
A Paternò–Büchi Reaction of Aromatics with Quinones under Visible Light Irradiation

 , and
, and
Abstract
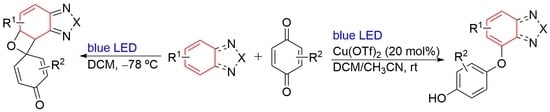
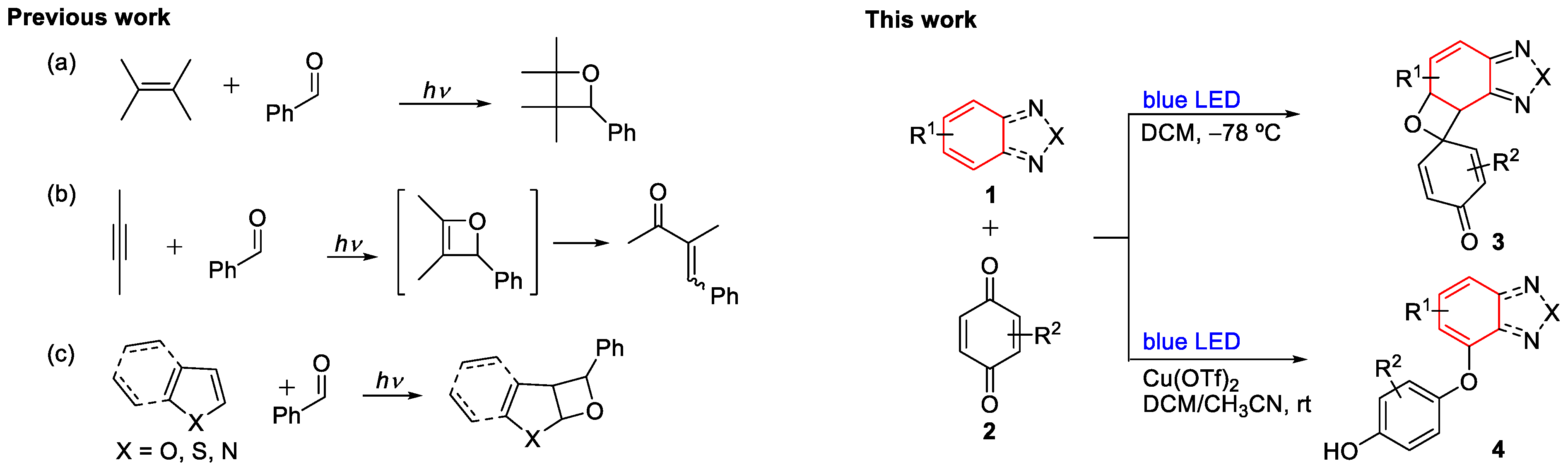
1. Introduction
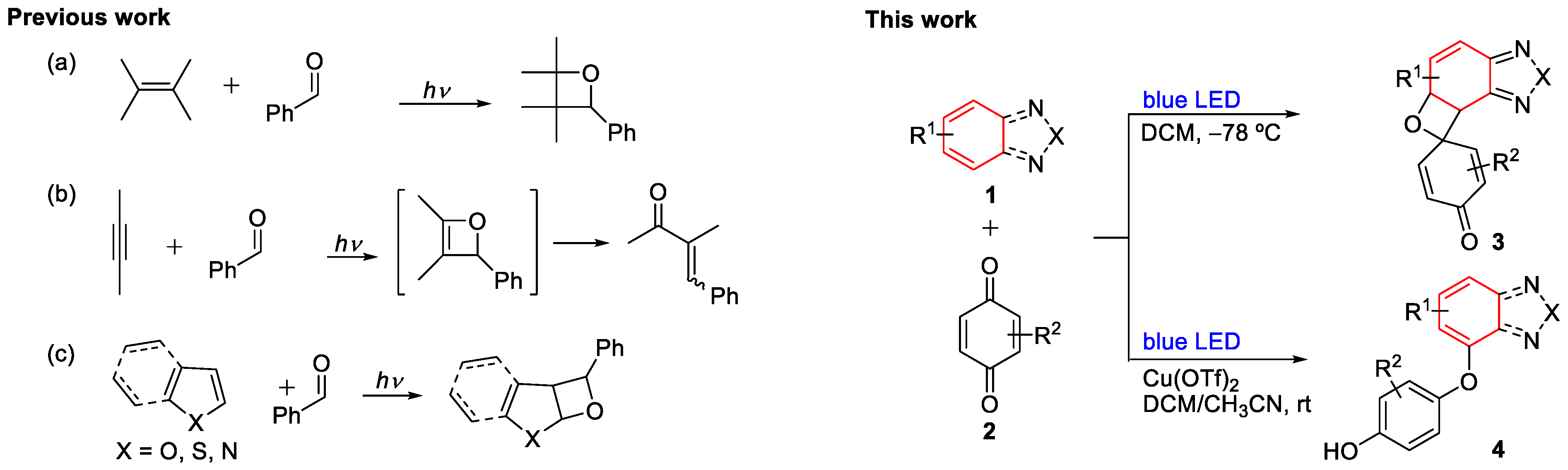
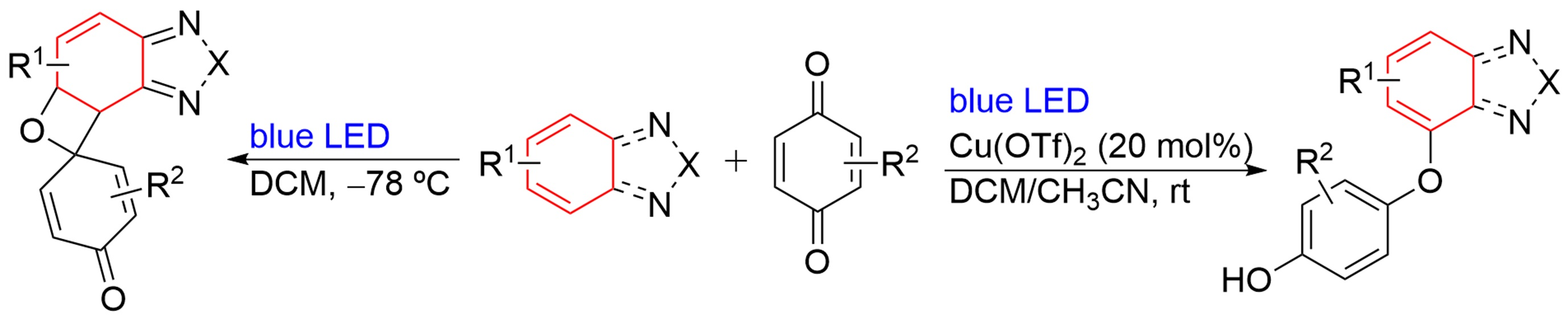
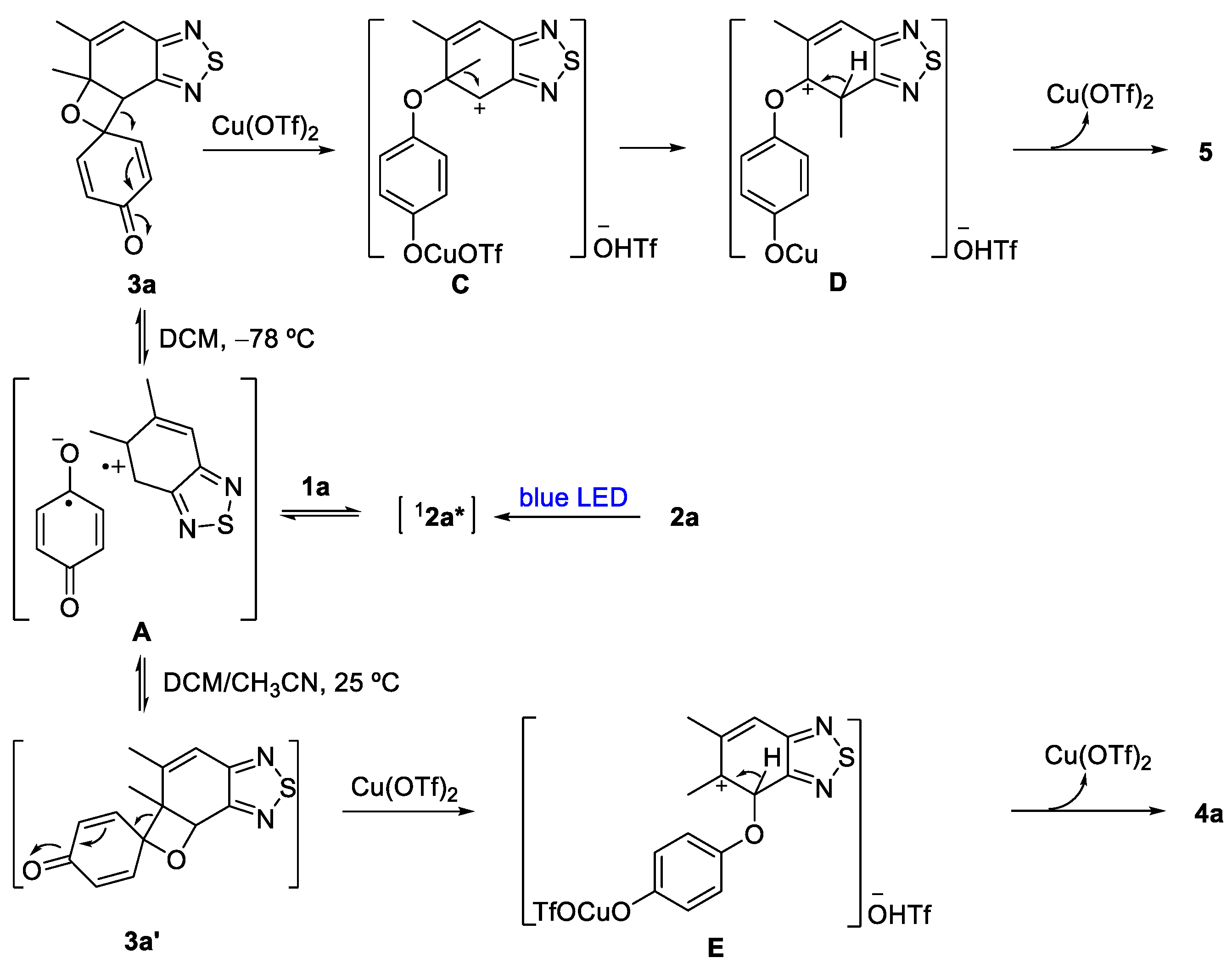
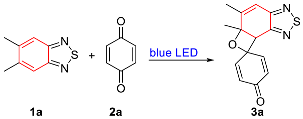
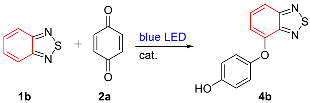
2. Results
3. Experimental Section
3.1. Synthetic Procedures
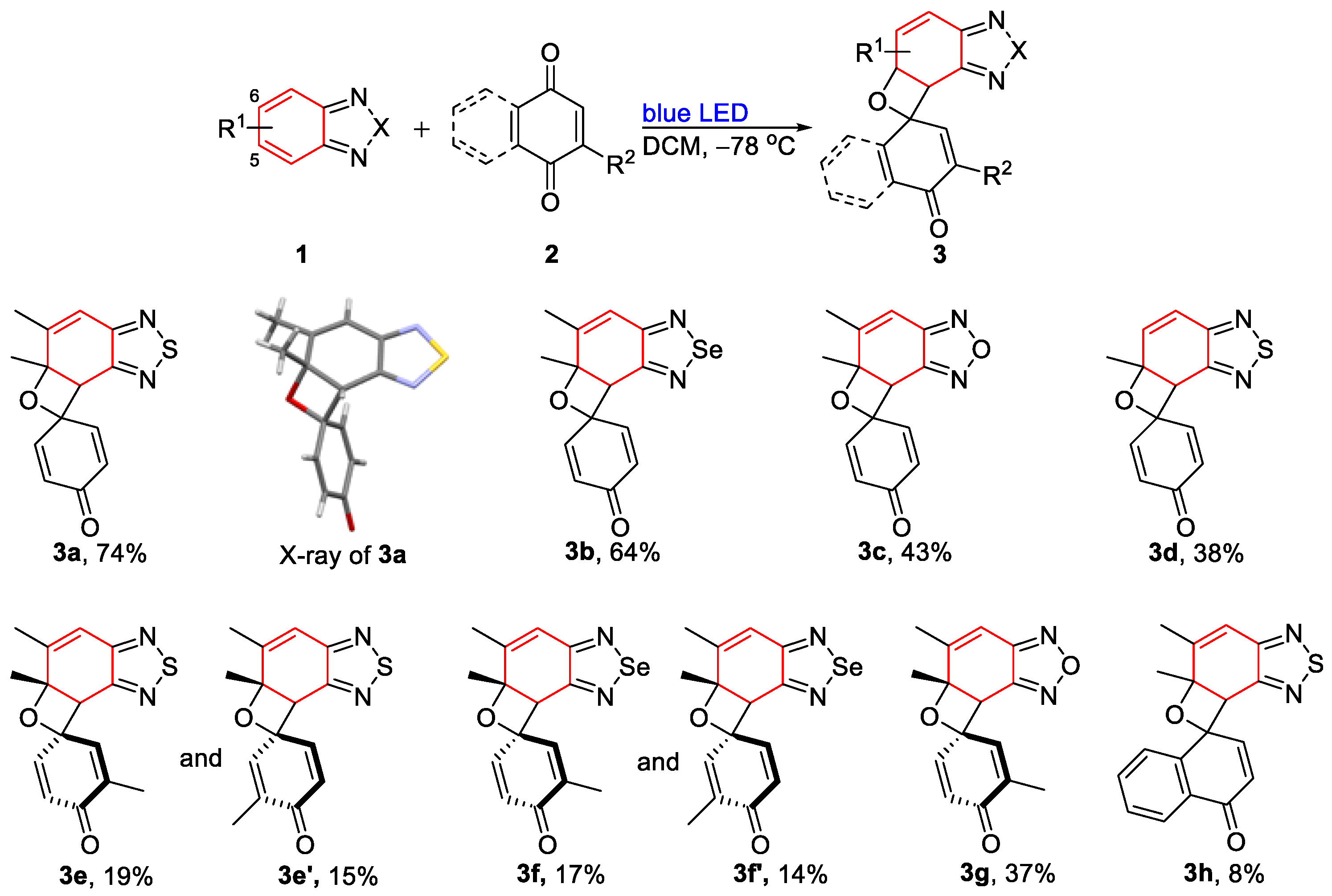
3.1.1. General Process I: Synthesis of Oxetanes (3a–3h)
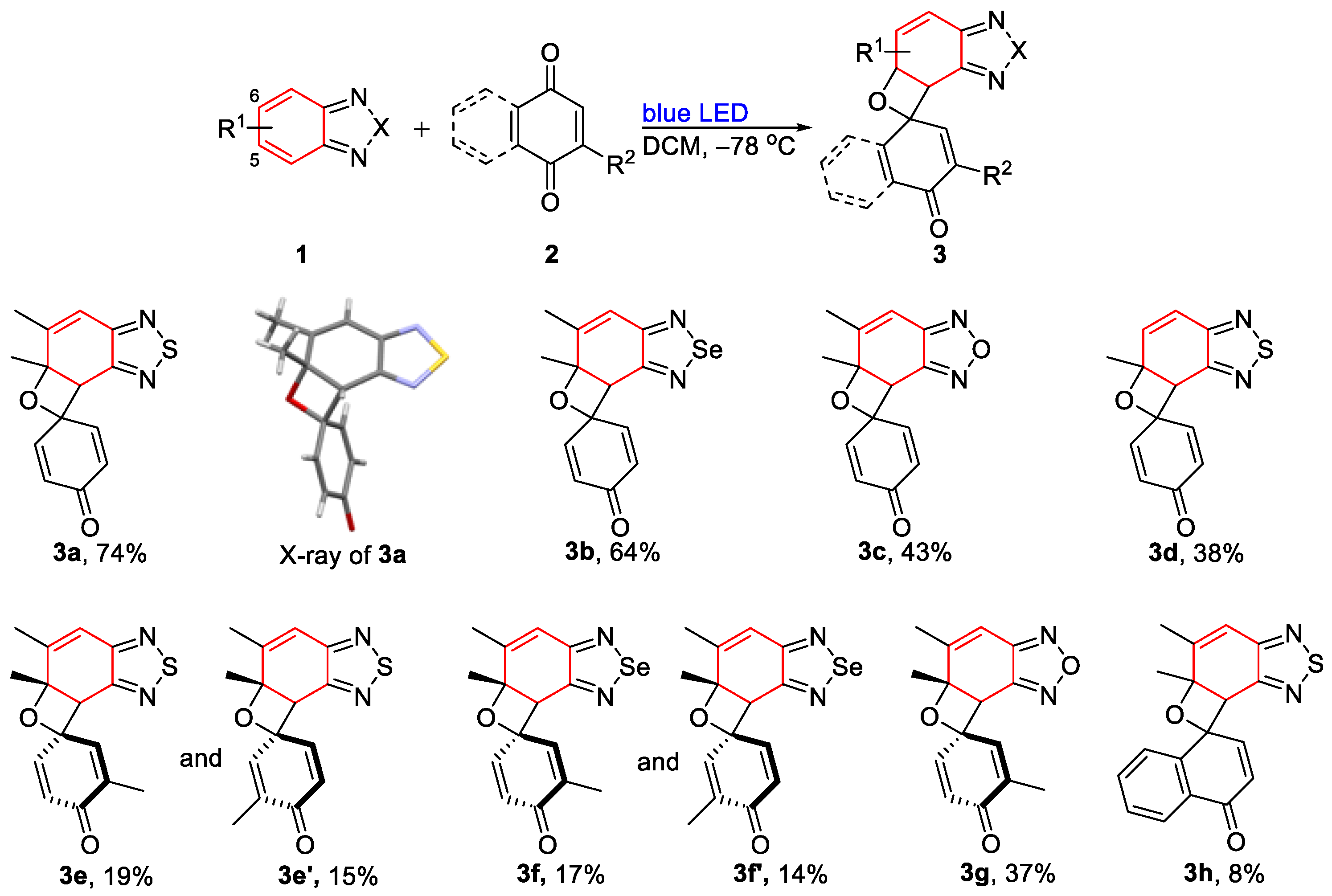
- 5′,5a′-Dimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3a). Colorless solid (61 mg, 74%); M.p. 108–110 °C; 1H NMR (600 MHz, CDCl3) δ 7.16 (dd, J = 10.1 Hz, 3.0 Hz, 1H), 6.84 (dd, J = 10.2 Hz, 2.9 Hz, 1H), 6.64 (s, 1H), 6.21 (d, J = 10.0 Hz, 1H), 5.98 (d, J = 10.3 Hz, 1H), 4.44 (s, 1H), 2.06 (s, 3H), 1.84 (d, J = 0.7 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 184.2 (C), 155.7 (C), 153.5 (C), 147.7 (CH), 146.7 (C), 144.6 (CH), 129.7 (CH), 128.4 (CH), 118.3 (CH), 82.6 (C), 78.8 (C), 50.6 (CH), 28.8 (CH3), 16.6 (CH3); HRMS (ESI) m/z calcd for C14H13N2O2S [M + H]+: 273.0692, found: 273.0694.
