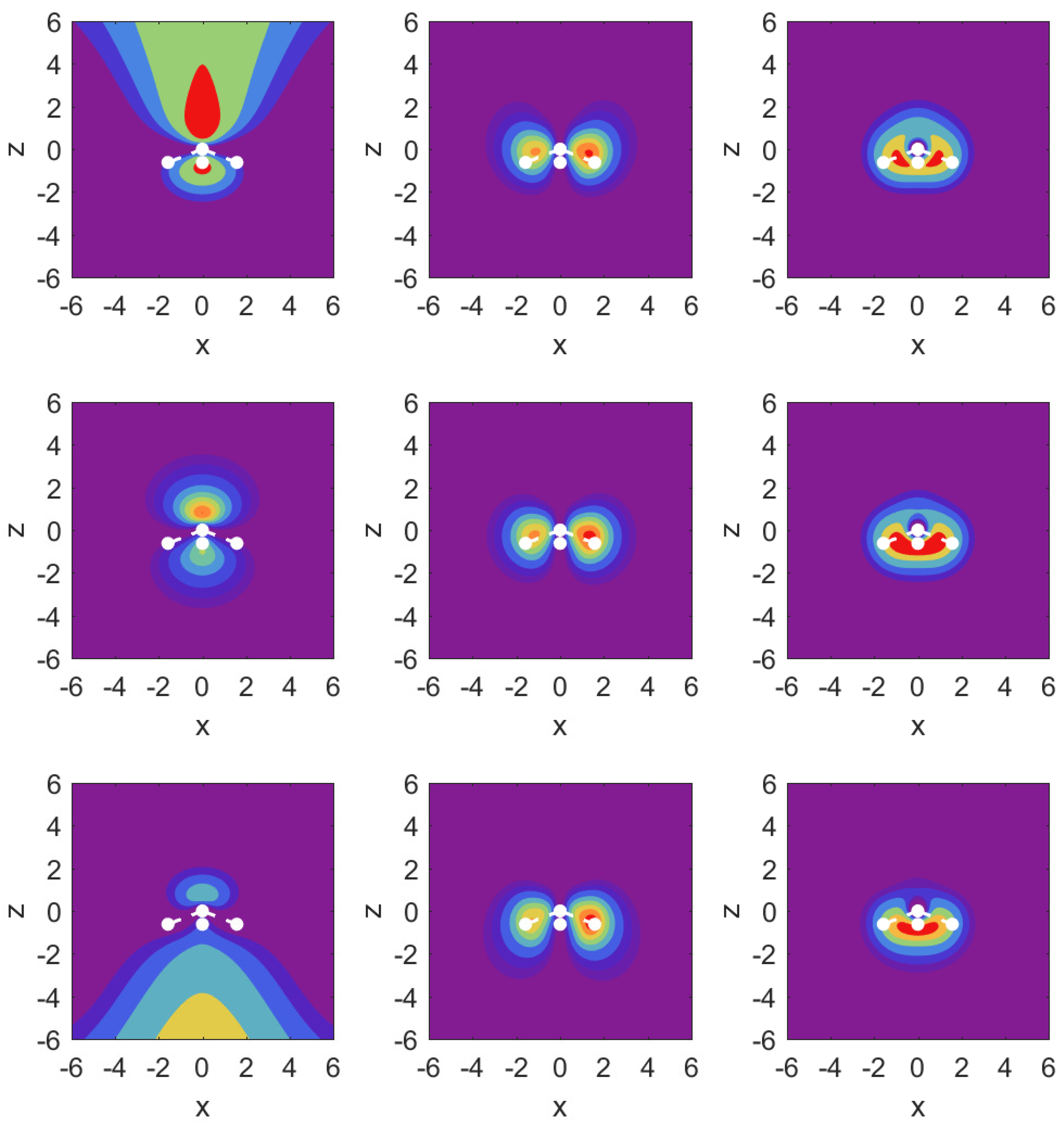
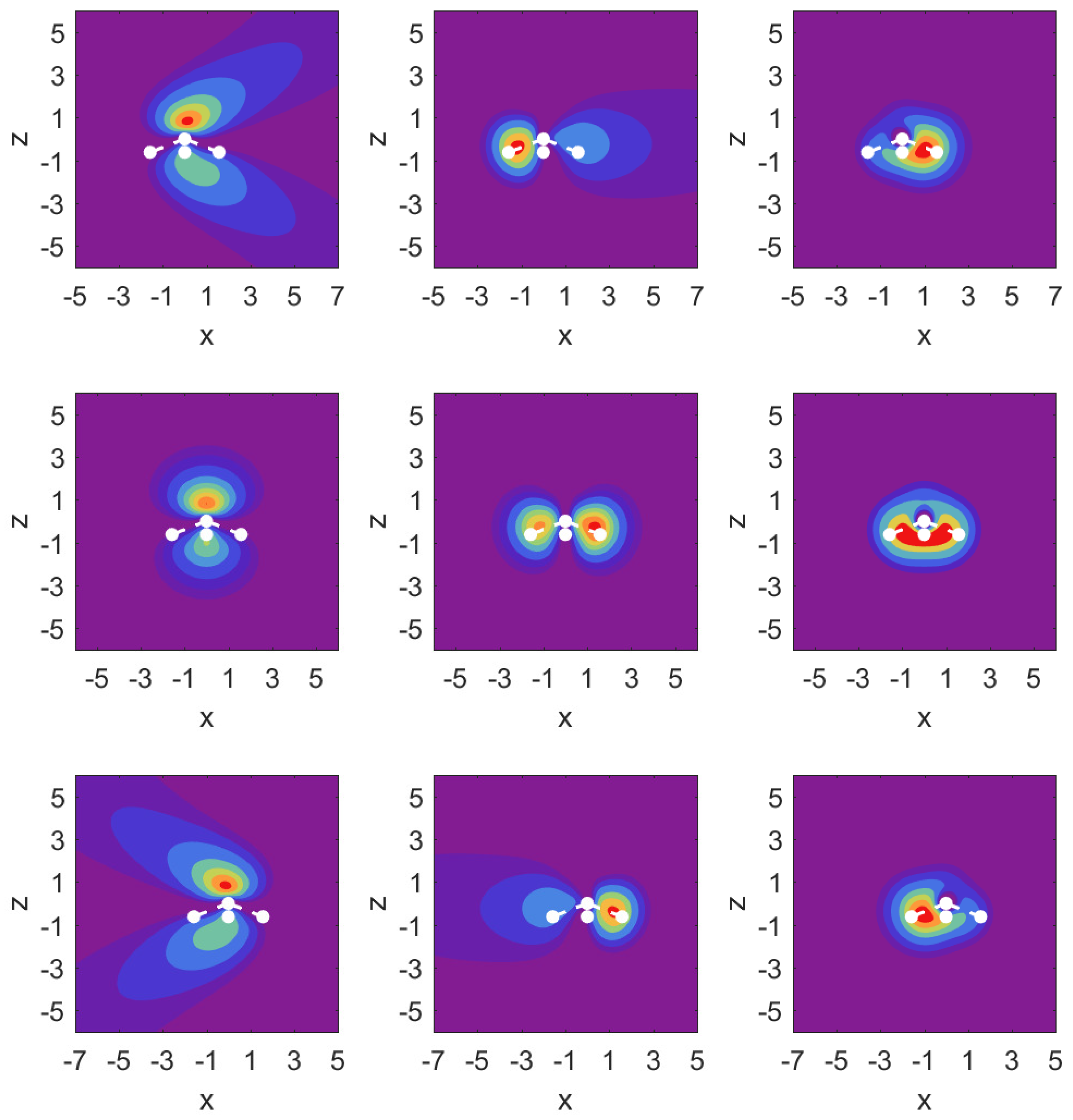
In
Figure 3, we illustrate the situation with probability density contour plots of the MOs. The field-free case is shown in the middle row. The outermost MO (
) shown on the left has an asymmetric probability density with respect to
with higher probability values on the nitrogen side. When the DC field is pushing electrons out on this side, the nitrogen potential provides attraction and causes some concentration of probability in this distribution, as shown in the top left panel (strong red drop-like shape at
and also at
). The interpretation of the density plots is that they describe steady-state decay.
The bottom left panel shows the case of a strong field pushing in the direction past the hydrogen atoms. The probability distribution is more diffuse, showing that the outflow on the side of the hydrogen plane is hindered less. This observation is consistent with the decay rate results shown in the top right panel of
Figure 2. The outflow of probability density is consistent with above-the-barrier ionization for both the top and bottom rows, i.e.,
The other two MOs show much less outflow at comparable fields, and are clearly in the tunneling regime. For the MO (middle column) with field turned on in either direction, there is a limited amount of density change compared to the MO. For the MO (right column), we observe symmetry in the field-free case, and shifting of probability density in the direction of the applied force, but in the tunneling regime not much probability density appears far away from the molecule.
