
Lattice Model Results for Pattern Formation in a Mixture with Competing Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
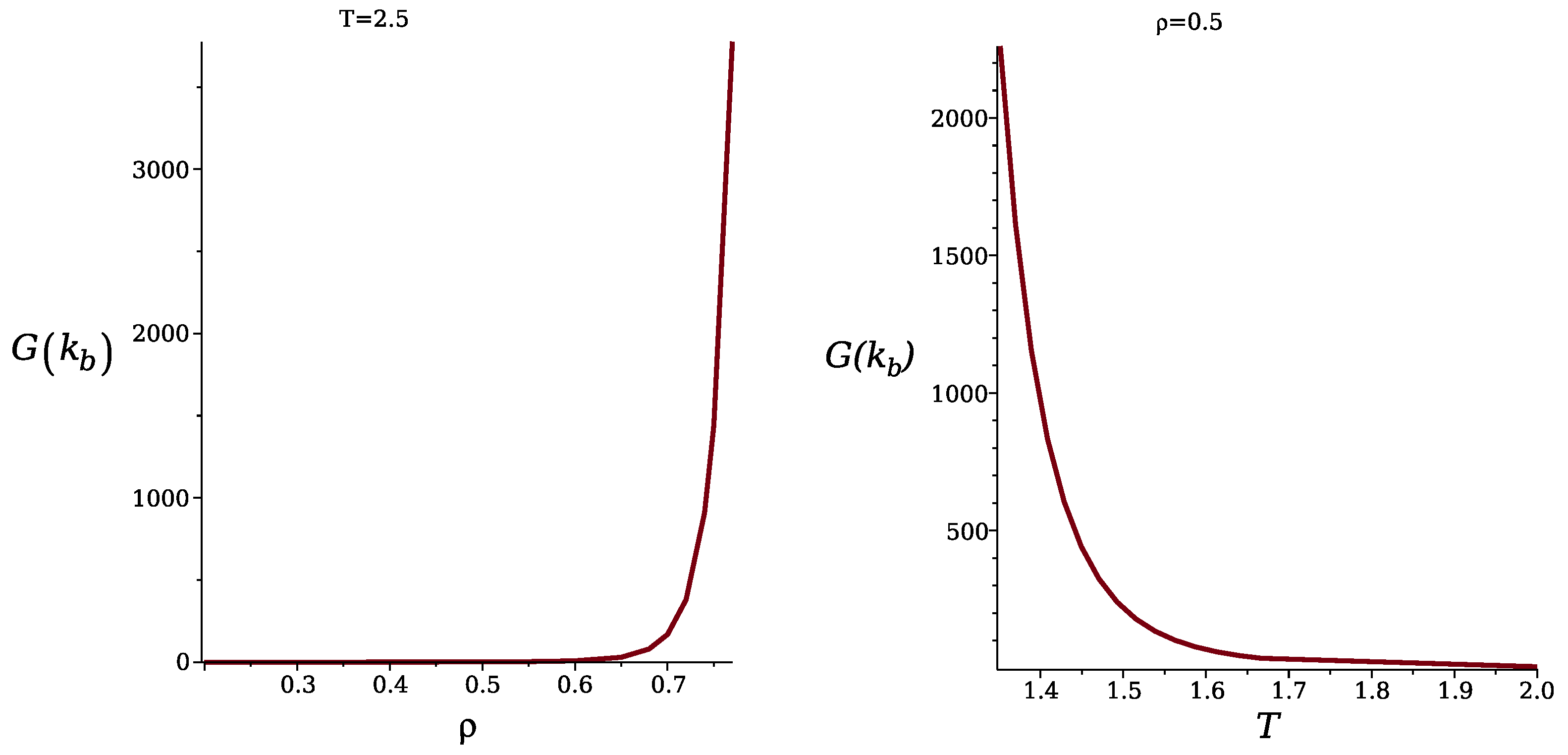{kind=link}
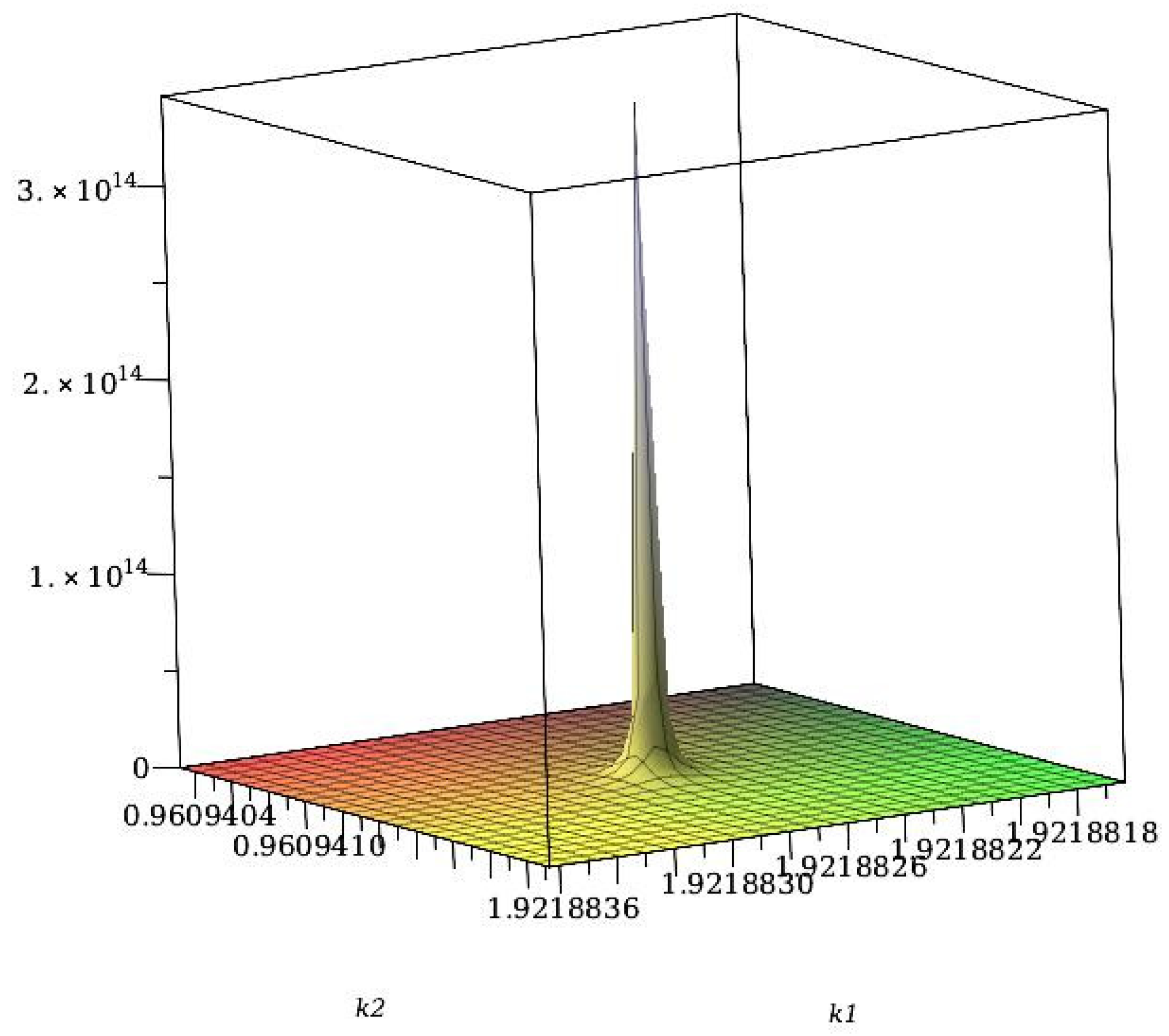{kind=link}
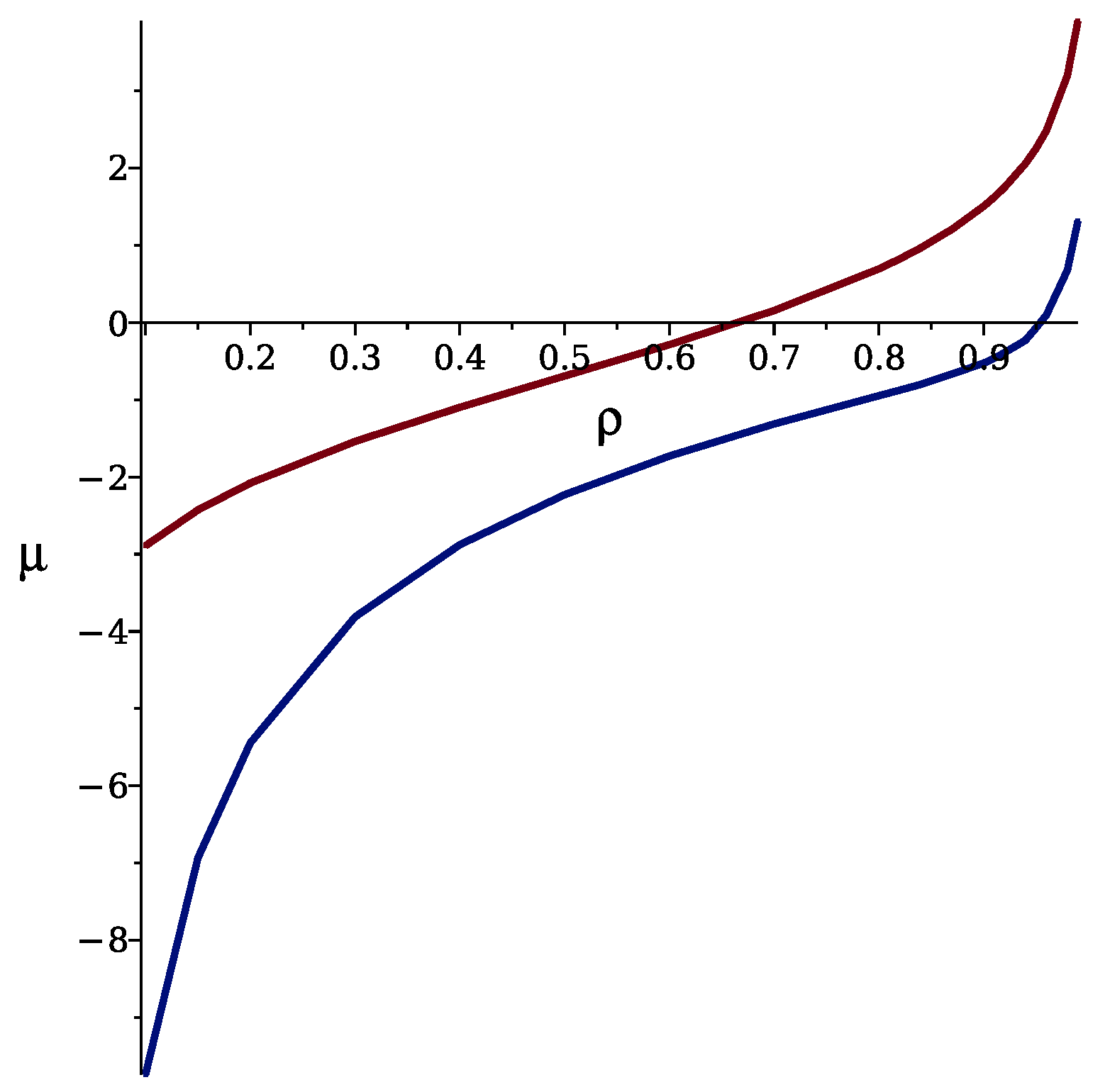{kind=link}