- 5′,5a′-Dimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]selenadiazole]-2,5-dien-4-one (3b). Colorless solid (61 mg, 64%); M.p. 107–109 °C; 1H NMR (400 MHz, CDCl3) δ 7.15 (dd, J = 10.0 Hz, 3.2 Hz, 1H), 6.84 (dd, J = 10.0 Hz, 3.2 Hz, 1H), 6.65 (d, J = 1.6 Hz, 1H), 6.20 (dd, J = 10.0 Hz, 2.0 Hz, 1H), 5.97 (dd, J = 10.4 Hz, 2.0 Hz, 1H), 4.43 (s, 1H), 2.04 (d, J = 1.6 Hz, 3H), 1.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 184.4 (C), 159.7 (C), 158.4 (C), 148.0 (CH), 147.3 (C), 144.7 (CH), 130.0 (CH), 128.7 (CH), 122.0 (CH), 82.8 (C), 78.7 (C), 53.7 (CH), 28.7 (CH3), 16.7 (CH3); HRMS (ESI) m/z calcd for C14H13N2O2Se [M + H]+: 321.0137, found: 321.0135.
- 5′,5a′-Dimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]oxadiazole]-2,5-dien-4-one (3c). Colorless solid (33 mg, 43%); M.p. 113–115 °C; 1H NMR (400 MHz, CDCl3) δ 7.15 (dd, J = 10.1 Hz, 2.8 Hz, 1H), 6.75 (dd, J = 10.4 Hz, 3.2 Hz, 1H), 6.62 (d, J = 1.6 Hz, 1H), 6.21 (dd, J = 10.2 Hz, 2.0 Hz, 1H), 6.05 (dd, J = 10.0 Hz, 2.0 Hz, 1H), 4.46 (s, 1H), 2.07 (d, J = 1.6 Hz, 3H), 1.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 184.0 (C), 149.8 (C), 148.8 (C), 147.1 (CH), 146.9 (C), 143.6 (CH), 130.4 (CH), 129.1 (CH), 110.4 (CH), 81.1 (C), 77.6 (C), 44.4 (CH), 28.5 (CH3), 17.2 (CH3); HRMS (ESI) m/z calcd for C14H13N2O3 [M + H]+: 257.0921, found: 257.0920.