3.2. Resonance Parameters for Fields along
the -Direction
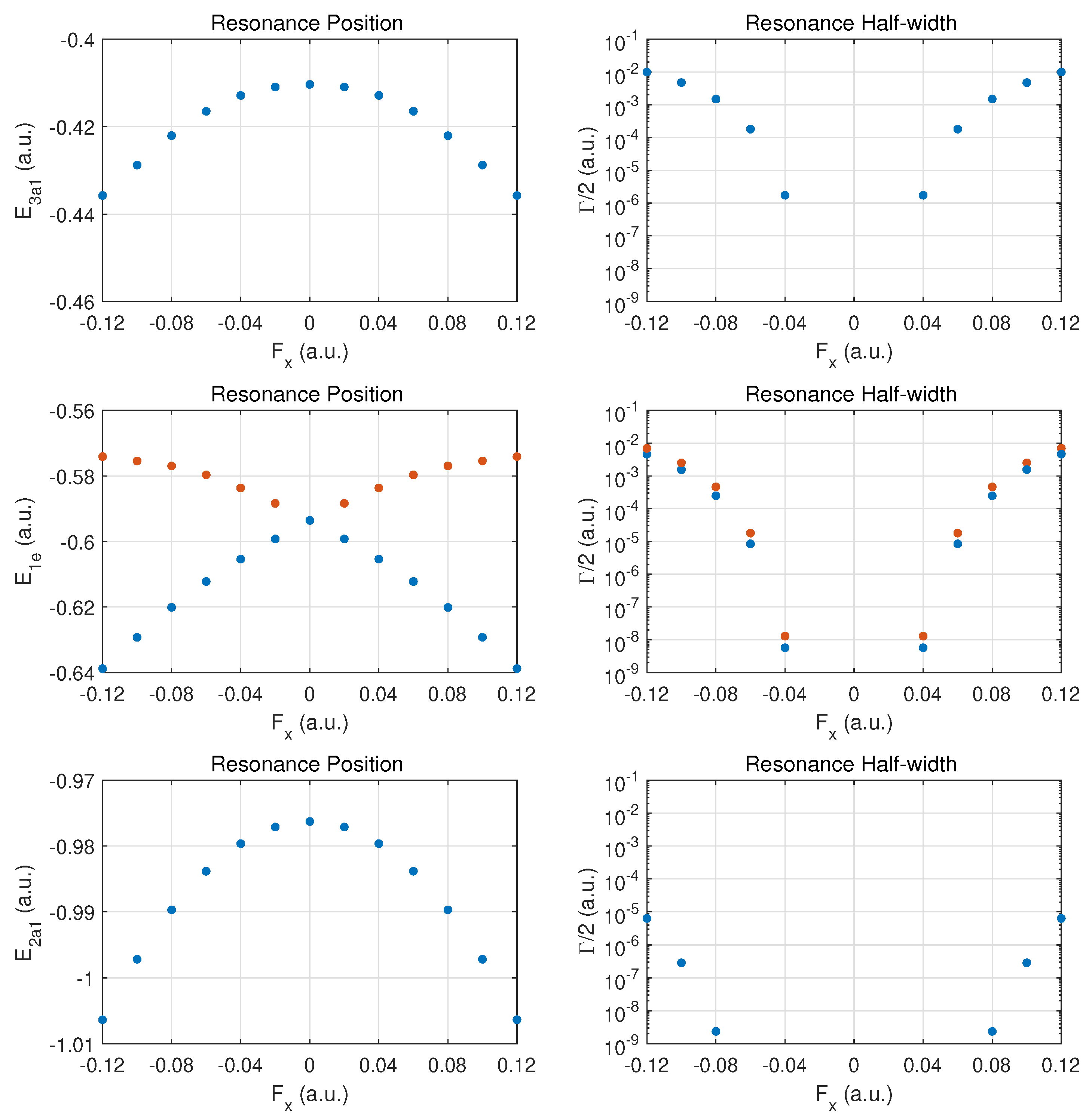
In
Figure 4, results are presented for fields in a perpendicular direction relative to the axis connecting the N atom with the hydrogen atom plane. The arrangement of the three hydrogen atoms is such that one resides on the
y-axis; i.e., field emission occurs along the direction of the H-H bond perpendicular to this axis. The degeneracy between the
and
MO energies is expected to be broken when a DC field is applied in this
(or the perpendicular
) direction.