{kind=link}
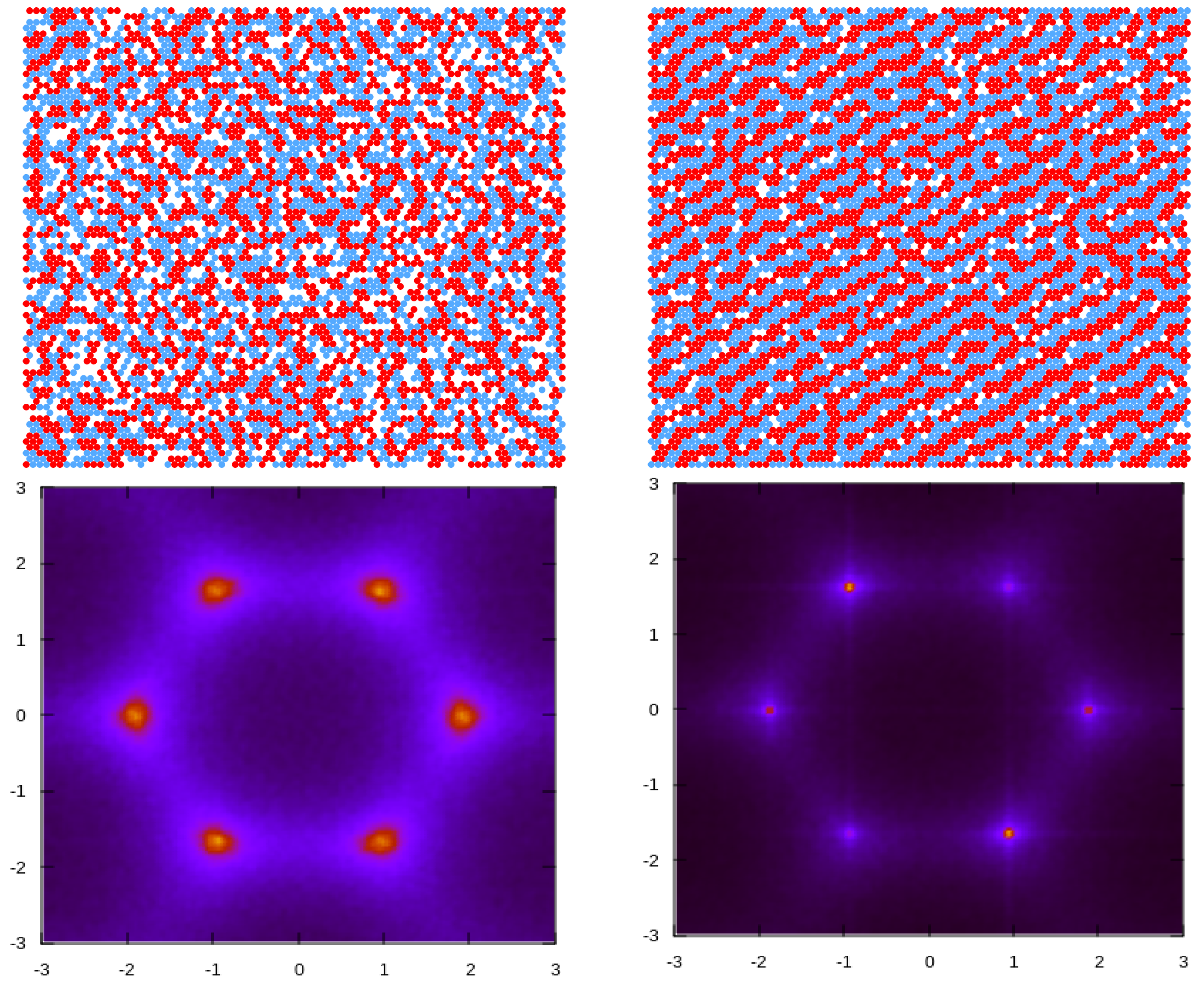{kind=link}
{kind=link}
{kind=link}
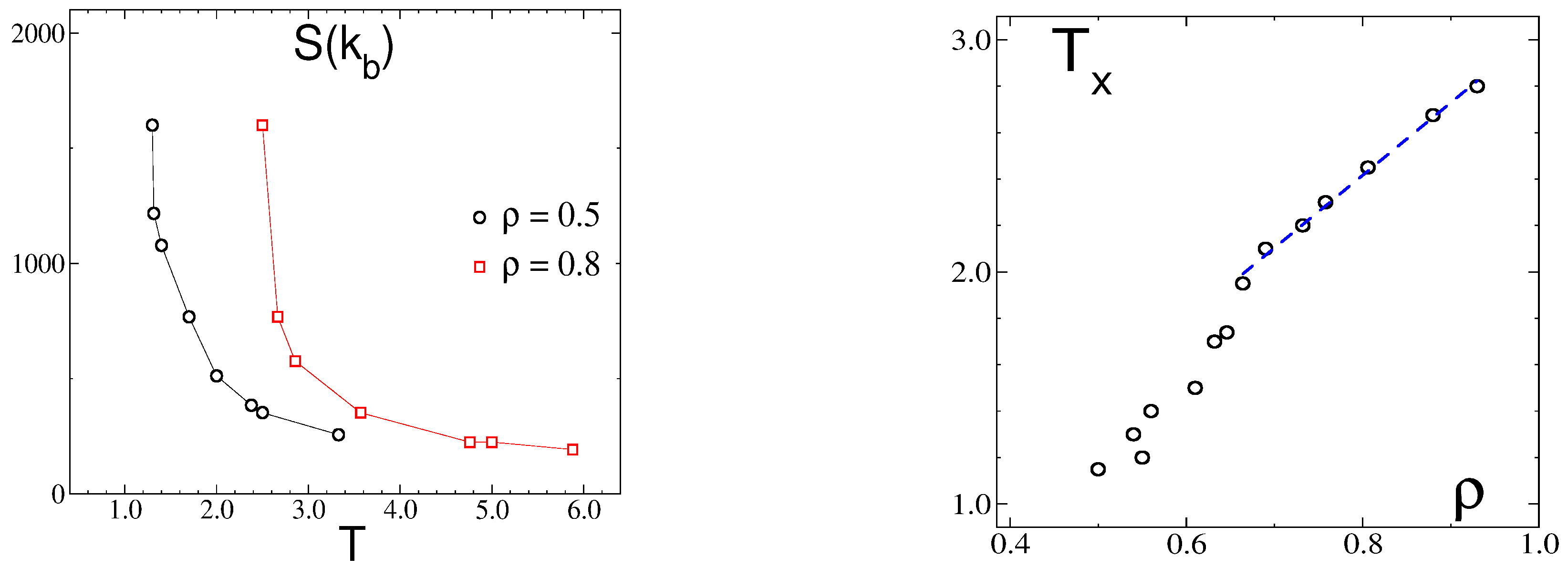{