- 5a′-Methyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3d). Light-yellow liquid (30 mg, 38%); 1H NMR (400 MHz, CDCl3) δ 7.18 (dd, J = 10.0 Hz, 3.2 Hz, 1H), 6.87 (d, J = 10.0 Hz, 1H), 6.76 (dd, J = 10.0 Hz, 2.8 Hz, 1H), 6.29 (d, J = 10.0 Hz, 1H), 6.20 (dd, J = 10.0 Hz, 2.0 Hz, 1H), 5.97 (dd, J = 10.4 Hz, 2.0 Hz, 1H), 4.46 (s, 1H), 1.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 184.3 (C), 154.8 (C), 154.2 (C), 147.6 (CH), 144.6 (CH), 137.5 (CH), 129.9 (CH), 128.4 (CH), 121.4 (CH), 80.9 (C), 79.8 (C), 49.9 (CH), 30.3 (CH3); HRMS (ESI) m/z calcd for C13H11N2O2S [M + H]+: 259.0536, found: 259.0535.
- 3,5′,5a′-Trimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3e). Light-yellow liquid (13 mg, 15%); 1H NMR (400 MHz, CDCl3) δ 6.93 (d, J = 1.6 Hz, 1H), 6.79 (dd, J = 10.0 Hz, 2.8 Hz, 1H), 6.63 (d, J = 1.2 Hz, 1H), 5.96 (d, J = 10.4 Hz, 1H), 4.42 (s, 1H), 2.05 (d, J = 1.6 Hz, 3H), 1.93 (d, J = 1.6 Hz, 3H), 1.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 185.2 (C), 155.9 (C), 154.0 (C), 147.1 (CH), 144.3 (C), 143.4 (CH), 135.9 (C), 129.9 (CH), 118.3 (CH), 82.3 (C), 79.2 (C), 50.9 (CH), 29.0 (CH3), 16.8 (CH3), 15.5 (CH3); HRMS (ESI) m/z calcd for C15H15N2O2S [M + H]+: 287.0849, found: 287.0847.
- 3,5′,5a′-Trimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3e’). Light-yellow liquid (16 mg, 19%); 1H NMR (400 MHz, CDCl3) δ 7.12 (dd, J = 10.0 Hz, 3.2 Hz, 1H), 6.65 (d, J = 1.6 Hz, 1H), 6.56 (d, J = 1.6 Hz, 1H), 6.20 (d, J = 10.0 Hz, 1H), 4.41 (s, 1H), 2.06 (d, J = 1.2 Hz, 3H), 1.82 (s, 3H), 1.67 (d, J = 1.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 185.1 (C), 155.9 (C), 154.0 (C), 147.6 (CH), 147.0 (C), 139.5 (CH), 137.2 (C), 128.5 (CH), 118.4 (CH), 82.4 (C), 79.4 (C), 50.7 (CH), 29.0 (CH3), 16.8 (CH3), 15.6 (CH3); HRMS (ESI) m/z calcd for C15H15N2O2S [M + H]+: 287.0849, found: 287.0847.
- 3,5′,5a′-Trimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3f). Light-yellow liquid (17 mg, 17%); 1H NMR (400 MHz, CDCl3) δ 6.94 (dd, J = 3.2 Hz, 1.6 Hz, 1H), 6.82 (dd, J = 10.0 Hz, 3.2 Hz, 1H), 6.66 (d, J = 1.2 Hz, 1H), 5.97 (d, J = 10.0 Hz, 1H), 4.41 (s, 1H), 2.055 (d, J = 1.6 Hz, 3H), 1.935 (d, J = 1.2 Hz, 3H), 1.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 185.1 (C), 159.8 (C), 158.7 (C), 147.6 (CH), 144.3 (CH), 143.6 (CH), 135.8 (C), 129.9 (CH), 121.8 (CH), 82.2 (C), 79.0 (C), 53.8 (CH), 28.7 (CH3), 16.7 (CH3), 15.5 (CH3); HRMS (ESI) m/z calcd for C15H15N2O2Se [M + H]+: 335.0293, found: 335.0293.
- 3,5′,5a′-Trimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3f′). Light-yellow liquid (14 mg, 14%); 1H NMR (400 MHz, CDCl3) 1H NMR (400 MHz, CDCl3) δ 7.13 (dd, J = 10.0 Hz, 2.8 Hz, 1H), 6.68 (d, J = 1.2 Hz, 1H), 6.58 (dd, J = 2.8 Hz, 1.6 Hz, 1H), 6.21 (d, J = 10.0 Hz, 1H), 4.41 (s, 1H), 2.06 (d, J = 0.8 Hz, 3H), 1.82 (d, 3H), 1.67 (d, J = 1.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 185.1 (C), 159.9 (C), 158.9 (C), 147.9 (CH), 147.4 (CH), 139.6 (CH), 137.2 (C), 128.5 (CH), 121.9 (CH), 82.4 (C), 79.2 (C), 53.5 (CH), 28.8 (CH3), 16.7 (CH3), 15.6 (CH3); HRMS (ESI) m/z calcd for C15H15N2O2Se [M + H]+: 335.0293, found: 335.0294.