The top row for the outermost MO shows a symmetric behaviour in the DC Stark shift (left panel) and likewise a symmetric ionization rate with respect to reversal of the field direction. The change in the rise of the ionization rate at strong fields indicates that one is only approaching the over-the-barrier regime, i.e., saturation has not set in yet at The increase in binding is at the level for the strongest fields. The ionization rates for these fields are smaller than for ionization along the axis by a substantial factor (about three or six, depending on the field direction ).
The middle row shows the different behaviors for the and MOs. We classify the two orbitals as fast- vs. slow-ionizing under oriented fields (orange vs. blue markers). The behavior is symmetric with respect to field orientation. The MO (orange dots) shifts towards less binding for both field orientations, and the (blue dots) is bound more deeply as the field is increased in either direction.
The ionization rates (right panel) are also symmetrical with respect to field reversal. The ionizes more readily by almost a factor of two for this field orientation. It is remarkable that these MOs ionize easily at strong fields with the MO displaying a rate which is moving towards that of the more weakly bound MO. Comparing the ionization rates for the and orbitals for fields along versus , we notice an order-of-magnitude increase. The reverse trend is true for the MO.
The bottom row shows that the results for the MO are symmetric with respect to field orientation (as for ). The decay rates are somewhat larger than in the case of z-oriented fields, even though the MO is bound more deeply with increasing field strength.
We support our resonance parameter values again with selective plots of probability densities from the ECS approach in
Figure 5. The middle row is identical with that in
Figure 5, but is included for direct comparison of the cases with electric field. It is immediately apparent that three MOs (
,
, and also
, which is not shown) contribute strongly to ionization of the molecule for this field orientation.
For the MO, we observe outflow in the form of two jets directed above and below the hydrogen atom plane. For the MO, we find that the apparent asymmetry in the x-z plane for the field-free case has no repercussions for the outflow in the case with fields of either direction: both cases show very symmetric probabilities under these conditions, which is consistent with the findings for the resonance parameters.
For the MO shown in the right column, symmetry with respect to field orientation is expected. This is evident from the density plots by comparing the top and bottom panels. We note the relatively strong effect the field has on this relatively deeply bound orbital.
3.3. Resonance Parameters for Fields along the -Direction
In
Figure 6, results are given for field orientation along
. Given the triangular nature of the hydrogen atom plane, these results differ strongly from those in the previous section. By choice of azimuthal angles
degrees, a proton is located on the positive
y axis, and asymmetry is expected when reversing the field direction.
The top row shows that this asymmetry plays a very small role for the outermost MO
at weak fields, and is barely noticeable. The tabulated data (cf.
Appendix A.3) show that the shifts differ by less than a percent.
The and MOs (blue and orange dots, middle row) show markedly different behavior when compared to oriented fields. The shifts follow monotonic curves, as there is no longer symmetry under field orientation reversal. The MO is ionizing more rapidly as compared to by about two orders of magnitude. The variation in resonance position is clearly at odds with this result; i.e., it apparently does not play a role here. As discussed before, the actual value of the MO binding energy is not the deciding factor, but rather how the electron density is driven towards the potential barrier by the external field.
The MO ionizes very weakly, and its rate is comparable to those obtained for oriented fields. Thus, one may conclude that the MO is affected by this field orientation dramatically.
In the third row, we give results for the deeply bound MO which remains deeply in the tunneling regime for the given field strengths. It shows a small amount of asymmetry in the DC Stark shifts and in the decay rates.
