
Solvation of Model Biomolecules in Choline-Aminoate Ionic Liquids: A Computational Simulation Using Polarizable Force Fields



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
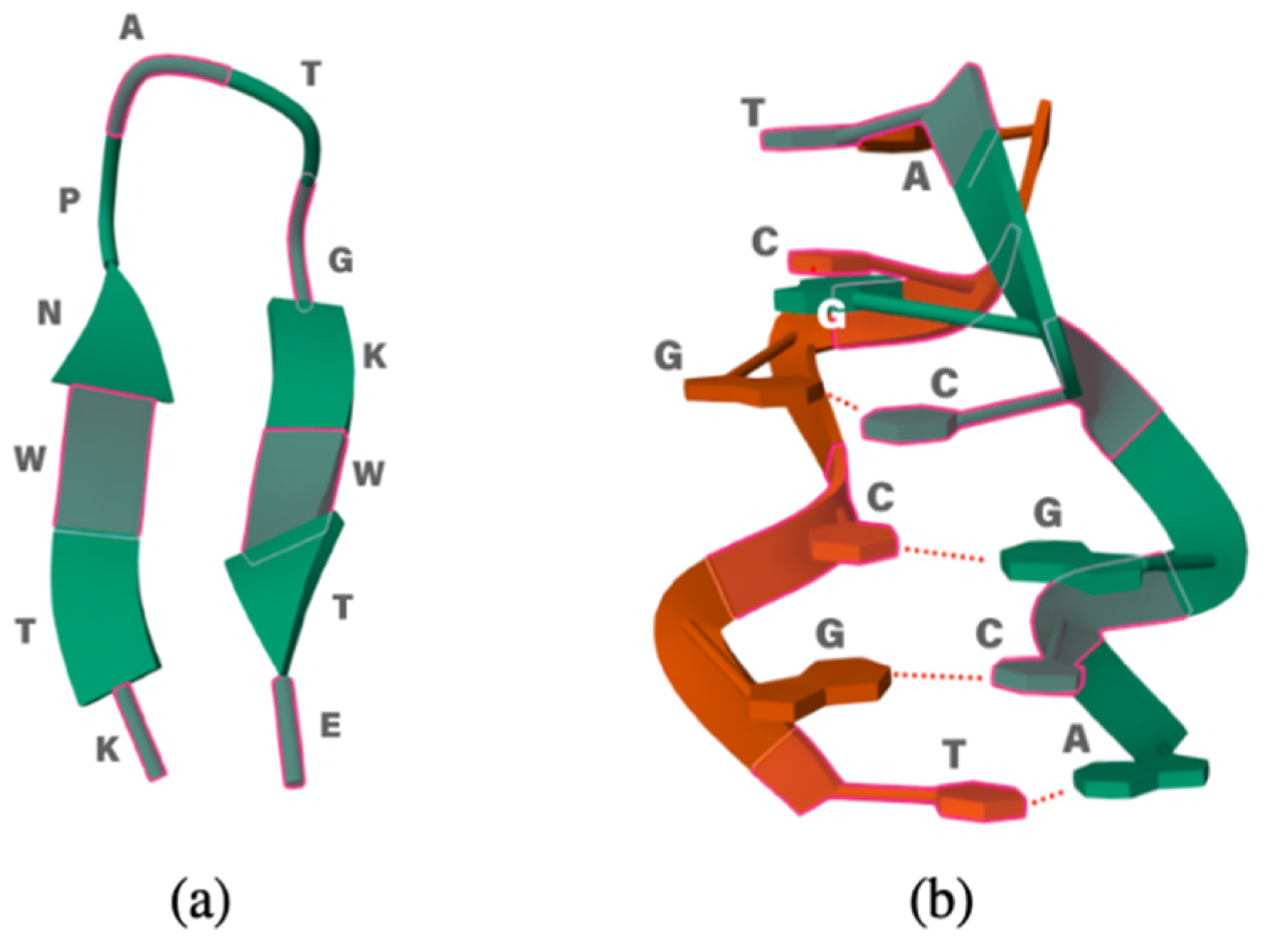
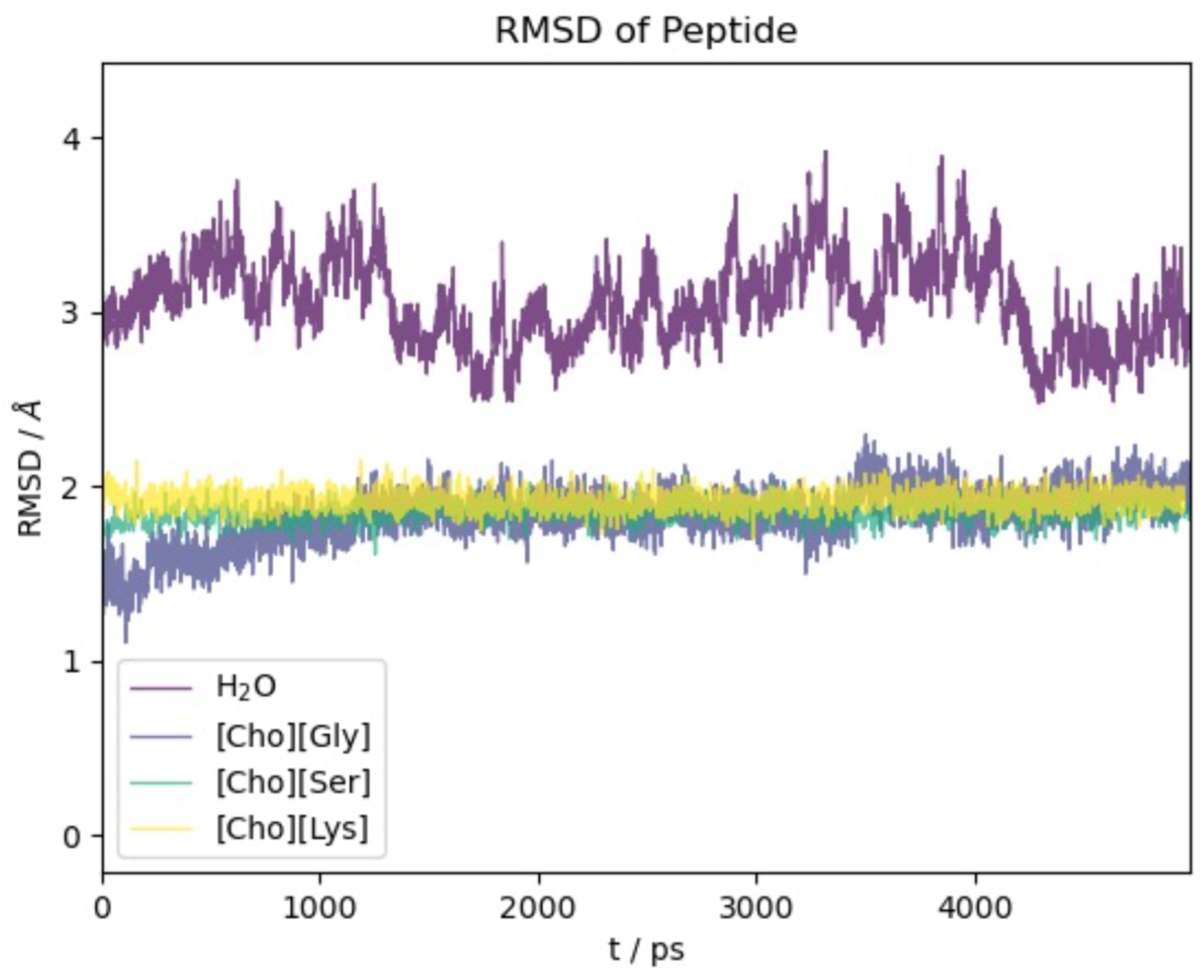
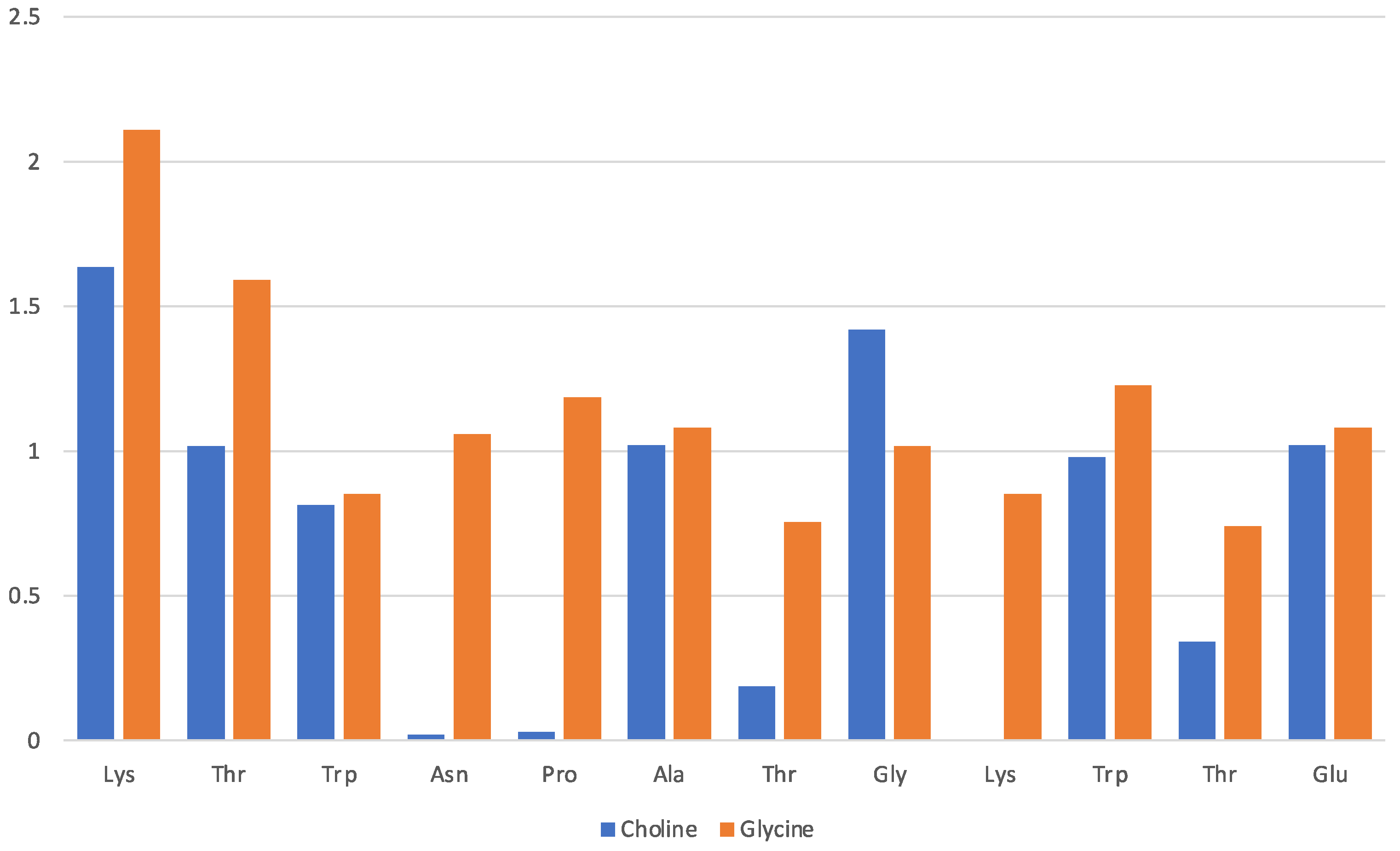
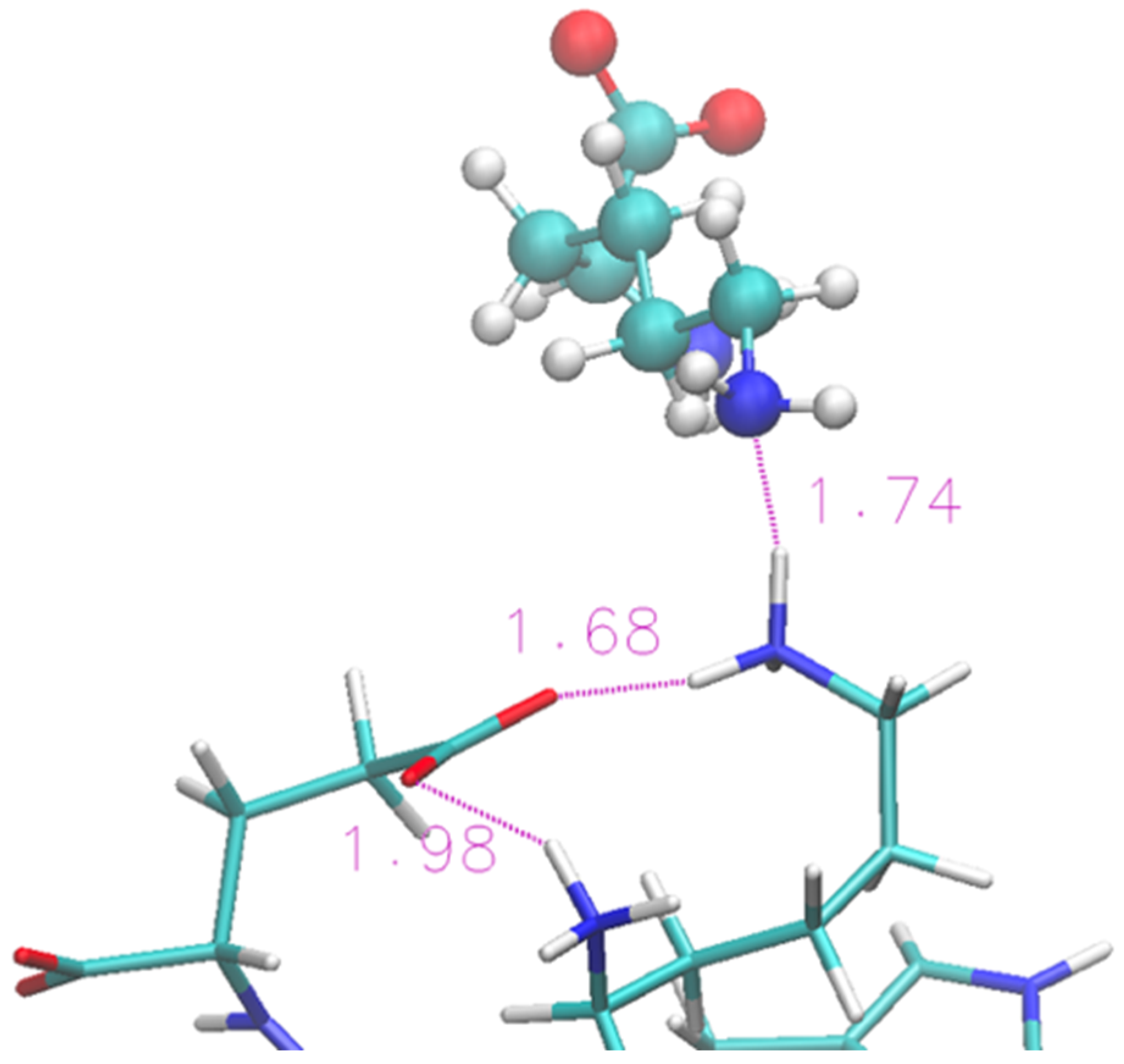
2.1. 12-Residue Oligopetide
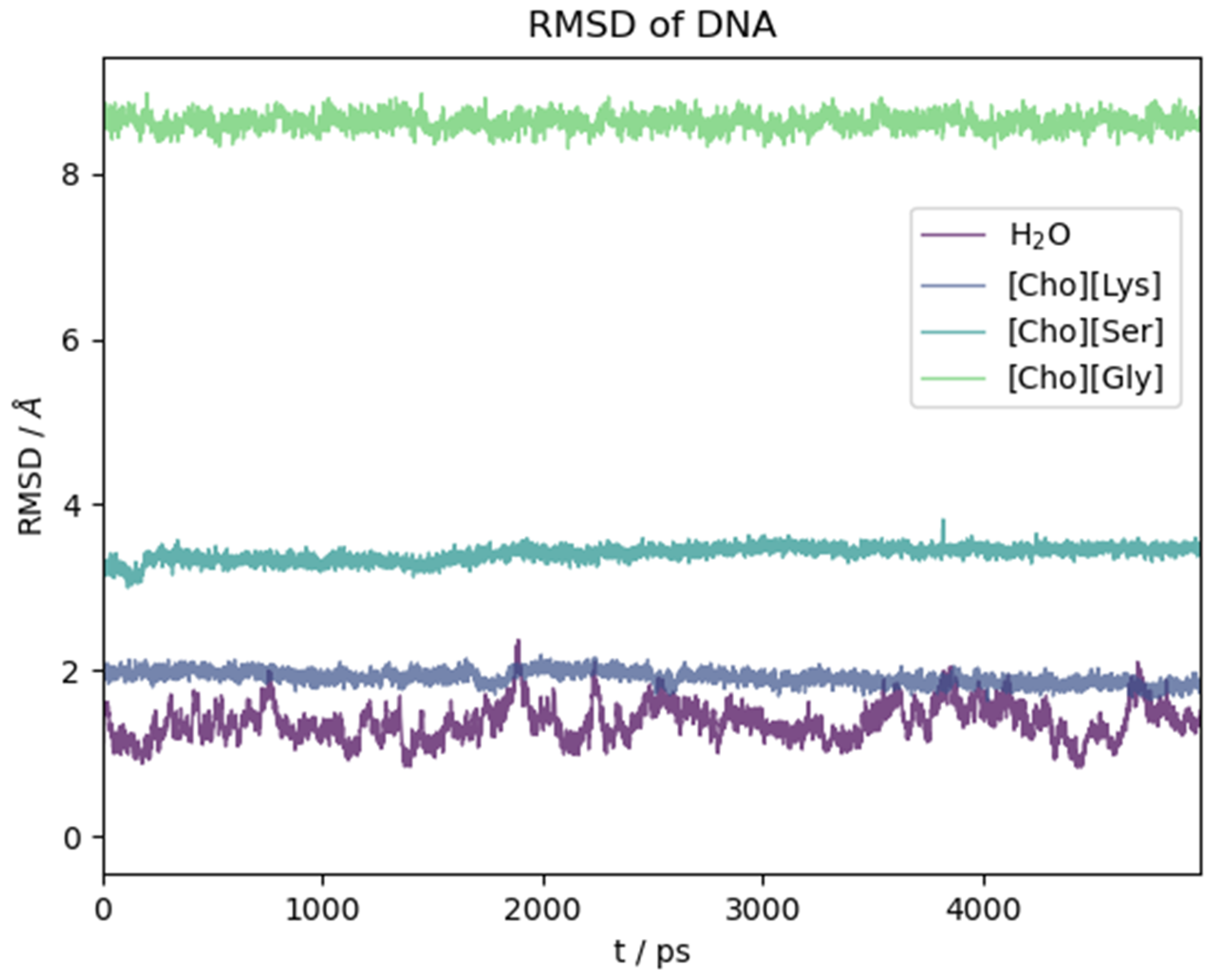
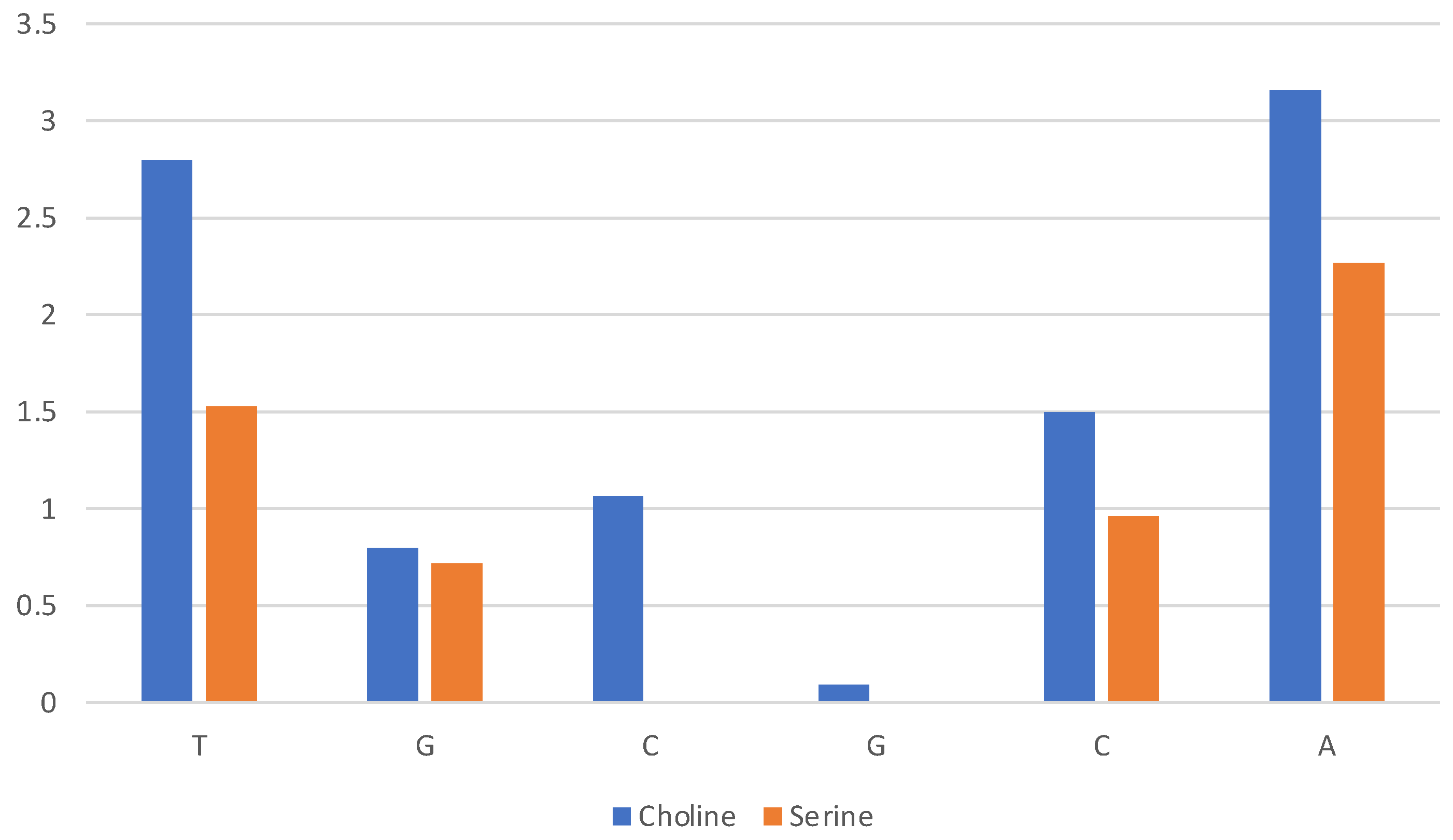
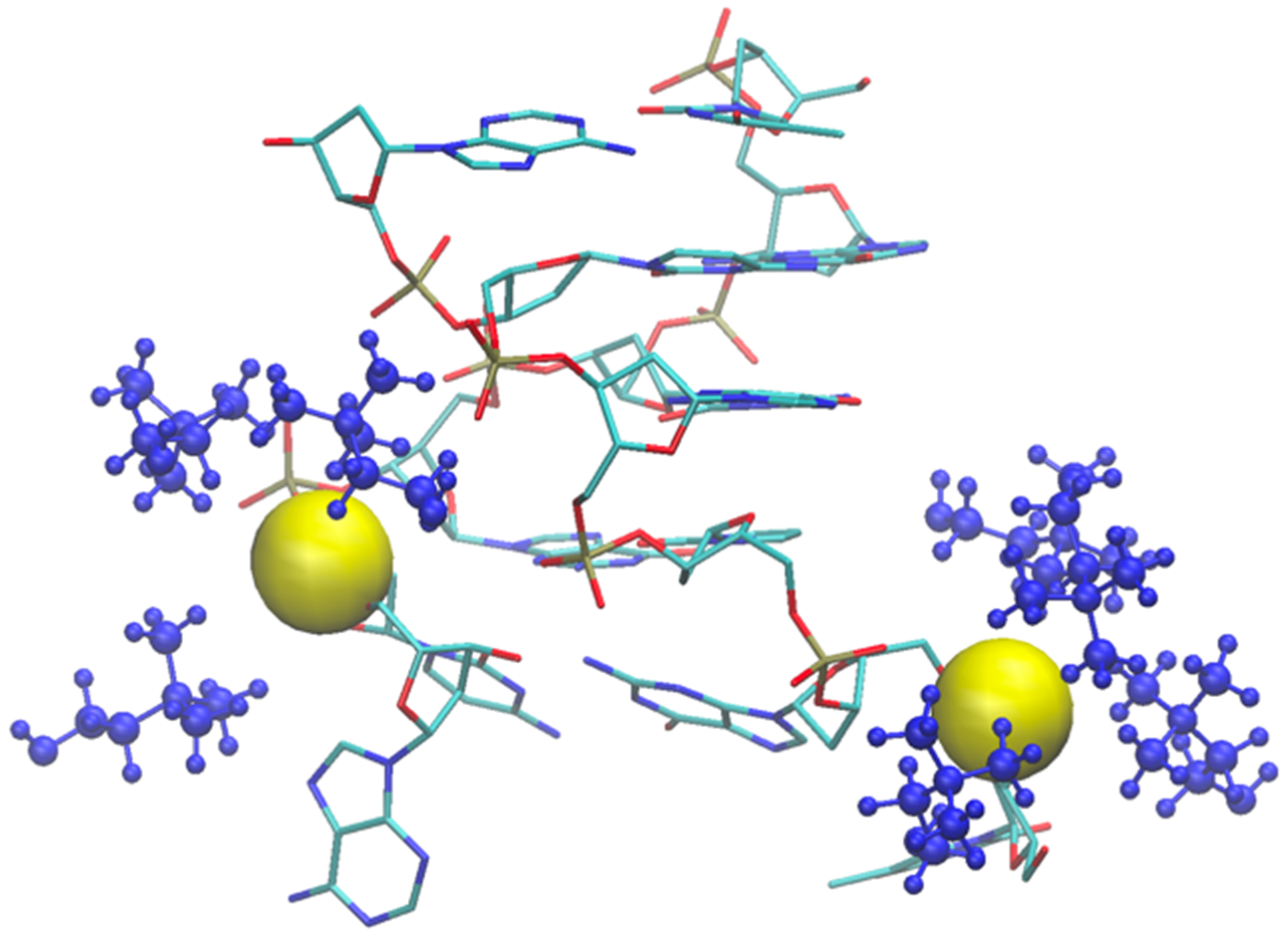
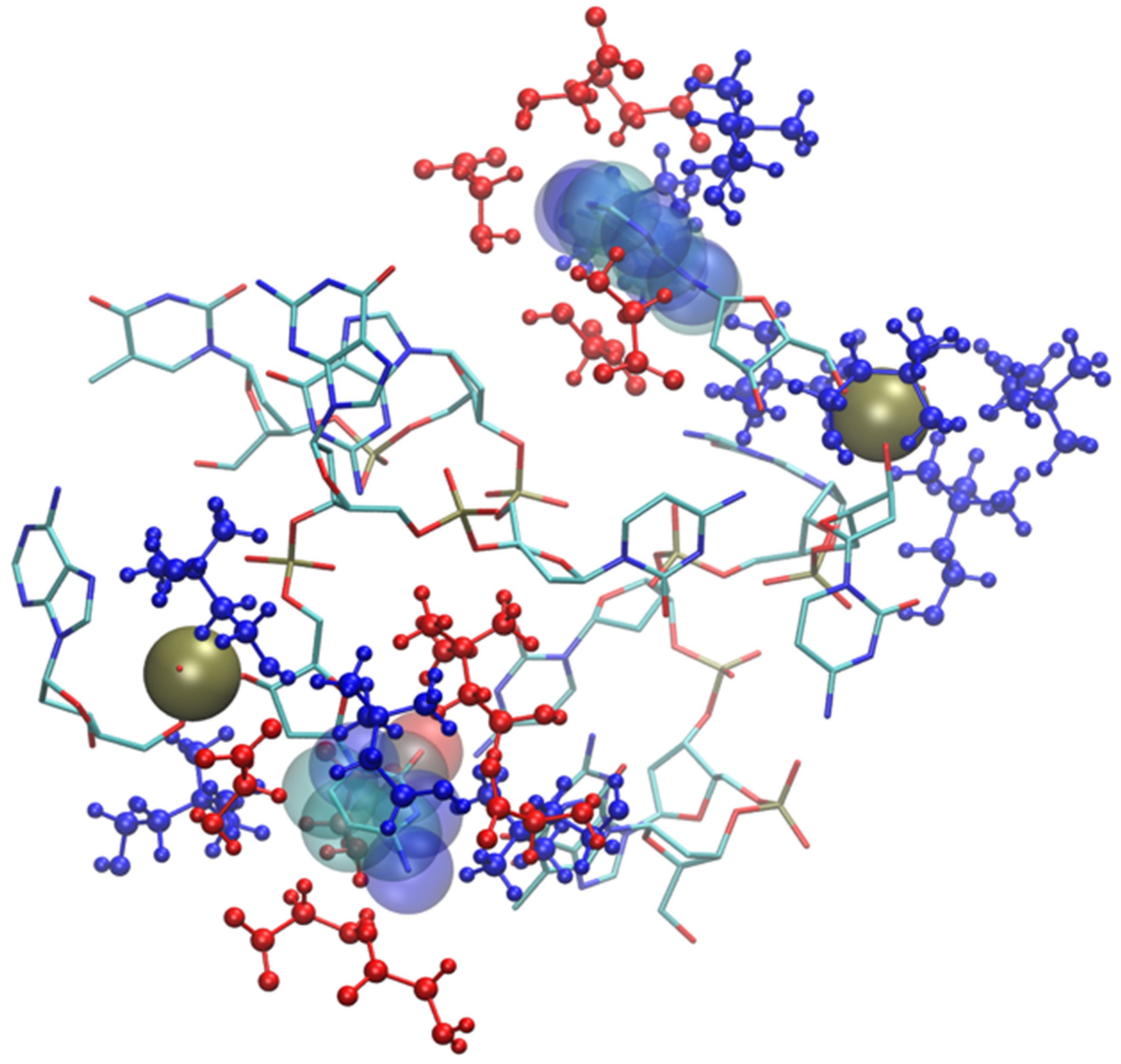
2.2. Six Base Pairs DNA Double Strand
3. Methods and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumari, P.; Pillai, V.V.S.; Benedetto, A. Mechanisms of Action of Ionic Liquids on Living Cells: The State of the Art. Biophys. Rev. 2020, 12, 1187–1215. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Hozaifa, M.; Ali, S.; Malik, A.; Locatelli, M. Advances in Ionic Liquids as Future Anti-Cancer Drugs. J. Mol. Liq. 2023, 388, 122823. [Google Scholar] [CrossRef]
- Balk, A.; Holzgrabe, U.; Meinel, L. ‘Pro et Contra’ Ionic Liquid Drugs—Challenges and Opportunities for Pharmaceutical Translation. Eur. J. Pharm. Biopharm. 2015, 94, 291–304. [Google Scholar] [CrossRef]
- Pillai, V.V.S.; Kumari, P.; Kolagatla, S.; Garcia Sakai, V.; Rudić, S.; Rodriguez, B.J.; Rubini, M.; Tych, K.M.; Benedetto, A. Controlling Amyloid Fibril Properties Via Ionic Liquids: The Representative Case of Ethylammonium Nitrate and Tetramethylguanidinium Acetate on the Amyloidogenesis of Lysozyme. J. Phys. Chem. Lett. 2022, 13, 7058–7064. [Google Scholar] [CrossRef]
- Cho, C.-W.; Pham, T.P.T.; Zhao, Y.; Stolte, S.; Yun, Y.-S. Review of the Toxic Effects of Ionic Liquids. Sci. Total Environ. 2021, 786, 147309. [Google Scholar] [CrossRef] [PubMed]
- Thuy Pham, T.P.; Cho, C.-W.; Yun, Y.-S. Environmental Fate and Toxicity of Ionic Liquids: A Review. Water Res. 2010, 44, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Sanches, M.V.; Freitas, R.; Oliva, M.; Cuccaro, A.; Monni, G.; Mezzetta, A.; Guazzelli, L.; Pretti, C. Toxicity of Ionic Liquids in Marine and Freshwater Microorganisms and Invertebrates: State of the Art. Environ. Sci. Pollut. Res. 2023, 30, 39288–39318. [Google Scholar] [CrossRef]