- 3,5′,5a′-Trimethyl-5a′,7a′-dihydrospiro[cyclohexane-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazole]-2,5-dien-4-one (3g). Light-yellow liquid (30 mg, 39%); 1H NMR (400 MHz, CDCl3) δ 6.92 (d, J = 1.2 Hz, 1H), δ 6.72 (dd, J = 10.0 Hz, 2.8 Hz, 1H), 6.63 (s, 1H), 6.05 (d, J = 10.4 Hz, 1H), 4.44 (s, 1H), 2.07 (d, J = 1.3 Hz, 3H), 1.93 (s, 3H), 1.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 184.8 (C), 15.0 (C), 148.9 (C), 147.2 (CH), 143.2 (C), 142.6 (CH), 136.5 (C), 130.4 (CH), 110.4 (CH), 80.7 (C), 78.1 (C), 44.7 (CH), 28.6 (CH3), 17.2 (CH3), 15.5 (CH3); HRMS (ESI) m/z calcd for C15H14N2NaO3 [M + Na]+: 293.0897, found: 293.0896.
- 5′,5a′-Dimethyl-5a′,7a′-dihydro-4H-spiro[naphthalene-1,7′-oxeto[3′,2′:3,4]benzo[1,2-c][1,2,5]thiadiazol]-4-one (3h). Light-brown solid (8 mg, 8%); M.p. 119–121 °C; 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 8.0 Hz 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 10.4 Hz, 1H), 6.69 (s, 1H), 6.11 (d, J = 10.4 Hz, 1H), 4.57 (s, 1H), 2.16 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 183.3 (C), 155.8 (C), 153.8 (C), 147.4 (C), 146.3 (CH), 143.5 (C), 133.6 (CH), 130.0 (CH), 129.8 (C), 129.1 (CH), 126.7 (CH), 126.4 (CH), 118.4 (CH), 82.2 (C), 81.3 (C), 56.4 (CH), 27.7 (CH3), 16.8 (CH3); HRMS (ESI) m/z calcd for C18H15N2O2S [M + H]+: 323.0849, found: 323.0847.
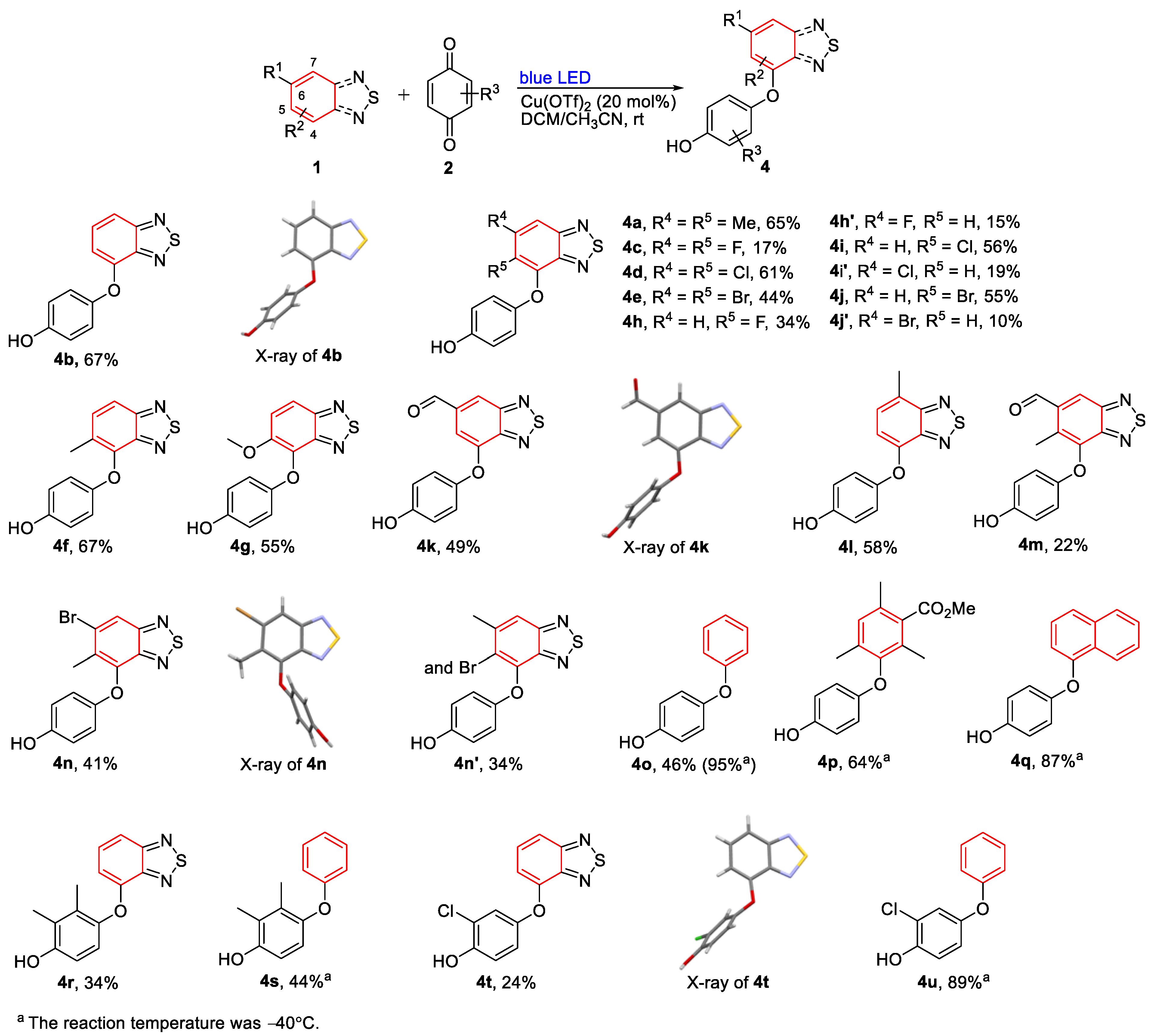
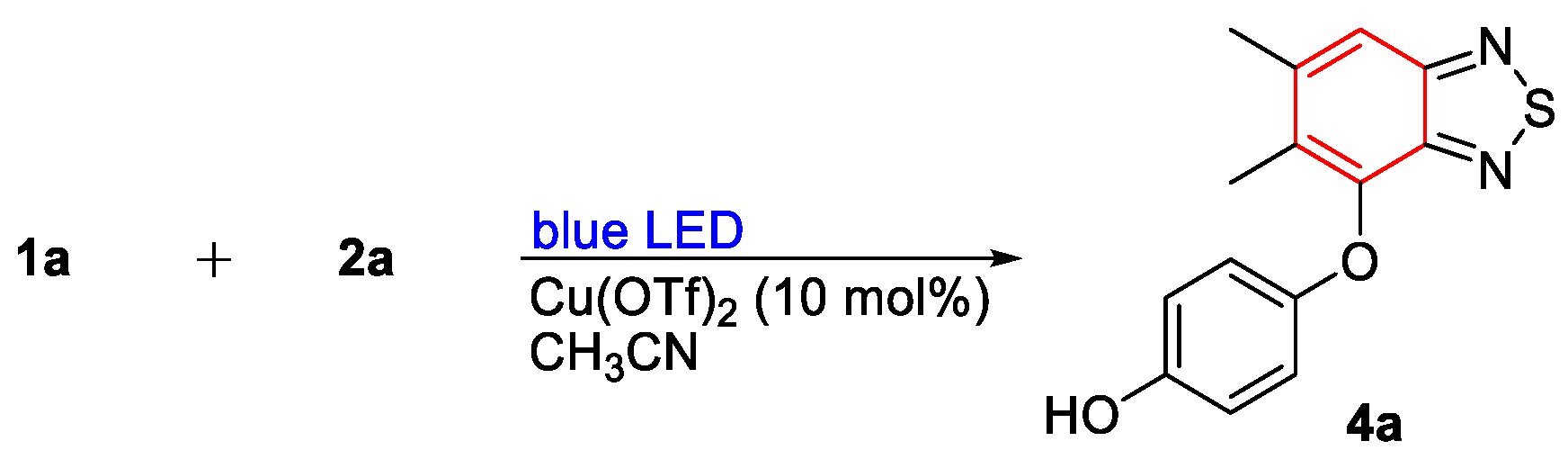
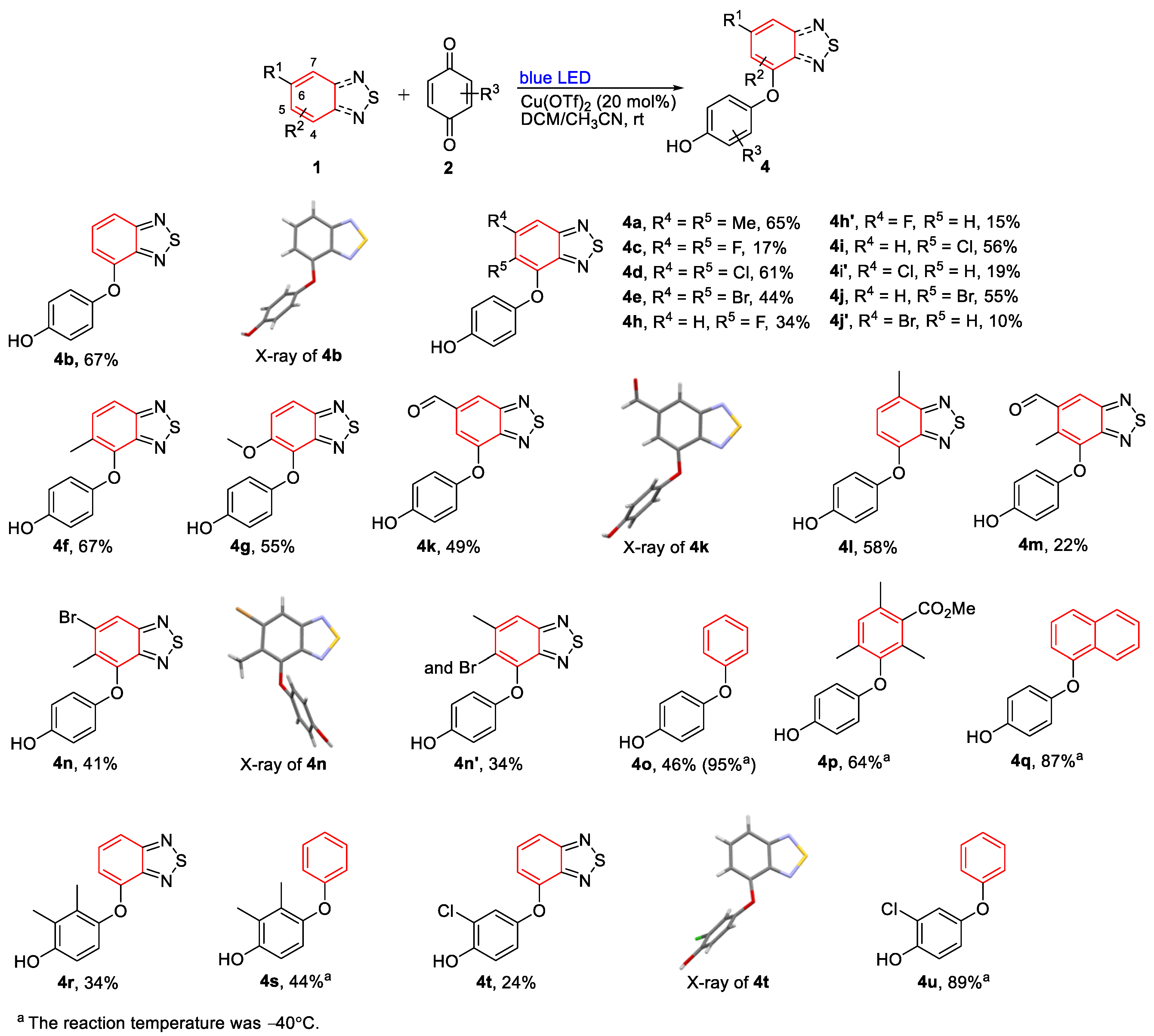
3.1.2. General Process II: Synthesis of Diaryl Ethers (4a–4n)
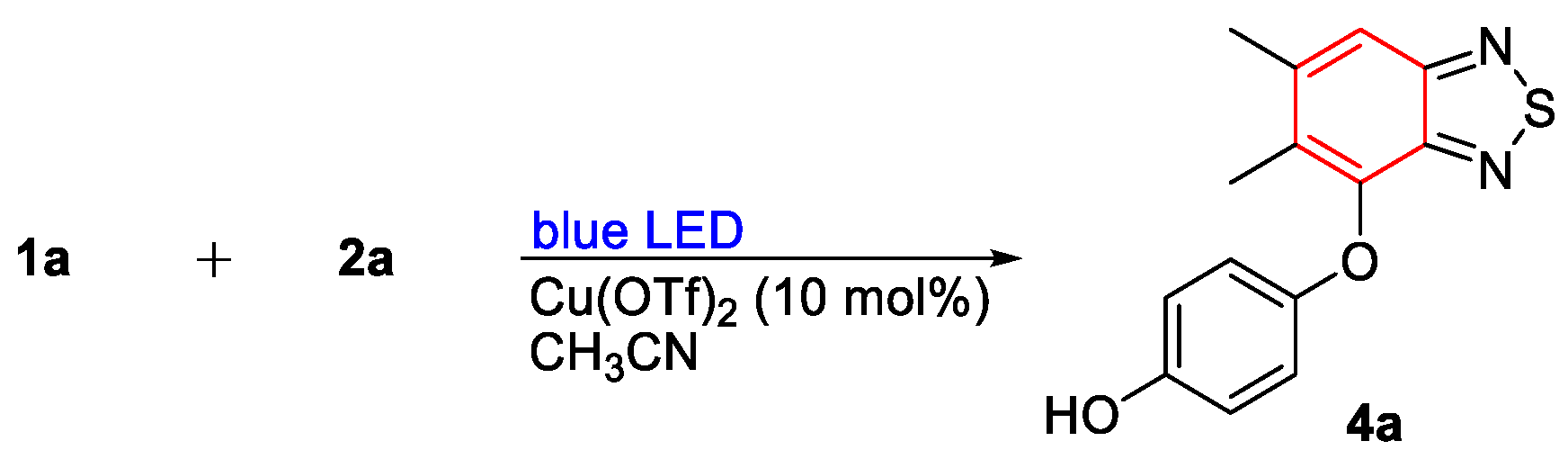
- 4-((5,6-Dimethylbenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4a). Light-yellow solid (53 mg, 65%); M.p. 187–188 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.05 (s, 1H), 7.71 (s, 1H), 6.76–6.71 (m, 4H), 2.523 (d, J = 0.8 Hz, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 156.1 (C), 153.3 (C), 152.4 (C), 149.7 (C), 143.8 (C), 143.0 (C), 132.0 (C), 117.5 (CH), 117.2 (CH), 116.7 (CH), 21.2 (CH3), 12.9 (CH3); HRMS (ESI) m/z calcd for C14H13N2O2S [M + H]+: 273.0692, found: 273.0692.
- 4-(Benzo[c][1,2,5]thiadiazol-4-yloxy)phenol (4b). Green solid (49 mg, 67%); M.P. 178–179 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.43 (s, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.60–7.56 (m, 1H), 7.09–7.07 (m, 2H), 6.95–6.93 (m, 2H), 6.77 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CD3COCD3) δ 157.6 (C), 155.6 (C), 151.9 (C), 149.1 (C), 148.6 (C), 131.2 (CH), 122.4 (CH), 117.3 (CH), 115.5 (CH), 111.6 (CH); HRMS (ESI) m/z calcd for C12H9N2O2S [M + H]+: 245.0379, found: 245.0378.
- 4-((5,6-Difluorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4c). Colorless solid (14 mg, 17%); M.P. 168–169 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.23 (s, 1H), 7.90–7.85 (m, 1H), 6.99 (d, J = 12.8 Hz, 2H), 6.80 (d, J = 10.8 Hz, 2H); 13C NMR (100 MHz, CD3COCD3) δ 155.5 (C, dd, J = 253.0 Hz, 16.0 Hz), 154.6 (C), 151.7 (C), 151.5 (C, d, J = 14.0 Hz), 148.0 (C), 146.5 (C, dd, J = 256.0 Hz, 20.0 Hz), 133.5 (C, d, J = 16.0 Hz), 118.5 (CH), 116.8 (CH), 103.0 (CH, d, J = 21.0 Hz); 19F NMR (100 MHz, CD3COCD3) δ −129.7 (F, dd, J = 4.6 Hz, 2.7 Hz), −149.8 (F, dd, J = 4.5 Hz, 1.8 Hz); HRMS (ESI) m/z calcd for C12H7F2N2O2S [M + H]+: 281.0191, found: 281.0190.