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Model and Methods
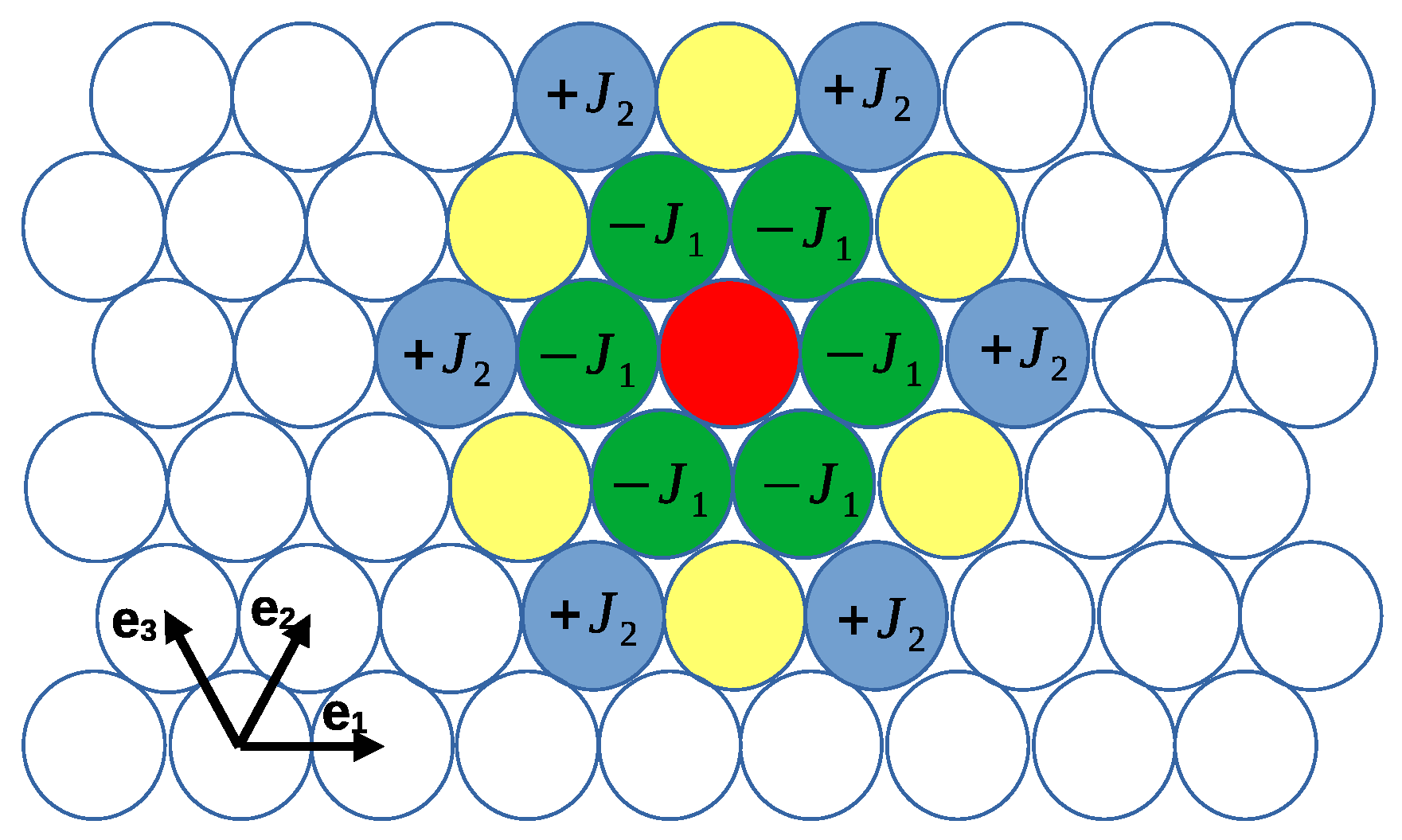
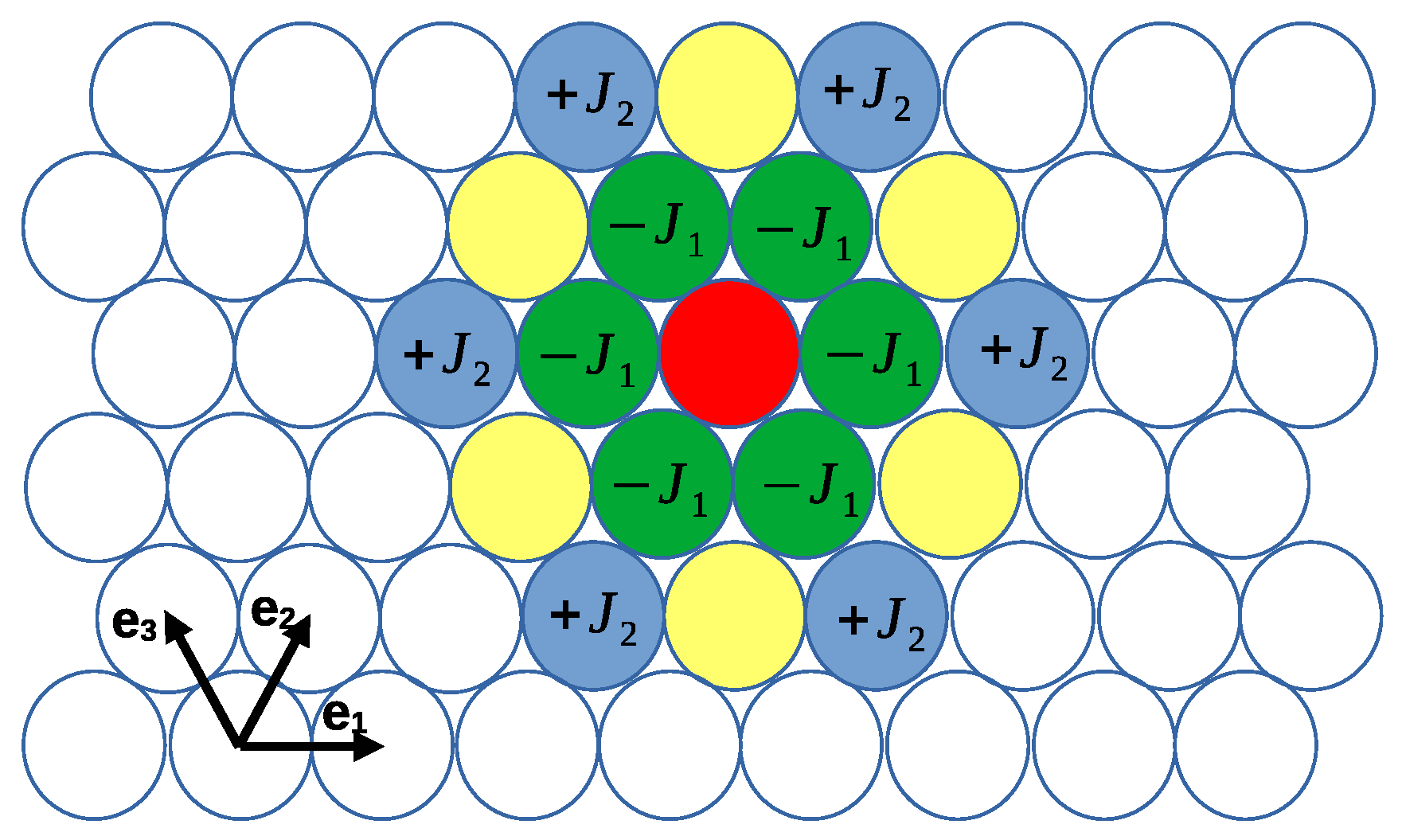