- Himani; Pratap Singh Raman, A.; Babu Singh, M.; Jain, P.; Chaudhary, P.; Bahadur, I.; Lal, K.; Kumar, V.; Singh, P. An Update on Synthesis, Properties, Applications and Toxicity of the ILs. J. Mol. Liq. 2022, 364, 119989. [Google Scholar] [CrossRef]
- Kuroda, K. A Simple Overview of Toxicity of Ionic Liquids and Designs of Biocompatible Ionic Liquids. New J. Chem. 2022, 46, 20047–20052. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef] [PubMed]
- Le Donne, A.; Bodo, E. Cholinium Amino Acid-Based Ionic Liquids. Biophys. Rev. 2021, 13, 147–160. [Google Scholar] [CrossRef] [PubMed]
- del Olmo, L.; Lage-Estebanez, I.; López, R.; García de la Vega, J.M. Understanding the Structure and Properties of Cholinium Amino Acid Based Ionic Liquids. J. Phys. Chem. B 2016, 120, 10327–10335. [Google Scholar] [CrossRef] [PubMed]
- Dhattarwal, H.S.; Kashyap, H.K. Unique and Generic Structural Features of Cholinium Amino Acid-Based Biocompatible Ionic Liquids. Phys. Chem. Chem. Phys. 2021, 23, 10662–10669. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.-D.; Liu, Q.-P.; Smith, T.J.; Li, N.; Zong, M.-H. Evaluation of Toxicity and Biodegradability of Cholinium Amino Acids Ionic Liquids. PLoS ONE 2013, 8, e59145. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Kashyap, H.K. Sensitivity and Resilience of Phosphatidylcholine and Phosphatidylethanolamine Lipid Membranes against Cholinium Glycinate Biocompatible Ionic Liquid. J. Phys. Chem. B 2019, 123, 4550–4561. [Google Scholar] [CrossRef] [PubMed]
- Pedro, A.Q.; Pereira, P.; Quental, M.J.; Carvalho, A.P.; Santos, S.M.; Queiroz, J.A.; Sousa, F.; Freire, M.G. Cholinium-Based Good’s Buffers Ionic Liquids as Remarkable Stabilizers and Recyclable Preservation Media for Recombinant Small RNAs. ACS Sustain. Chem. Eng. 2018, 6, 16645–16656. [Google Scholar] [CrossRef] [PubMed]
- Morandeira, L.; Álvarez, M.S.; Markiewicz, M.; Stolte, S.; Rodríguez, A.; Sanromán, M.Á.; Deive, F.J. Testing True Choline Ionic Liquid Biocompatibility from a Biotechnological Standpoint. ACS Sustain. Chem. Eng. 2017, 5, 8302–8309. [Google Scholar] [CrossRef]