- 4-((5,6-Dichlorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4d). Green solid (57 mg, 61%); M.P. 182–183 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.25 (s, 1H), 8.22 (s, 1H), 6.88 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 9.2 Hz, 2H); 13C NMR (100 MHz, CD3COCD3) δ 154.7 (C), 154.2 (C), 151.4 (C), 149.2 (C), 144.7 (C), 135.8 (C), 127.0 (C), 118.8 (CH), 118.1 (CH), 116.7 (CH); HRMS (ESI) m/z calcd for C12H7Cl2N2O2S [M + H]+: 312.9600, found: 312.9591.
- 4-((5,6-Dibromobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4e). Yellow solid (53 mg, 44%); M.P. 183–184 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.41 (s, 1H), 8.21 (s, 1H), 6.85 (d, J = 9.2 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CD3COCD3) δ 156.0 (C), 154.2 (C), 151.4 (C), 149.4 (C), 146.0 (C), 127.9 (C), 122.3 (CH), 120.5 (C), 118.1 (CH), 116.7 (CH); HRMS (ESI) m/z calcd for C12H7Br2N2O2S [M + H]+: 402.8569, found: 402.8567.
- 4-((5-Methylbenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4f). Colorless solid (52 mg, 67%); M.P. 133–134 °C; 1H NMR (400 MHz, CDCl3), δ 7.79 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 9.2 Hz, 1H), 6.76–6.71 (m, 4H), 5.05 (s, 1H) 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.5 (C), 152.0 (C), 150.7 (C), 149.9 (C), 143.1 (C), 133.8 (CH), 130.4 (C), 117.6 (CH), 116.5 (CH), 116.2 (CH), 15.8 (CH3); HRMS (ESI) m/z calcd for C13H11N2O2S [M + H]+: 259.0536, found: 259.0534.
- 4-((5-Methoxybenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4g). Green solid (45 mg, 55%); M.P. 149–150 °C; 1H NMR (400 MHz, CDCl3), δ 7.85 (d, J = 9.2 Hz, 1H), 7.56 (d, J = 9.6 Hz, 1H), 6.86–6.82 (m, 2H), 6.75–6.71 (m, 2H), 4.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.2 (C), 152.1 (C), 151.5 (C), 150.7 (C), 132.8 (C), 121.9 (CH), 118.0 (CH), 116.7 (CH), 116.1 (CH), 58.3 (CH3); HRMS (ESI) m/z calcd for C13H11N2O3S [M + H]+: 275.0485, found: 275.0483.
- 4-((5-Fluorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4h). Yellow solid (27 mg, 34%); M.P. 159–160 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.20 (s, 1H), 7.96 (dd, J = 9.2 Hz, 4.4 Hz, 1H), 7.78 (dd, J = 10.8 Hz, 10.0 Hz, 1H), 6.94–6.90 (m, 2H), 6.81–6.77 (m, 2H); 13C NMR (100 MHz, CD3COCD3) δ 155.4 (C, d, J = 249.0 Hz), 154.2 (C), 154.1 (C), 152.1 (C), 151.3 (C, d, J = 7.0 Hz), 133.5 (C, d, J = 14.0 Hz), 122.9 (CH, d, J = 26.0 Hz), 119.0 (CH, d, J = 10.0 Hz), 118.0 (CH), 116.7 (CH); 19F NMR (100 MHz, CD3COCD3) δ −130.8 (F, d, J = 2.0 Hz); HRMS (ESI) m/z calcd for C12H8FN2O2S [M + H]+: 263.0285, found: 263.0284.
- 4-((6-Fluorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4h′). Colorless solid (12 mg, 15%); M.P. 200–202 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.59 (s, 1H), 7.38 (dd, J = 9.2 Hz, 2.4 Hz, 1H), 7.17–7.15 (m, 2H), 7.00–6.98 (m, 2H), 6.57 (dd, J = 11.2 Hz, 2.4 Hz, 1H); 13C NMR (100 MHz, CD3COCD3) δ 165.4 (C, d, J = 248.0 Hz), 156.7 (C, d, J = 14.0 Hz), 156.3 (C), 152.8 (C, d, J = 15.0 Hz), 147.7 (C), 146.7 (C), 122.8 (CH), 117.6 (CH), 102.9 (d, J = 34.0 Hz, CH), 98.7 (d, J = 25.0 Hz, CH); 19F NMR (100 MHz, CD3COCD3) δ −108.4 (F); HRMS (ESI) m/z calcd for C12H8FN2O2S [M + H]+: 263.0285, found: 263.0284.
- 4-((5-Chlorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4i). Green solid (47 mg, 56%); M.P. 154–156 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.13 (s, 1H), 7.94 (d, J = 9.2 Hz, 1H), 7.82 (d, J = 9.2 Hz, 1H), 6.86–6.82 (m, 2H), 6.79–6.75 (m, 2H); 13C NMR (100 MHz, CD3COCD3) δ 156.4 (C), 154.7 (C), 151.8 (C), 150.9 (C), 143.8 (C), 132.5 (CH), 127.6 (C), 119.3 (CH), 118.1 (CH), 116.8 (CH); HRMS (ESI) m/z calcd for C12H8ClN2O2S [M + H]+: 278.9990, found: 278.9987.
- 4-((6-Chlorobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4i′). Green solid (16 mg, 19%); M.P. 160–162 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.59 (s, 1H), 7.73 (d, J = 2 Hz, 1H), 7.18–7.13 (m, 2H), 7.00–6.97 (m, 2H), 6.64 (d, J = 2 Hz, 1H); 13C NMR (100 MHz, CD3COCD3) δ 156.9 (C), 156.2 (C), 152.2 (C), 147.8 (C), 147.7 (C), 137.3 (C), 122.7 (CH), 117.6 (CH), 114.3 (CH), 112.3 (CH); HRMS (ESI) m/z calcd for C12H8ClN2O2S [M + H]+: 278.9990, found: 278.9987.
- 4-((5-Bromobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4j). Brown solid (53 mg, 55%); M.P. 174–175 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.17 (s, 1H), 7.95 (d, J = 9.2 Hz, 1H), 7.88 (d, J = 9.2 Hz, 1H), 6.85–6.81 (m, 2H), 6.79–6.75 (m, 2H); 13C NMR (100 MHz, CD3COCD3) δ 156.8 (C), 154.0 (C), 151.7 (C), 150.8 (C), 145.2 (C), 134.9 (CH), 119.5 (CH), 118.0 (CH), 117.1 (C), 116.7 (CH); HRMS (ESI) m/z calcd for C12H8BrN2O2S [M + H]+: 322.9484, found: 322.9483.
- 4-((6-Bromobenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4j′). Light-yellow solid (10 mg, 10%); M.P. 173–174 °C; 1H NMR (400 MHz, CD3COCD3), δ 8.61 (s, 1H), 7.926 (d, J = 1.6 Hz, 1H), 7.18–7.14 (m, 2H), 7.01–6.97 (m, 2H), 6.748 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, CD3COCD3) δ 157.5 (C), 156.2 (C), 152.2 (C), 148.0 (C), 147.7 (C), 125.2 (C), 122.8 (CH), 117.7 (CH), 117.6 (CH), 114.6 (CH); HRMS (ESI) m/z calcd for C12H8BrN2O2S [M + H]+: 322.9484, found: 322.9483.
- 7-(4-Hydroxyphenoxy)benzo[c][1,2,5]thiadiazole-5-carbaldehyde (4k). Brown solid (40 mg, 49%); M.P. 199–201 °C; 1H NMR (400 MHz, CD3COCD3); δ 10.16 (s, 1H), 8.54 (s, 1H), 8.364 (d, J = 1.2 Hz, 1H), 7.17–7.13 (m, 2H), 7.105 (d, J = 1.2 Hz, 1H), 7.01–6.97 (m, 2H); 13C NMR (100 MHz, CD3COCD3) δ 192.3 (CH) 157.2 (C), 156.1 (C), 153.0 (C), 151.4 (C), 147.8 (C), 139.4 (C), 122.8 (CH), 122.3 (CH), 117.5 (CH), 105.8 (CH); HRMS (ESI) m/z calcd for C13H9N2O3S [M + H]+: 273.0328, found: 273.0327.
- 4-((7-Methylbenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4l). Yellow solid (45 mg, 58%); M.p. 204–206 °C; 1H NMR (400 MHz, CD3COCD3); δ 8.37 (s, 1H), 7.34 (d, J = 6.4 Hz, 1H), 7.04 (dd, J = 6.4 Hz, 2 Hz, 1H), 6.91 (d, J = 4.4 Hz, 1H), 6.74 (d, J = 7.6 Hz, 1H), 2.63 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 156.7 (C), 154.3 (C), 148.7 (C), 148.3 (C), 148.2 (C), 128.3 (CH), 124.6 (C), 121.0 (CH), 116.3 (CH), 111.9 (CH), 16.4 (CH3); HRMS (ESI) m/z calcd for C13H10N2O2S [M]+: 258.0457, found: 258.0461.
- 4-(4-Hydroxyphenoxy)-6-methylbenzo[c][1,2,5]thiadiazole-5-carbaldehyde (4m). Yellow solid (19 mg, 22%); M.p. 175–177 °C; 1H NMR (400 MHz, CD3COCD3); δ 10.46 (s, 1H), 8.51 (s, 1H), 8.07 (s, 1H), 6.79–6.74 (m, 4H), 2.68 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 193.5 (CH), 155.4 (C), 153.7 (C), 152.4 (C), 152.3 (C), 145.5 (C), 138.7 (C), 130.6 (C), 126.0 (CH), 117.6 (CH), 116.9 (CH), 12.5 (CH3); HRMS (ESI) m/z calcd for C14H11N2O3S [M + H]+: 287.0485, found: 287.0486.
- 4-((6-Bromo-5-methylbenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4n). Yellow solid (41 mg, 41%); M.P. 180–181 °C; 1H NMR (400 MHz, CD3COCD3); δ 8.22 (s, 1H), 6.75 (s, 4H), 4.62(s, 1H), 2.50 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 156.7 (C), 153.7 (C), 152.2 (C), 149.9 (C), 144.6 (C), 131.2 (C), 129.8 (C), 121.7 (CH), 117.6 (CH), 116.8 (CH), 16.9 (CH3); HRMS (ESI) m/z calcd for C13H10BrN2O2S [M + H]+: 336.9641, found: 336.9639.
- 4-((5-Bromo-6-methylbenzo[c][1,2,5]thiadiazol-4-yl)oxy)phenol (4n′). Colorless solid (34 mg, 34%); M.P. 173–174 °C; 1H NMR (400 MHz, CD3COCD3); δ 7.77 (s, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.8 Hz, 2H), 4.63 (s, 1H), 2.67 (d, J = 0.8 Hz, 3H); 13C NMR (100 MHz, CD3COCD3) δ 156.2 (C), 153.9 (C), 151.6 (C), 149.2 (C), 144.9 (C), 141.5 (C), 121.8 (C), 118.0 (CH), 117.9 (CH), 116.7 (CH), 24.3 (CH3);HRMS (ESI) m/z calcd for C13H10BrN2O2S [M + H]+: 336.9641, found: 336.9639.