2.1. The Model
2.2. The Theoretical Methods
2.3. Simulation Methods
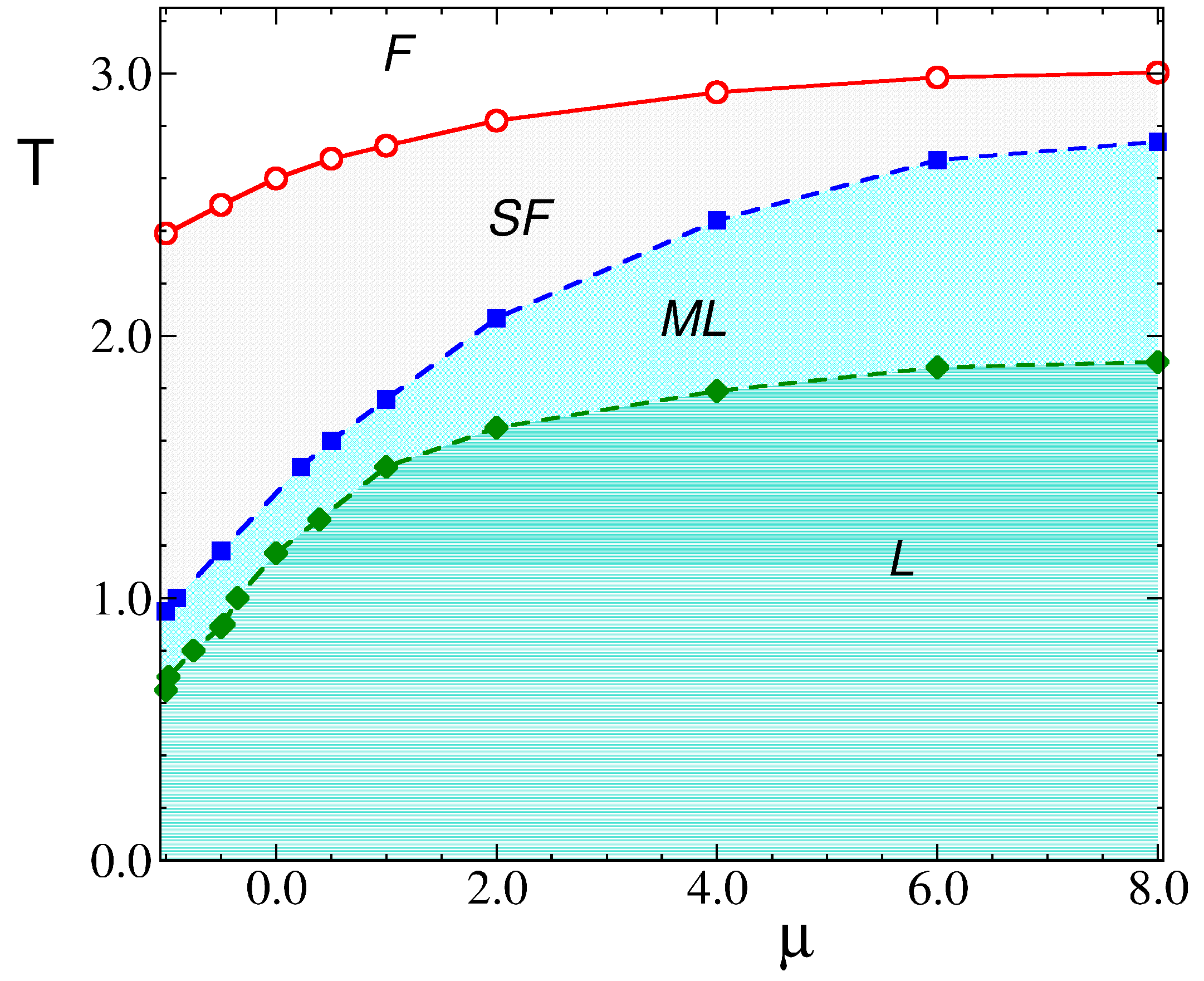
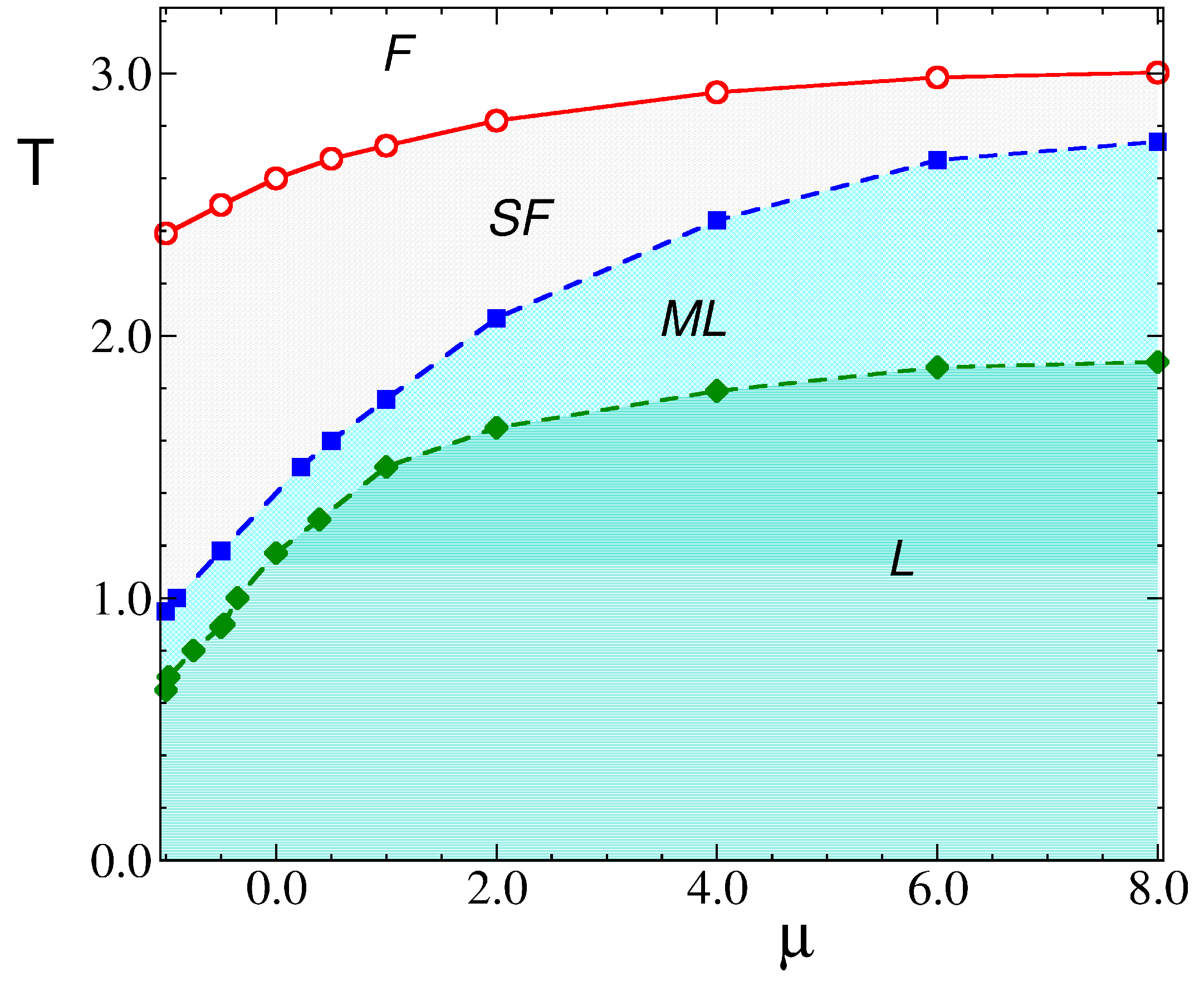