- Sindhu, A.; Kumar, S.; Venkatesu, P. Contemporary Advancement of Cholinium-Based Ionic Liquids for Protein Stability and Long-Term Storage: Past, Present, and Future Outlook. ACS Sustain. Chem. Eng. 2022, 10, 4323–4344. [Google Scholar] [CrossRef]
- Yazdani, A.; Sivapragasam, M.; Leveque, J.M.; Moniruzzaman, M. Microbial Biocompatibility and Biodegradability of Choline-Amino Acid Based Ionic Liquids. J. Microb. Biochem. Technol. 2016, 8, 415–421. [Google Scholar] [CrossRef]
- De Santis, S.; Masci, G.; Casciotta, F.; Caminiti, R.; Scarpellini, E.; Campetella, M.; Gontrani, L. Cholinium-Amino Acid Based Ionic Liquids: A New Method of Synthesis and Physico-Chemical Characterization. Phys. Chem. Chem. Phys. 2015, 17, 20687–20698. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.; Chowdhury, U.D.; Barik, S.; Parida, S.; Bhargava, B.L.; Sarkar, M. Deciphering the Role of Anions of Ionic Liquids in Modulating the Structure and Stability of Ct -DNA in Aqueous Solutions. Langmuir 2023, 39, 17318–17332. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.; Barik, S.; Satish, L.; Chakraborty, M.; Sarkar, M. Assessing the Suitability of a Dicationic Ionic Liquid as a Stabilizing Material for the Storage of DNA in Aqueous Medium. Langmuir 2022, 38, 14857–14868. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bisht, M.; Venkatesu, P. Biocompatibility of Ionic Liquids towards Protein Stability: A Comprehensive Overview on the Current Understanding and Their Implications. Int. J. Biol. Macromol. 2017, 96, 611–651. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bhakuni, K.; Venkatesu, P. Strategic Planning of Proteins in Ionic Liquids: Future Solvents for the Enhanced Stability of Proteins against Multiple Stresses. Phys. Chem. Chem. Phys. 2019, 21, 23269–23282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H. Protein Stabilization and Enzyme Activation in Ionic Liquids: Specific Ion Effects. J. Chem. Technol. Biotechnol. 2016, 91, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C. Proteins in Ionic Liquids: Current Status of Experiments and Simulations. Top. Curr. Chem. 2017, 375, 25. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Mikkola, J.-P. Use of Ionic Liquids in Protein and DNA Chemistry. Front. Chem. 2020, 8, 598662. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, R.; Izgorodin, A.; Ganesh, V.; Surianarayanan, M.; MacFarlane, D.R. Long-Term Structural and Chemical Stability of DNA in Hydrated Ionic Liquids. Angew. Chem. Int. Ed. 2010, 49, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Mukesh, C.; Mondal, D.; Sharma, M.; Prasad, K. Rapid Dissolution of DNA in a Novel Bio-Based Ionic Liquid with Long-Term Structural and Chemical Stability: Successful Recycling of the Ionic Liquid for Reuse in the Process. Chem. Commun. 2013, 49, 6849. [Google Scholar] [CrossRef]
- Kalhor, S.; Fattahi, A. Design of Ionic Liquids Containing Glucose and Choline as Drug Carriers, Finding the Link between QM and MD Studies. Sci. Rep. 2022, 12, 21941. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wu, Q.; Jiang, L.; Liu, M.; Li, C. Protein Stability Analysis in Ionic Liquids by 19F NMR. Anal. Bioanal. Chem. 2019, 411, 4929–4935. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.L.; Vieira, N.S.M.; Araújo, J.M.M.; Pereiro, A.B. Unveiling the Influence of Non-Toxic Fluorinated Ionic Liquids Aqueous Solutions in the Encapsulation and Stability of Lysozyme. Sustain. Chem. 2021, 2, 149–166. [Google Scholar] [CrossRef]
- Rakowska, P.W.; Kloskowski, A. Impact of the Alkyl Side Chains of Cations and Anions on the Activity and Renaturation of Lysozyme: A Systematic Study Performed Using Six Amino-Acid-Based Ionic Liquids. ChemistrySelect 2021, 6, 3089–3095. [Google Scholar] [CrossRef]
- Smiatek, J. Aqueous Ionic Liquids and Their Effects on Protein Structures: An Overview on Recent Theoretical and Experimental Results. J. Phys. Condens. Matter 2017, 29, 233001. [Google Scholar] [CrossRef] [PubMed]
- Zeindlhofer, V.; Schröder, C. Computational Solvation Analysis of Biomolecules in Aqueous Ionic Liquid Mixtures: From Large Flexible Proteins to Small Rigid Drugs. Biophys. Rev. 2018, 10, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Lesch, V.; Heuer, A.; Tatsis, V.A.; Holm, C.; Smiatek, J. Peptides in the Presence of Aqueous Ionic Liquids: Tunable Co-Solutes as Denaturants or Protectants? Phys. Chem. Chem. Phys. 2015, 17, 26049–26053. [Google Scholar] [CrossRef] [PubMed]