3.1.3. General Process III: Synthesis of Diaryl Ethers (4o–4u)
- 4-Phenoxyphenol (4o) [50]. Colorless solid (54 mg, 95%); M.P. 83–84 °C; 1H NMR (400 MHz, CDCl3) δ 7.33–7.29 (m, 2H), 7.05 (t, J = 7.4 Hz, 1H), 6.96–6.93 (m, 4H), 6.84–6.80 (m, 2H), 4.85 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 158.4 (C), 151.6 (C), 150.2 (C), 129.6 (CH), 122.5 (CH), 121.0 (CH), 117.6 (CH), 116.3 (CH); HRMS (ESI) m/z calcd for C12H11O2 [M + H]+: 187.0754, found: 187.0751.
- 3-(4-Hydroxyphenoxy)-2,4,6-trimethylbenzoate (4p). Colorless solid (55 mg, 64%); M.P. 153–154 °C; 1H NMR (400 MHz, CDCl3) δ 6.93 (s, 1H), 6.69 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 8.8 Hz, 2H), 5,09 (s, 1H), 3.91 (s, 3H), 2.28 (s, 3H), 2.075 (s, 3H), 2.071 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4 (C), 151.6 (C), 150.0 (C), 149.3 (C), 133.1 (C), 131.4 (C), 130.6 (CH), 128.8 (C), 116.1 (CH), 115.3 (CH), 52.1 (CH3), 19.3(CH3), 16.4 (CH3), 13.5 (CH3); HRMS (ESI) m/z calcd for C17H19O4 [M + H]+: 287.1278, found: 287.1277.
- 4-(Naphthalen-1-yloxy)phenol (4q). Colorless liquid (61 mg, 87%); 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.58–7.52 (m, 3H), 7.35 (t, J = 8.0 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 6.85–6.81 (m, 3H), 4.32 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 154.3 (C), 151.6 (C), 150.7 (C), 134.8 (C), 127.6 (CH), 126.6 (CH), 126.3 (C), 125.8 (CH), 125.7 (CH), 122.4 (CH), 122.0 (CH), 120.7 (CH), 116.4 (CH), 111.3 (CH); HRMS (ESI) m/z calcd for C16H13O2 [M + H]+: 237.0910, found: 237.0908.
- 4-(Benzo[c][1,2,5]thiadiazol-4-yloxy)-2-methylphenol (4r). Light-yellow solid (28 mg, 34%); M.P. 142–143 °C; 1H NMR (400 MHz, CDCl3), δ 7.62 (dd, J = 8.8 Hz, 0.8 Hz, 1H), 7.41–7.37 (m, 1H), 6.87 (d, J = 8.4 Hz 1H), 6.72 (d, J = 8.8 Hz, 1H), 6.43 (dd, J = 7.6 Hz, 0.8 Hz, 1H), 5.25 (s, 1H), 2.23 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.5 (C), 151.1 (C), 150.9 (C), 147.9 (C), 146.0 (C), 130.7 (C), 130.1 (CH), 124.6 (C), 119.2 (CH), 114.2 (CH), 113.3 (CH), 109.0 (CH), 12.8 (CH3), 12.2 (CH3). HRMS (ESI) m/z calcd for C14H12N2O2S [M + H]+: 273.0692, found: 273.0692.
- 2,3-Dimethyl-4-phenoxyphenol (4s). Colorless solid (28 mg, 44%); M.P. 79–80 °C; 1H NMR (400 MHz, CDCl3) δ 7.28–7.24 (m, 1H), 6.98 (t, J = 7.6 Hz, 1H), 6.82 (d, J = 8.0 Hz, 2H), 6.74 (d, J = 8.4 Hz, 1H), 6.63 (d, J = 8.4 Hz, 1H), 4.63 (s, 1H), 2.21 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.0 (C), 150.2 (C), 147.2 (C), 130.8 (C), 129.5 (CH), 124.2 (C), 121.5 (CH), 119.0 (CH), 116.0 (CH), 113.0 (CH), 12.8 (CH3), 12.1 (CH3); HRMS (ESI) m/z calcd for C14H15O2 [M + H]+: 215.1067, found: 215.1064.
- 4-(Benzo[c][1,2,5]thiadiazol-4-yloxy)-2-chlorophenol (4t). Light-yellow solid (20 mg, 24%); M.P. 140–142 °C; 1H NMR (400 MHz, CDCl3), δ 7.71 (d, J = 8.8 Hz, 1H), 7.49–7.45 (m, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.09–7.03 (m, 2H), 6.78 (d, J = 7.2 Hz, 1H), 5.51 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 150.0 (C) δ 148.7 (C), δ 148.5 (C), δ 129.8 (CH), δ 121.0 (CH), δ 120.7 (CH), δ 120.2 (C), δ 117.0 (CH), δ 115.8 (CH), δ 111.5 (CH); HRMS (ESI) m/z calcd for C12H8ClN2O2S [M + H]+: 278.9990, found: 278.9986.
- 2-Chloro-4-phenoxyphenol (4u) [51]. Red liquid (59 mg, 89%); 1H NMR (400 MHz, CDCl3) δ 7.36–7.32 (m, 2H), 7.11 (t, J = 7.2 Hz, 1H), 7.055 (d, J = 2.4 Hz, 1H), 7.03–6.97 (m, 3H), 6.915 (dd, J = 8.8 Hz, 2.8 Hz, 1H), 5.48 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 157.6 (C), 150.2 (C), 147.6 (C), 129.7 (CH), 123.1 (CH), 120.0 (CH), 119.97 (CH), 119.7 (C), 117.9 (CH), 116.7 (CH); HRMS (ESI) m/z calcd for C12H10ClO2 [M + H]+: 221.0364, found: 221.0360.
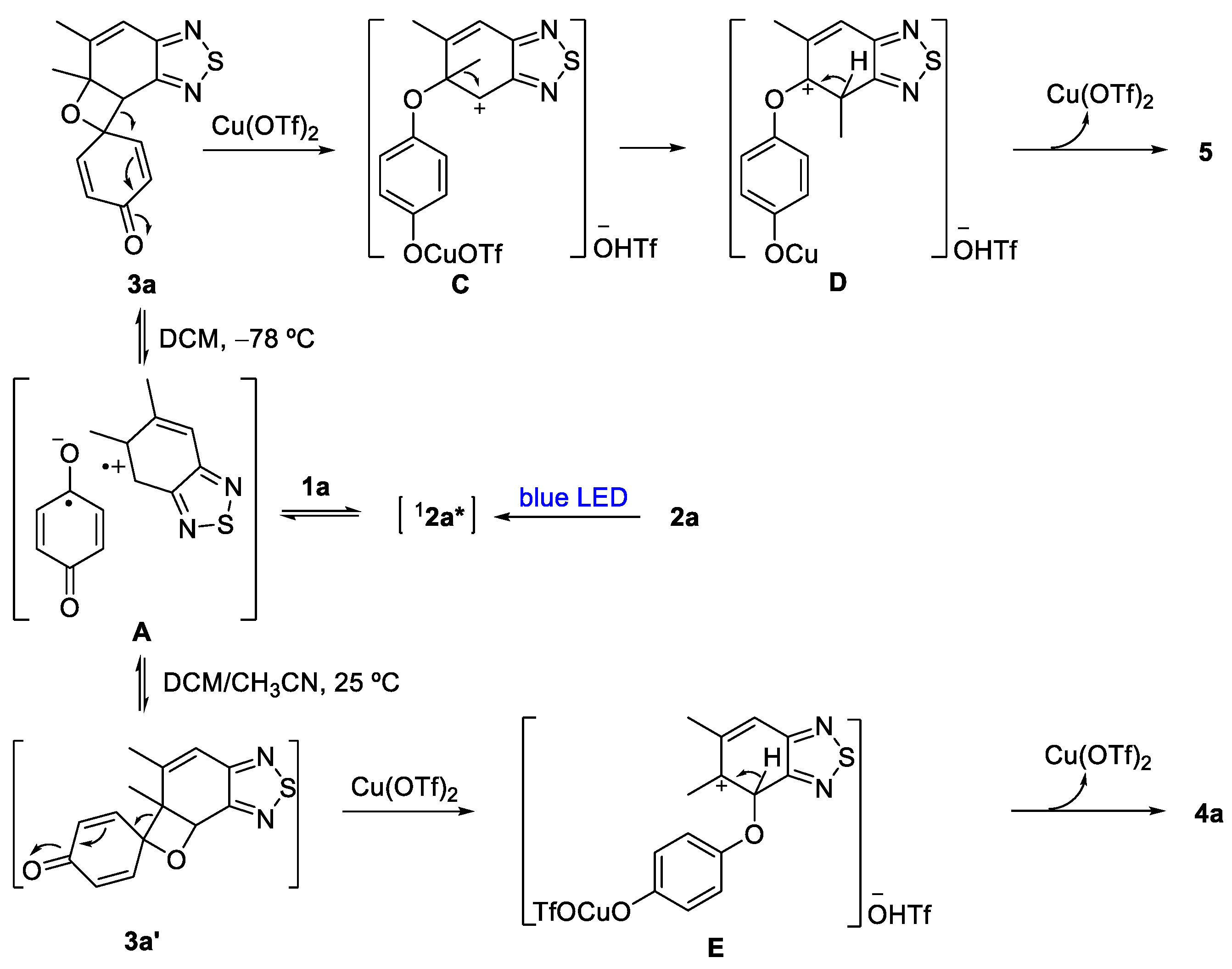
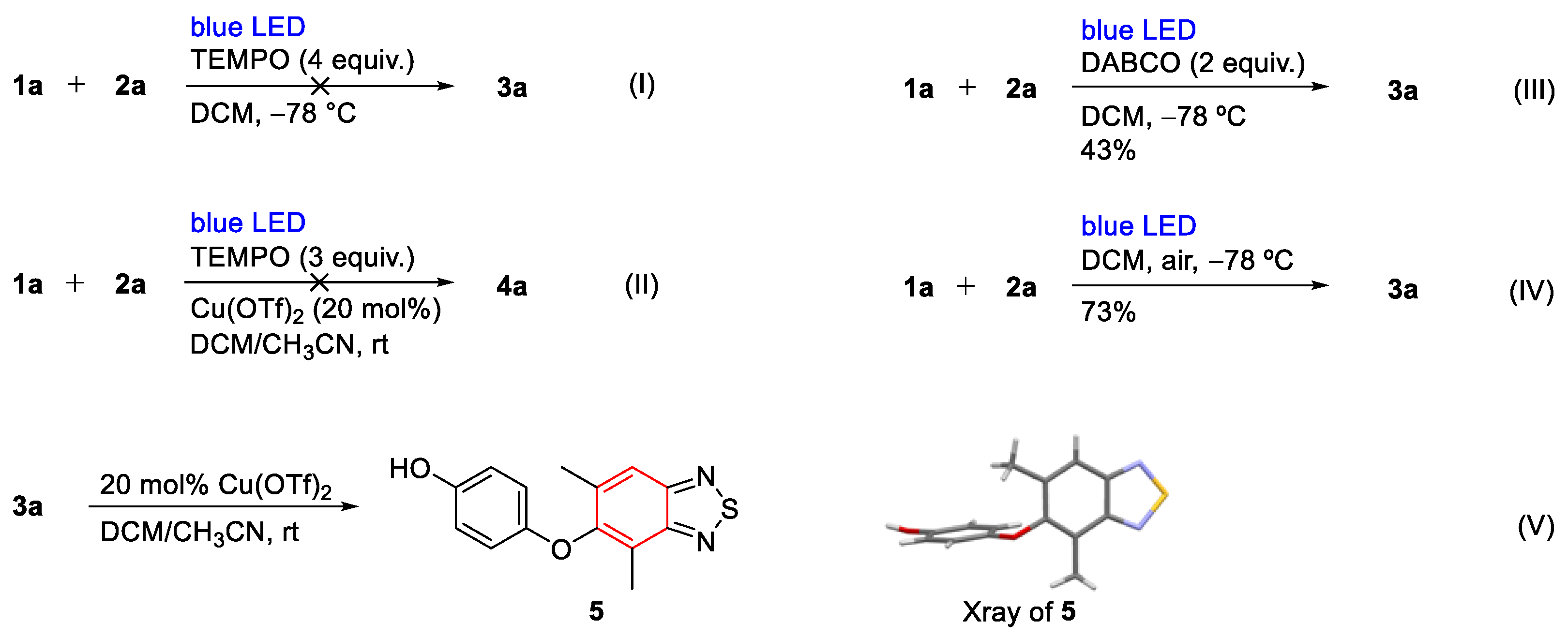
3.1.4. Synthesis of 5
- 4-((4,6-Dimethylbenzo[c][1,2,5]thiadiazol-5-yl)oxy)phenol (5). Compound 3a (82 mg, 0.30 mmol) was dissolved in a mixed solvent of acetonitrile and dichloromethane and then copper triflate (22 mg, 0.06 mmol) was added in an argon atmosphere. After TLC monitoring, the raw material disappeared. Then, the solvent was directly removed for column chromatography (PE/EtOAc = 10:1). Green solid (8 mg, 10%); M.P. 142–144 °C; 1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H), 6.75 (d, J = 8.8 Hz, 2H),6.69 (d, J = 8.8 Hz, 2H), 4.71 (s, 1H), 2.53 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.4, 152.7, 152.4, 151.7, 150.4, 137.2, 122.4, 118.6, 116.4, 115.6, 18.2, 11.9; HRMS (ESI) m/z calcd for C14H13N2O2S [M + H]+: 273.0692, found: 273.0692.