3. Results
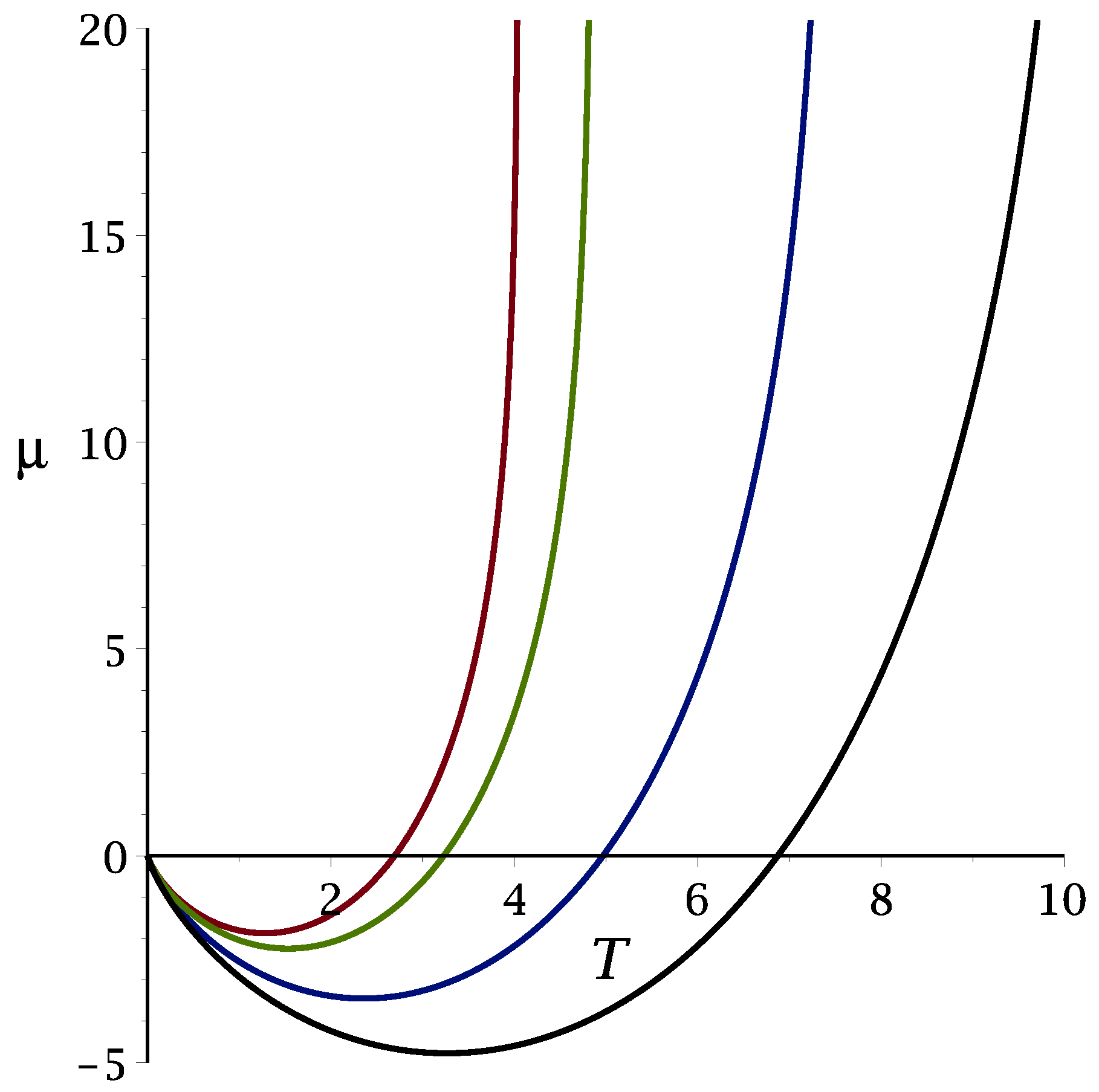
3.1. Theoretical Results
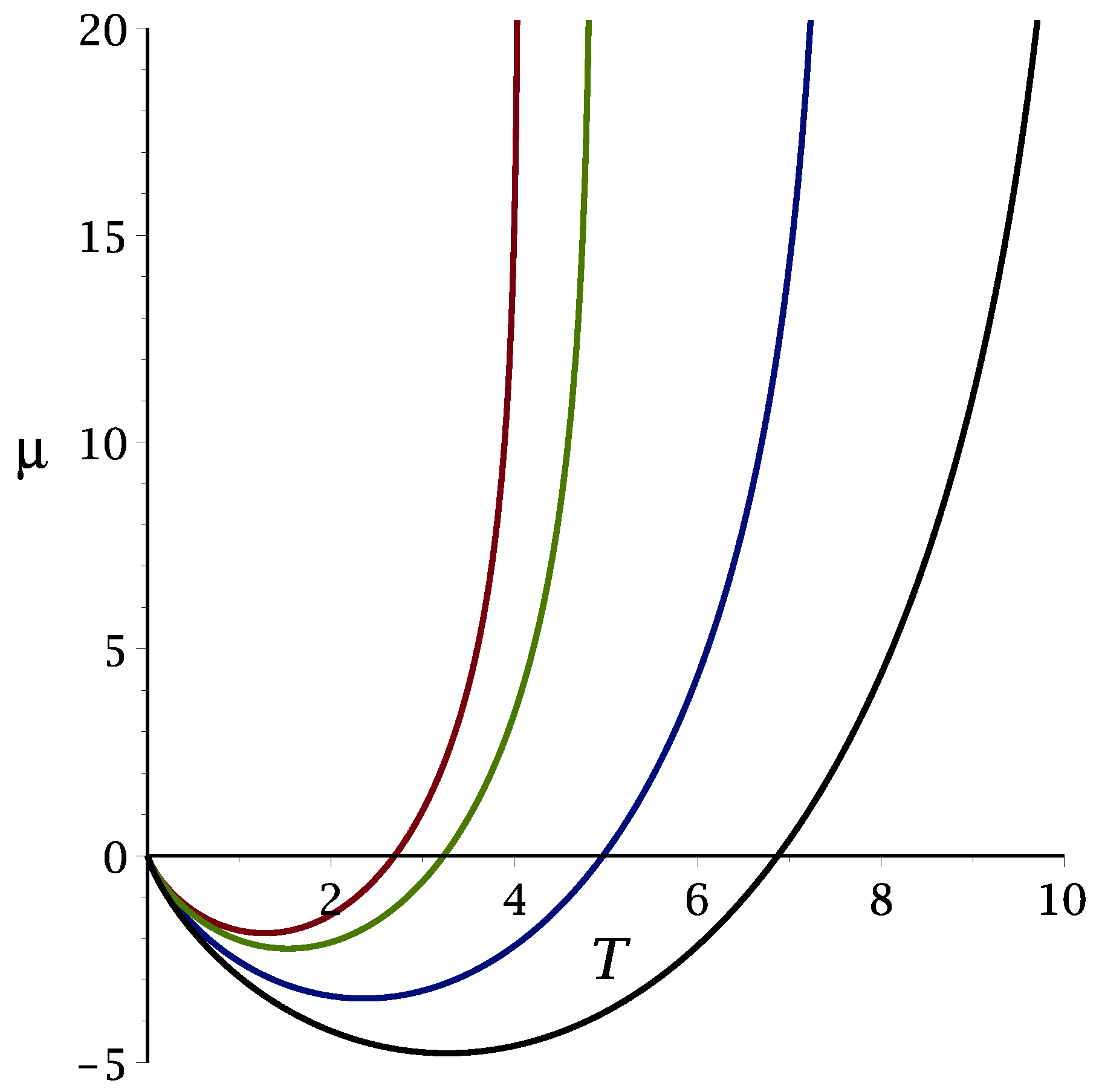