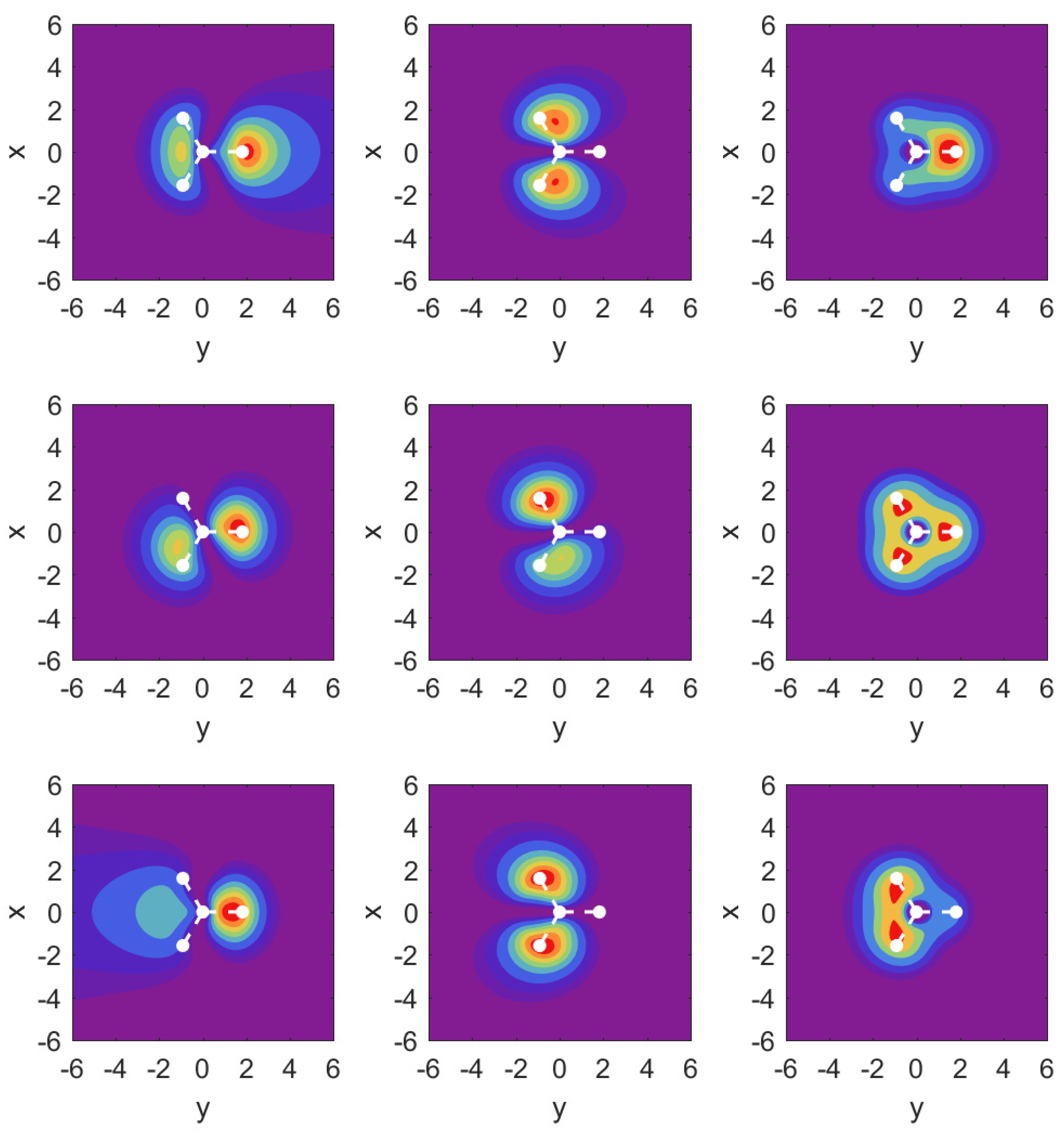
The probability density plots shown in
Figure 7 again help to understand the finding for the parameter values. The results for MO
are very close to the corresponding plots in
Figure 5 and are not shown. Instead, we show over the
plane probability densities for the strongly ionizing
MO (left column), the much more weakly ionizing
MO (middle column), and
in the right column.
For the field-free case, the density plots show that the MOs and are actually not perfectly aligned with our x- and y-axes. This is related to the fact that they share the same probability density shapes, except that they are rotated by 90 degrees, and this is incompatible with the three-fold symmetry. Once the strong field is turned on along either the x- or the y-axis, however, the densities respect the symmetry of the external field, and one can identify and .
The results for the
MO (left column) show an asymmetry in the outflow for the case
vs.
. It is interesting to observe that while the shape of the outflow is very different for both cases, the ionization rates actually differ only at the level of up to
(cf.
Appendix A.3).
In the middle column for MO , we observe a symmetry in the outflows despite the fact that the arrangement of hydrogen atoms is asymmetric. For the case of MO , we find that the central parts of the density are very different for the two field directions, but the parts showing the outflow are again very similar, and this is similar to what one observes for MO .











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}