3.2. X-ray Crystallographic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimada, N.; Hasegawa, S.; Harada, T.; Tomisawa, T.; Fujii, A.; Takita, T. Oxetanocin, a novel nucleoside from bacteria. J. Antibiot. 1986, 39, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.S.; Hamann, P.R.; Still, W.C. Synthesis of thromboxane A2. J. Am. Chem. Soc. 1985, 107, 6372–6376. [Google Scholar] [CrossRef]
- Omura, S.; Murata, M.; Imamura, N.; Iwai, Y.; Tanaka, H.; Furusaki, A.; Matsumoto, H. Oxetin, a new antimetabolite from an actinomycete fermentation, isolation, structure and biological activity. J. Antibiot. 1984, 37, 1324–1332. [Google Scholar] [CrossRef]
- Loh, J.; Carlson, R.W.; York, W.S.; Stacey, G. Bradyoxetin, a unique chemical signal involved in symbiotic gene regulation. Proc. Natl. Acad. Sci. USA 2002, 99, 14446–14451. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-P.; Shu, Y.; Liu, S.-X.; Hu, J.-T.; Sun, C.-T.; Zhou, H.; Gan, D.; Cai, X.-Y.; Pu, W.; Cai, L.; et al. Expanstines A–D: Four unusual isoprenoid epoxycyclohexenones generated by Penicillium expansum YJ-15 fermentation and photopromotion. Org. Chem. Front. 2019, 6, 3839–3846. [Google Scholar] [CrossRef]
- Becker, M.R.; Wearing, E.R.; Schindler, C.S. Synthesis of azetidines via visible-light-mediated intermolecular [2+2] photocycloadditions. Nat. Chem. 2020, 12, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.D.; Becker, M.R.; Schindler, C.S. Synthesis of azetidines by aza Paternò-Büchi reactions. Chem. Sci. 2020, 11, 7553–7561. [Google Scholar] [CrossRef]
- Kandappa, S.K.; Valloli, L.K.; Ahuja, S.; Parthiban, J.; Sivaguru, J. Taming the excited state reactivity of imines—From non-radiative decay to aza Paternò-Büchi reaction. Chem. Soc. Rev. 2021, 50, 1617–1641. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Grosskopf, J.; Jandl, C.; Bach, T. Enantioselective, visible light mediated aza Paternò-Büchi reactions of quinoxalinones. Angew. Chem. Int. Ed. 2021, 60, 2684–2688. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M. The Paternò-Büchi reaction–a comprehensive review. Photochem. Photobiol. Sci. 2019, 18, 2297–2362. [Google Scholar] [CrossRef]
- Flores, D.M.; Schmidt, V.A. Intermolecular 2 + 2 Carbonyl-Olefin Photocycloadditions Enabled by Cu(I)-Norbornene MLCT. J. Am. Chem. Soc. 2019, 141, 8741–8745. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, P.; Mateos, J.; Vega-Penaloza, A.; Dell’Amico, L. Microfluidic visible-light Paternò-Büchi reaction of oxindole enol ethers. Eur. J. Org. Chem. 2020, 2020, 6718–6722. [Google Scholar] [CrossRef]
- Fréneau, M.; Hoffmann, N. The Paternò-Büchi reaction–Mechanisms and application to organic synthesis. J. Photochem. Photobiol. C Photochem. Rev. 2017, 33, 83–108. [Google Scholar] [CrossRef]
- Buchi, G.; Ihman, C.G.; Lipinsky, E.S. Light-catalyzed organic reactions. I. The reaction of carbonyl compounds with 2-methyl-2-butene in the presence of ultraviolet light. J. Am. Chem. Soc. 1954, 76, 4327–4331. [Google Scholar] [CrossRef]
- Paterno, E.; Chieffi, G. Sintesi in chimica organica per mezzo della luce. Composti degli idrocarbun non saturi con aldeidi e chetoni. Gazz. Chim. Ital. 1909, 39, 341–361. [Google Scholar]
- Friedrich, L.E.; Bower, J.D. Detection of an oxetene intermediate in the photoreaction of benzaldehyde with 2-butyne. J. Am. Chem. Soc. 1973, 95, 6869–6870. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R.; Romaniello, G. The Paternò-Büchi reaction of 2-furylmethanols. Eur. J. Org. Chem. 2000, 2000, 3265–3272. [Google Scholar] [CrossRef]
- D’Auria, M.; Racioppi, R. Paterno-Büchi reaction on 5-methyl-2-furylmethanol derivatives. Arkivoc 2000, 2000, 133–140. [Google Scholar] [CrossRef]
- Vargas, F.; Rivas, C.; Navarro, M.; Alvarado, Y. Synthesis of an elusive oxetane by photoaddition of benzophenone to thiophene in the presence of a Lewis acid. J. Photochem. Photobiol. A Chem. 1996, 93, 169–171. [Google Scholar] [CrossRef]
- Capozzo, M.; D’Auria, M.; Emanuele, L.; Racioppi, R. Synthesis and photochemical reactivity towards the Paternò-Büchi reaction of benzo[b]furan derivatives: Their use in the preparation of 3-benzofurylmethanol derivatives. J. Photochem. Photobiol. A Chem. 2007, 185, 38–43. [Google Scholar] [CrossRef]
- Rivas, C.; Velez, M.; Crescente, O. Synthesis of an oxetan by photoaddition of benzophenone to a thiophen derivative. Chem. Commun. 1970, 1474. [Google Scholar] [CrossRef]
- Jones, G.; Gilow, H.M.; Low, J. Regioselective photoaddition of pyrroles and aliphatic carbonyl compounds. A new synthesis of 3(4)-substituted pyrroles. J. Org. Chem. 1979, 44, 2949–2951. [Google Scholar] [CrossRef]
- Rivas, C.; Bolivar, R.A. Synthesis of oxetanes by photoaddition of carbonyl compounds to pyrrole derivatives. J. Heterocycl. Chem. 1976, 13, 1037–1040. [Google Scholar] [CrossRef]
- Matsuura, T.; Banba, A.; Ogura, K. Photoinduced reactions–XLV: Photoaddition of ketones to methylimidazoles. Tetrahedron 1971, 27, 1211–1219. [Google Scholar] [CrossRef]
- Nakano, T.; Rodriguez, W.; De Roche, S.Z.; Larrauri, J.M.; Rivas, C.; Perez, C. Photoaddition of ketones to imidazoles, thiazoles, isothiazoles and isoxazoles. Synthesis of their oxetanes. J. Heterocycl. Chem. 1980, 17, 1777–1780. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Fiege, M.; Lex, J. Oxazole–Carbonyl photocycloadditions: Selectivity pattern and synthetic route to erythro α-amino, β-hydroxy ketones. Chem. Commun. 2000, 7, 589–590. [Google Scholar] [CrossRef]
- Mateos, J.; Vega-Penaloza, A.; Franceschi, P.; Rigodanza, F.; Andreetta, P.; Companyo, X.; Pelosi, G.; Bonchio, M.; Dell’Amico, L. A visible-light Paternò-Büchi dearomatisation process towards the construction of oxeto-indolinic polycycles. Chem. Sci. 2020, 11, 6532–6538. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Stein, P.M.; Shi, H.; Hu, C.; Rudolph, M.; Hashmi, A.S.K. Hydroxylamine-mediated C–C amination via an aza-hock rearrangement. Nat. Commun. 2021, 12, 7029. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hoffmann, M.; Dreuw, A.; Hasagić, E.; Hu, C.; Stein, P.M.; Witzel, S.; Shi, H.; Yang, Y.; Rudolph, M.; et al. A Metal-Free Direct Arene C−H Amination. Adv. Synth. Catal. 2021, 363, 2783–2795. [Google Scholar] [CrossRef]
- Becker, M.R.; Richardson, A.D.; Schindler, C.S. Functionalized azetidines via visible light-enabled aza Paternò-Büchi reactions. Nat. Commun. 2019, 10, 5095. [Google Scholar] [CrossRef] [PubMed]
- Blum, T.R.; Miller, Z.D.; Bates, D.M.; Guzei, I.A.; Yoon, T.P. Enantioselective photochemistry through Lewis acid-catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Strieth-Kalthoff, F.; James, M.J.; Teders, M.; Pitzer, L.; Glorius, F. Energy transfer catalysis mediated by visible light: Principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; LaPointe, A.M.; Shaffer, T.D.; Coates, G.W. Nature-inspired methylated polyhydroxybutyrates from C1 and C4 feedstocks. Nat. Chem. 2023, 15, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Getzler, Y.D.Y.L.; Kundnani, V.; Lobkovsky, E.B.; Coates, G.W. Catalytic Carbonylation of β-Lactones to Succinic Anhydrides. J. Am. Chem. Soc. 2004, 126, 6842–6843. [Google Scholar] [CrossRef]