- Haberler, M.; Steinhauser, O. On the Influence of Hydrated Ionic Liquids on the Dynamical Structure of Model Proteins: A Computational Study. Phys. Chem. Chem. Phys. 2011, 13, 17994. [Google Scholar] [CrossRef] [PubMed]
- Haberler, M.; Schröder, C.; Steinhauser, O. Solvation Studies of a Zinc Finger Protein in Hydrated Ionic Liquids. Phys. Chem. Chem. Phys. 2011, 13, 6955. [Google Scholar] [CrossRef]
- Klähn, M.; Lim, G.S.; Wu, P. How Ion Properties Determine the Stability of a Lipase Enzyme in Ionic Liquids: A Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2011, 13, 18647. [Google Scholar] [CrossRef] [PubMed]
- Diddens, D.; Lesch, V.; Heuer, A.; Smiatek, J. Aqueous Ionic Liquids and Their Influence on Peptide Conformations: Denaturation and Dehydration Mechanisms. Phys. Chem. Chem. Phys. 2017, 19, 20430–20440. [Google Scholar] [CrossRef]
- Sundaram, V.; Ramanan, R.N.; Selvaraj, M.; Vijayaraghavan, R.; MacFarlane, D.R.; Ooi, C.W. Structural Stability of Insulin Aspart in Aqueous Cholinium Aminoate Ionic Liquids Based on Molecular Dynamics Simulation Studies. J. Mol. Liq. 2021, 322, 114501. [Google Scholar] [CrossRef]
- Chevrot, G.; Fileti, E.E.; Chaban, V.V. Enhanced Stability of the Model Mini-protein in Amino Acid Ionic Liquids and Their Aqueous Solutions. J. Comput. Chem. 2015, 36, 2044–2051. [Google Scholar] [CrossRef]
- Bisht, M.; Jha, I.; Venkatesu, P. Does Choline-Based Amino Acid Ionic Liquid Behave as a Biocompatible Solvent for Stem Bromelain Structure? Process Biochem. 2018, 74, 77–85. [Google Scholar] [CrossRef]
- Tateishi-Karimata, H.; Sugimoto, N. Structure, Stability and Behaviour of Nucleic Acids in Ionic Liquids. Nucleic Acids Res. 2014, 42, 8831–8844. [Google Scholar] [CrossRef]