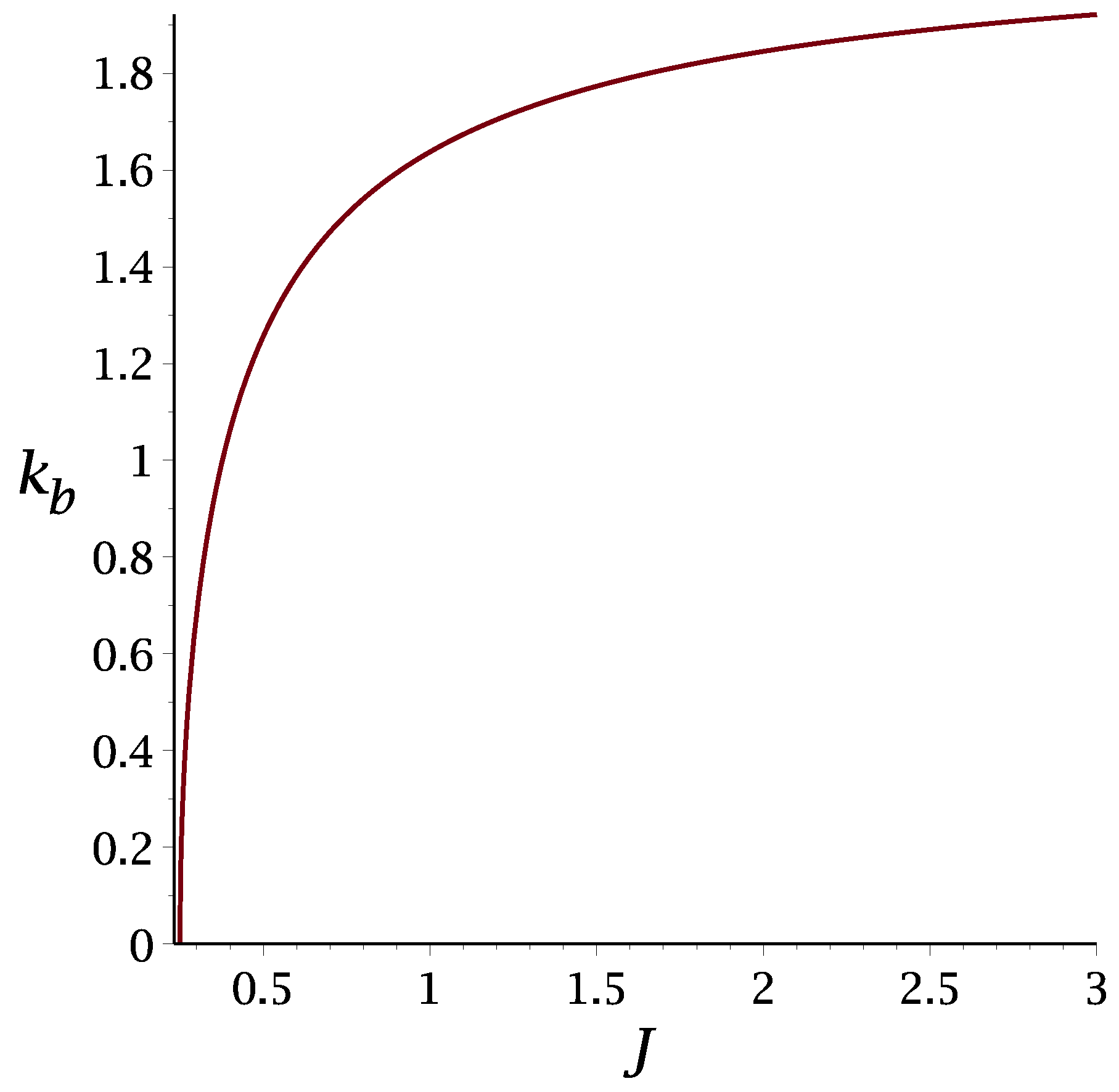
3.1.1. Mean-Field Approximation
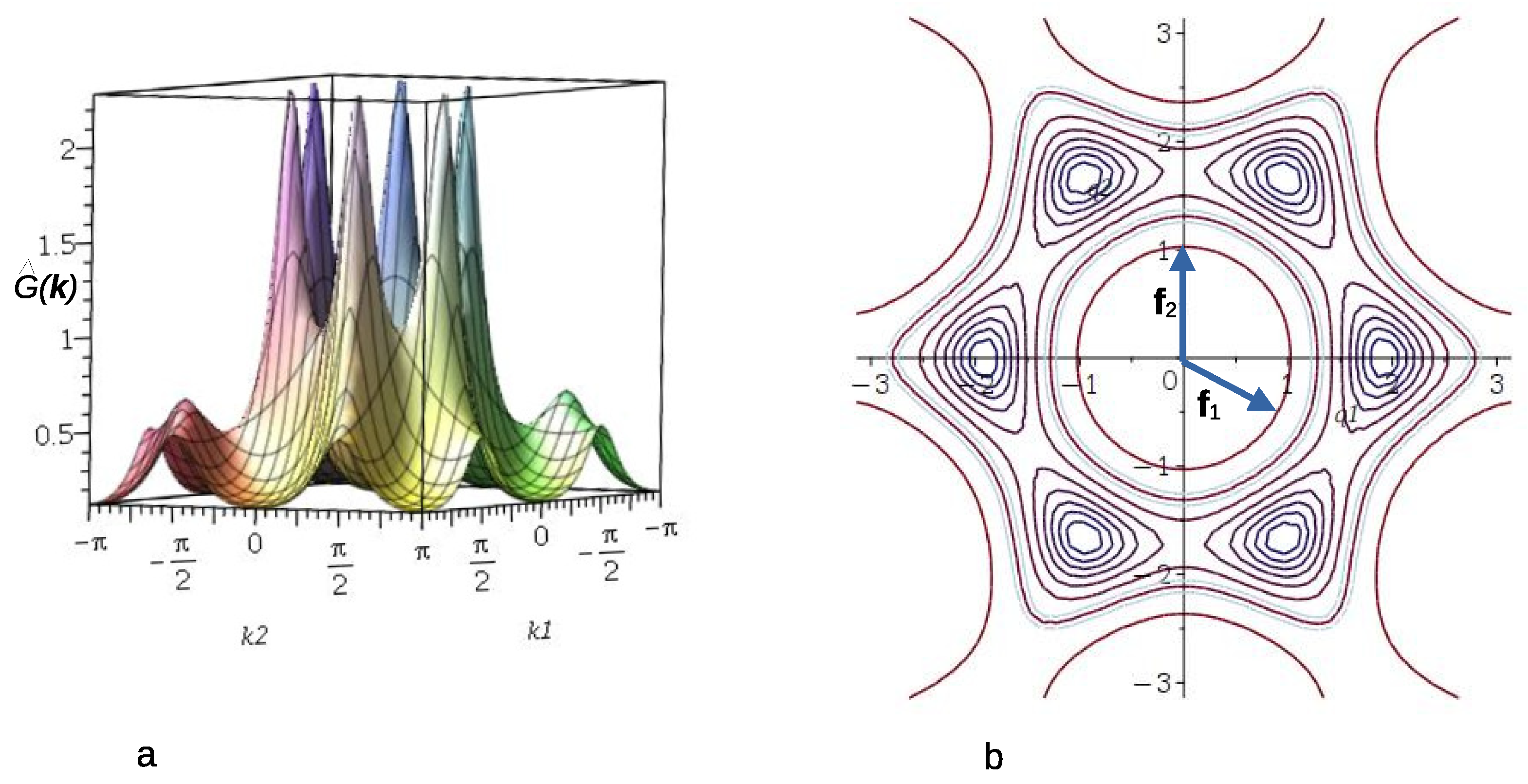
3.1.2. Self-Consistent Gaussian Approximation