- Theil, F. Synthesis of diaryl ethers: A long-standing problem has been solved. Angew. Chem. Int. Ed. 1999, 38, 2345–2347. [Google Scholar] [CrossRef]
- Rao, A.V.R.; Gurjar, M.K.; Reddy, K.L.; Rao, A.S. Studies directed toward the synthesis of vancomycin and related cyclic peptides. Chem. Rev. 1995, 95, 2135–2167. [Google Scholar] [CrossRef]
- Zhu, J. SNAr bases macrocyclization via biaryl ether formation: Application in natural product synthesis. Synlett 1997, 1997, 133–144. [Google Scholar] [CrossRef]
- Boger, D.L.; Borzilleri, R.M.; Nukui, S.; Beresis, R.T. Synthesis of the vancomycin CD and DE ring systems. J. Org. Chem. 1997, 62, 4721–4736. [Google Scholar] [CrossRef]
- Mulla, S.A.R.; Inamdar, S.M.; Pathan, M.Y.; Chavan, S.S. Ligand free, highly efficient synthesis of diaryl ether over copper fluorapatite as heterogeneous reusable catalyst. Tetrahedron Lett. 2012, 53, 1826–1829. [Google Scholar] [CrossRef]
- Zhang, Y.; Ni, G.; Li, C.; Xu, S.; Zhang, Z.; Xie, X. The coupling reactions of aryl halides and phenols catalyzed by palladium and MOP-type ligands. Tetrahedron 2015, 71, 4927–4932. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Cellier, P.P.; Hamada, S.; Spindler, J.-F.; Taillefer, M. A general and mild ullmann-type synthesis of fiaryl ethers. Org. Lett. 2004, 6, 913–916. [Google Scholar] [CrossRef]
- Munoz, A.; Leo, P.; Orcajo, G.; Martínez, F.; Calleja, G. URJC-1-MOF as new heterogeneous recyclable catalyst for C-Heteroatom coupling reactions. ChemCatChem 2019, 11, 3376–3380. [Google Scholar] [CrossRef]
- Cermak, J.K.; Cirkva, V. Copper-mediated synthesis of mono- and dichlorinated diaryl ethers. Tetrahedron Lett. 2014, 55, 4185–4188. [Google Scholar] [CrossRef]
- Chan, D.M.T.; Monaco, K.L.; Wang, R.-P.; Winters, M.P. New N- and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar] [CrossRef]
- Evans, D.A.; Katz, J.L.; West, T.R. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar] [CrossRef]
- Sang, R.; Korkis, S.E.; Su, W.; Ye, F.; Engl, P.S.; Berger, F.; Ritter, T. Site-selective C−H oxygenation via aryl sulfonium salts. Angew. Chem. Int. Ed. 2019, 58, 16161–16166. [Google Scholar] [CrossRef]
- Sharma, A.; Dixit, V.; Kumar, S.; Jain, N. Visible Light-Mediated In Situ Generation of δ,δ-Disubstituted p-Quinone Methides: Construction of a Sterically Congested Quaternary Stereocenter. Org. Lett. 2021, 23, 3409–3414. [Google Scholar] [CrossRef]
- Franceschi, P.; Cuadros, S.; Goti, G.; Dell’Amico, L. Mechanisms and Synthetic Strategies in Visible Light-Driven [2+2]-Heterocycloadditions. Angew. Chem. Int. Ed. 2023, 62, e202217210. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Rivera-Fortin, M.A.; Zuzukin, V.; Whitmire, K.H. Origin of Regioselectivity in Paterno-Buechi Reactions of Benzoquinones with Alkylidenecycloalkanes. J. Am. Chem. Soc. 1994, 116, 1272–1277. [Google Scholar] [CrossRef]
- Janssen, D.E.; VanAllan, J.; Wilson, C.V. The synthesis of 4-phenoxycatechol and 2-phenoxyhydroquinone. J. Org. Chem. 1955, 20, 1326–1329. [Google Scholar] [CrossRef]
- Koyama, H.; Miller, D.J.; Boueres, J.K.; Desai, R.C.; Jones, A.B.; Berger, J.P.; MacNaul, K.L.; Kelly, L.J.; Doebber, T.W.; Wu, M.S.; et al. (2R)-2-Ethylchromane-2-carboxylic Acids: Discovery of Novel PPARα/γ Dual Agonists as Antihyperglycemic and Hypolipidemic Agents. J. Med. Chem. 2004, 47, 3255–3263. [Google Scholar] [CrossRef]
- Sagadevan, A.; Ragupathi, A.; Hwang, K.C. Photoinduced Copper-Catalyzed Regioselective Synthesis of Indoles: Three-Component Coupling of Arylamines, Terminal Alkynes, and Quinones. Angew. Chem. Int. Ed. 2015, 54, 13896–13901. [Google Scholar] [CrossRef] [PubMed]
- Schnapp, K.A.; Wilson, R.M.; Ho, D.M.; Caldwell, R.A.; Creed, D. Benzoquinone-olefin exciplexes: The observation and chemistry of the p-benzoquinone-tetraphenylallene exciplex. J. Am. Chem. Soc. 1990, 112, 3700–3702. [Google Scholar] [CrossRef]
- Yang, J.; Duan, J.; Wang, G.; Zhou, H.; Ma, B.; Wu, C.; Xiao, J. Visible-Light-Promoted Site-Selective N(1)-Alkylation of Benzotriazoles with alpha-Diazoacetates. Org. Lett. 2020, 22, 7284–7289. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | 1a:2a (Equiv.) | Photosensitizers (5 mmol%) | Solvent | Temp. (°C) | Yield (%) b |
| 1 | 1:1 | - | CH3CN | 40 | 25 |
| 2 | 1:3 | - | CH3CN | 40 | 33 |
| 3 | 2:1 | - | CH3CN | 40 | 32 |
| 4 | 3:1 | - | CH3CN | 40 | 43 |
| 5 [c] | 3:1 | - | CH3CN | 40 | 7 |
| 6 | 3:1 | Ir(ppy)3 | CH3CN | 40 | np [d] |
| 7 | 3:1 | Ru(bpy)3Cl2·6H2O | CH3CN | 40 | 32 |
| 8 | 3:1 | Thioxanthen-9-one | CH3CN | 40 | 40 |
| 9 | 3:1 | - | CH3CN | –40 | 48 |
| 10 | 3:1 | - | Et2O | –78 | np |
| 11 | 3:1 | - | THF | –78 | np |
| 12 | 3:1 | - | DCM | –78 | 74 |
| 13 [e] | 3:1 | - | DCM | –78 | np |
 | |||||
|---|---|---|---|---|---|
| Entry | 1b:2a (Equiv.) | Catalyst (% b) | Solvent | Temp. (°C) | Yield (%) c |
| 1 | 3:1 | Cu(OTf)2 (10) | CH3CN | 40 | 50 |
| 2 d | 3:1 | Cu(OTf)2 (10) | CH3CN | 40 | np |
| 3 e | 3:1 | Cu(OTf)2 (10) | CH3CN | 40 | 23 |
| 4 f | 3:1 | Cu(OTf)2 (10) | CH3CN | 40 | 46 |
| 5 | 3:1 | Fe(OTf)3 (10) | CH3CN | 40 | 45 |
| 6 | 3:1 | Zn(OTf)2 (10) | CH3CN | 40 | 43 |
| 7 | 3:1 | Yb(OTf)3 (10) | CH3CN | 40 | 16 |
| 8 | 3:1 | Nd(OTf)3 (10) | CH3CN | 40 | 15 |
| 9 | 3:1 | Sc(OTf)3 (10) | CH3CN | 40 | 10 |
| 10 | 3:1 | Pd(OAc)2 (10) | CH3CN | 40 | np |
| 11 | 3:1 | AgOAc (10) | CH3CN | 40 | np |
| 12 | 3:1 | BF3·Et2O (10) | CH3CN | 40 | np |
| 13 | 3:1 | Cu(OTf)2 (10) | CH3CN | 25 | 53 |
| 14 | 3:1 | Cu(OTf)2 (10) | CH3CN | 10 | 44 |
| 15 | 1:1 | Cu(OTf)2 (10) | CH3CN | 25 | 28 |
| 16 | 1.5:1 | Cu(OTf)2 (10) | CH3CN | 25 | 37 |
| 17 | 2:1 | Cu(OTf)2 (10) | CH3CN | 25 | 39 |
| 18 | 1:1.5 | Cu(OTf)2 (10) | CH3CN | 25 | 39 |
| 19 | 3:1 | Cu(OTf)2 (20) | CH3CN | 25 | 65 |
| 20 | 3:1 | Cu(OTf)2 (20) | CH3OH | 25 | np |
| 21 | 3:1 | Cu(OTf)2 (20) | acetone | 25 | np |
| 22 | 3:1 | Cu(OTf)2 (20) | Et2O | 25 | np |
| 23 | 3:1 | Cu(OTf)2 (20) | THF | 25 | np |
| 24 | 3:1 | Cu(OTf)2 (20) | DCM | 25 | 45 |
| 25 | 3:1 | Cu(OTf)2 (20) | CH3CN/ DCM | 25 | 67 |
| 26 g | 3:1 | Cu(OTf)2 (20) | CH3CN/ DCM | 25 | np |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.-W.; Zhao, J.-L.; Wang, Z.-Y.; Li, P.-T.; Shi, Z.-F.; Cao, X.-P.; Liu, Q. A Paternò–Büchi Reaction of Aromatics with Quinones under Visible Light Irradiation. Molecules 2024, 29, 1513. https://doi.org/10.3390/molecules29071513
Li W-W, Zhao J-L, Wang Z-Y, Li P-T, Shi Z-F, Cao X-P, Liu Q. A Paternò–Büchi Reaction of Aromatics with Quinones under Visible Light Irradiation. Molecules. 2024; 29(7):1513. https://doi.org/10.3390/molecules29071513
Chicago/Turabian StyleLi, Wen-Wen, Jia-Lin Zhao, Ze-Yu Wang, Pei-Ting Li, Zi-Fa Shi, Xiao-Ping Cao, and Qiang Liu. 2024. "A Paternò–Büchi Reaction of Aromatics with Quinones under Visible Light Irradiation" Molecules 29, no. 7: 1513. https://doi.org/10.3390/molecules29071513
APA StyleLi, W.-W., Zhao, J.-L., Wang, Z.-Y., Li, P.-T., Shi, Z.-F., Cao, X.-P., & Liu, Q. (2024). A Paternò–Büchi Reaction of Aromatics with Quinones under Visible Light Irradiation. Molecules, 29(7), 1513. https://doi.org/10.3390/molecules29071513