- Zhao, H. DNA Stability in Ionic Liquids and Deep Eutectic Solvents: DNA Stability in Ionic Liquids and Deep Eutectic Solvents. J. Chem. Technol. Biotechnol. 2015, 90, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.; Micaelo, N.M. DNA Molecular Solvation in Neat Ionic Liquids. ChemPhysChem 2011, 12, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Fadaei, F.; Tortora, M.; Gessini, A.; Masciovecchio, C.; Catalini, S.; Vigna, J.; Mancini, I.; Mele, A.; Vacek, J.; Reha, D.; et al. Structural Specificity of Groove Binding Mechanism between Imidazolium-Based Ionic Liquids and DNA Revealed by Synchrotron-UV Resonance Raman Spectroscopy and Molecular Dynamics Simulations. J. Mol. Liq. 2022, 347, 118350. [Google Scholar] [CrossRef]
- Jumbri, K.; Abdul Rahman, M.B.; Abdulmalek, E.; Ahmad, H.; Micaelo, N.M. An Insight into Structure and Stability of DNA in Ionic Liquids from Molecular Dynamics Simulation and Experimental Studies. Phys. Chem. Chem. Phys. 2014, 16, 14036–14046. [Google Scholar] [CrossRef] [PubMed]
- Athira, K.K.; Gardas, R.L. Interactions of Ammonium Based Ionic Liquids with DNA in Aqueous Medium through Volumetric, Acoustic and Viscometric Properties. J. Mol. Liq. 2023, 389, 122858. [Google Scholar] [CrossRef]
- Chandran, A.; Ghoshdastidar, D.; Senapati, S. Groove Binding Mechanism of Ionic Liquids: A Key Factor in Long-Term Stability of DNA in Hydrated Ionic Liquids? J. Am. Chem. Soc. 2012, 134, 20330–20339. [Google Scholar] [CrossRef]
- Bedrov, D.; Piquemal, J.-P.; Borodin, O.; MacKerell, A.D.; Roux, B.; Schröder, C. Molecular Dynamics Simulations of Ionic Liquids and Electrolytes Using Polarizable Force Fields. Chem. Rev. 2019, 119, 7940–7995. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C. Comparing Reduced Partial Charge Models with Polarizable Simulations of Ionic Liquids. Phys. Chem. Chem. Phys. 2012, 14, 3089. [Google Scholar] [CrossRef] [PubMed]
- Klajmon, M.; Červinka, C. Does Explicit Polarizability Improve Simulations of Phase Behavior of Ionic Liquids? J. Chem. Theory Comput. 2021, 17, 6225–6239. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.-P.; Ren, P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef]
- Baker, C.M. Polarizable Force Fields for Molecular Dynamics Simulations of Biomolecules. WIREs Comput. Mol. Sci. 2015, 5, 241–254. [Google Scholar] [CrossRef]
- Nerenberg, P.S.; Head-Gordon, T. New Developments in Force Fields for Biomolecular Simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef]
- Russo, S.; Bodo, E. A Polarisable Force Field for Bio-Compatible Ionic Liquids Based on Amino Acids Anions. Mol. Simul. 2022, 48, 1650–1659. [Google Scholar] [CrossRef]
- Ponder, J.W.; Wu, C.; Ren, P.; Pande, V.S.; Chodera, J.D.; Schnieders, M.J.; Haque, I.; Mobley, D.L.; Lambrecht, D.S.; DiStasio, R.A.; et al. Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. [Google Scholar] [CrossRef]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef]
- Jing, Z.; Qi, R.; Liu, C.; Ren, P. Study of Interactions between Metal Ions and Protein Model Compounds by Energy Decomposition Analyses and the AMOEBA Force Field. J. Chem. Phys. 2017, 147, 161733. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, C.; Jing, Z.; Wu, C.; Piquemal, J.-P.; Ponder, J.W.; Ren, P. AMOEBA Polarizable Atomic Multipole Force Field for Nucleic Acids. J. Chem. Theory Comput. 2018, 14, 2084–2108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, T.; Li, L.; Zhang, D. Effects of PHB and SFC Charge Models on the Side Chain-Side Chain Interactions in the Simulation of β-Hairpins. Chem. Phys. Lett. 2019, 736, 136801. [Google Scholar] [CrossRef]
- Yang, S.; Onuchic, J.N.; García, A.E.; Levine, H. Folding Time Predictions from All-Atom Replica Exchange Simulations. J. Mol. Biol. 2007, 372, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Principles of Nucleic Acid Structure; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 978-0-12-369507-9.
- Sharma, M.; Mondal, D.; Singh, N.; Trivedi, N.; Bhatt, J.; Prasad, K. High Concentration DNA Solubility in Bio-Ionic Liquids with Long-Lasting Chemical and Structural Stability at Room Temperature. RSC Adv. 2015, 5, 40546–40551. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Jena, S.; Dutta, J.; Chakrabarty, S.; Biswal, H.S. Critical Assessment of the Interaction between DNA and Choline Amino Acid Ionic Liquids: Evidences of Multimodal Binding and Stability Enhancement. ACS Cent. Sci. 2018, 4, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Tulsiyan, K.D.; Jena, S.; González-Viegas, M.; Kar, R.K.; Biswal, H.S. Structural Dynamics of RNA in the Presence of Choline Amino Acid Based Ionic Liquid: A Spectroscopic and Computational Outlook. ACS Cent. Sci. 2021, 7, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.H.; Olsen, K.A.; Fesinmeyer, R.M.; Tan, X.; Hudson, F.M.; Eidenschink, L.A.; Farazi, S.R. Minimization and Optimization of Designed β-Hairpin Folds. J. Am. Chem. Soc. 2006, 128, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, S.; Satheesh Kumar, P.; Rajan, S.S.; Gautham, N. Structure of d(TGCGCA)2 at 293 K: Comparison of the Effects of Sequence and Temperature. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1381–1384. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.-H.; Duran, A.; Lagardère, L.; Ponder, J.W.; Ren, P.; Piquemal, J.-P. Raising the Performance of the Tinker-HP Molecular Modeling Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 10419. [Google Scholar] [CrossRef]
- Tung, H.-J.; Pfaendtner, J. Kinetics and Mechanism of Ionic-Liquid Induced Protein Unfolding: Application to the Model Protein HP35. Mol. Syst. Des. Eng. 2016, 1, 382–390. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, S.; Bodo, E. Solvation of Model Biomolecules in Choline-Aminoate Ionic Liquids: A Computational Simulation Using Polarizable Force Fields. Molecules 2024, 29, 1524. https://doi.org/10.3390/molecules29071524
Russo S, Bodo E. Solvation of Model Biomolecules in Choline-Aminoate Ionic Liquids: A Computational Simulation Using Polarizable Force Fields. Molecules. 2024; 29(7):1524. https://doi.org/10.3390/molecules29071524
Chicago/Turabian StyleRusso, Stefano, and Enrico Bodo. 2024. "Solvation of Model Biomolecules in Choline-Aminoate Ionic Liquids: A Computational Simulation Using Polarizable Force Fields" Molecules 29, no. 7: 1524. https://doi.org/10.3390/molecules29071524
APA StyleRusso, S., & Bodo, E. (2024). Solvation of Model Biomolecules in Choline-Aminoate Ionic Liquids: A Computational Simulation Using Polarizable Force Fields. Molecules, 29(7), 1524. https://doi.org/10.3390/molecules29071524