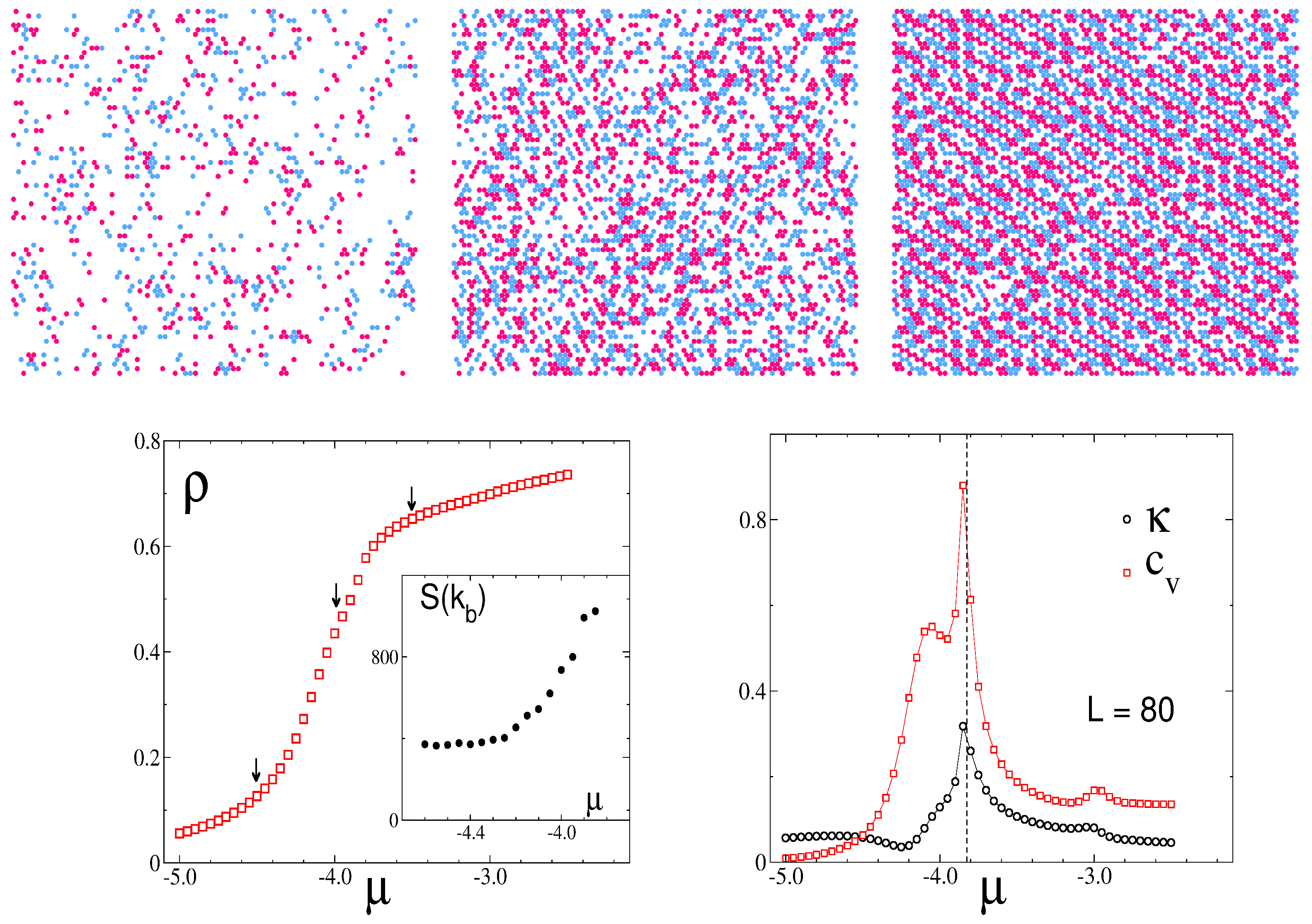
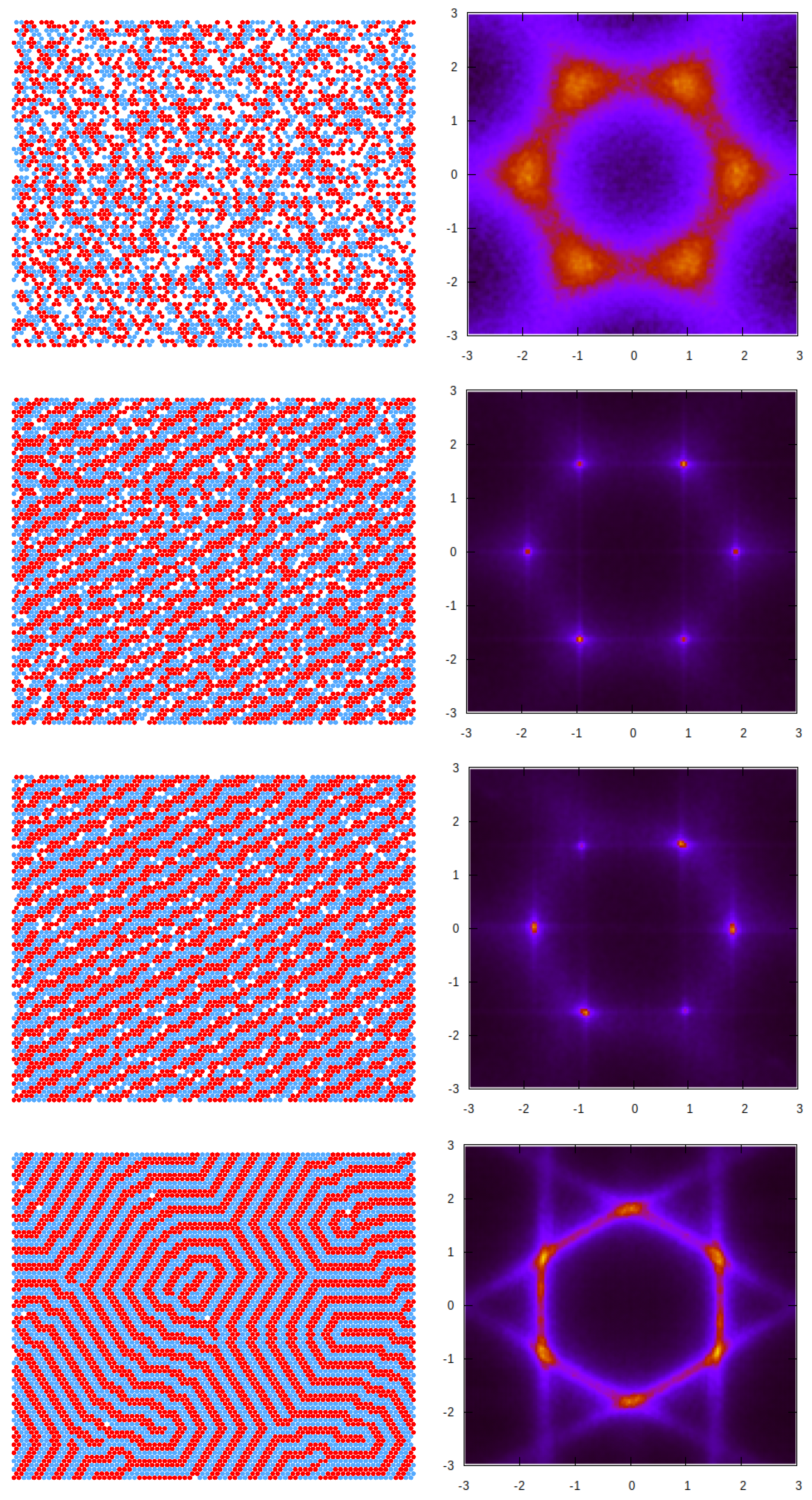
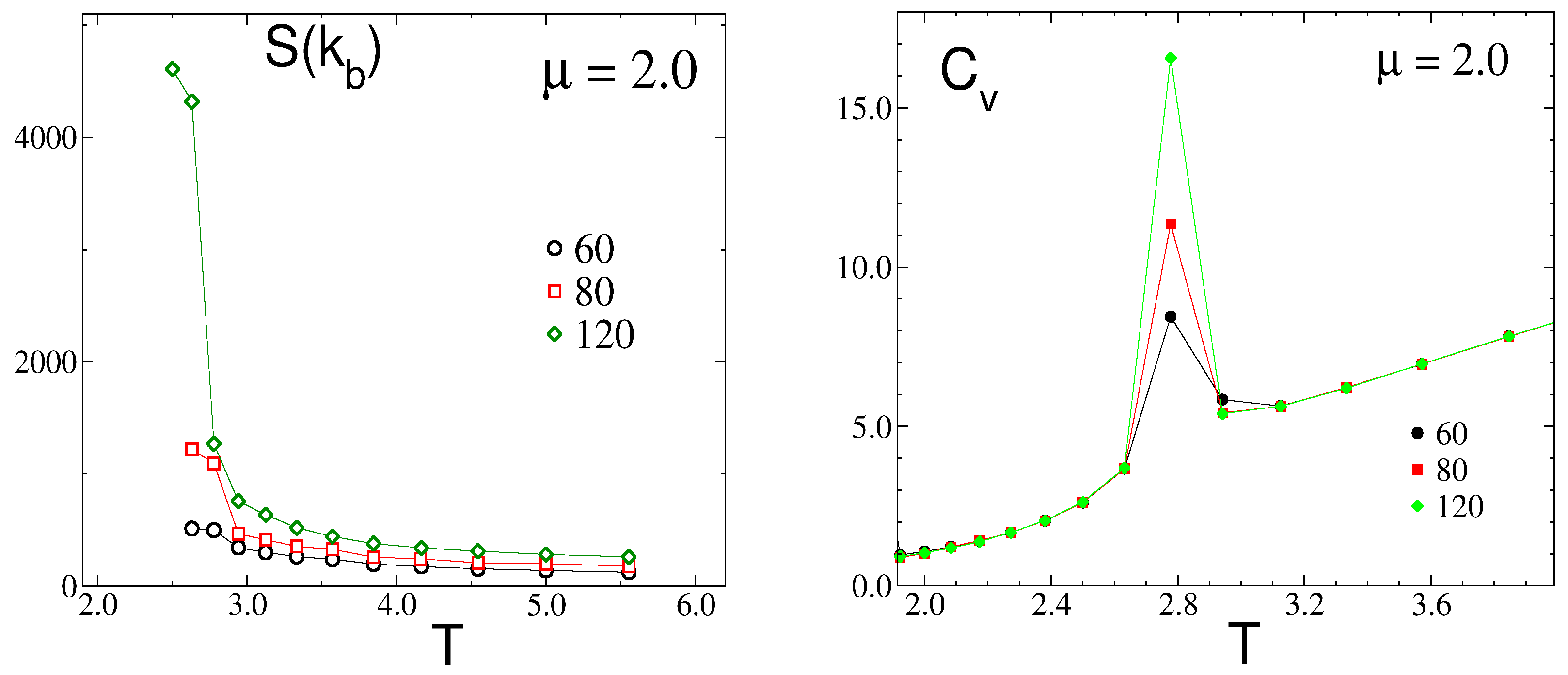
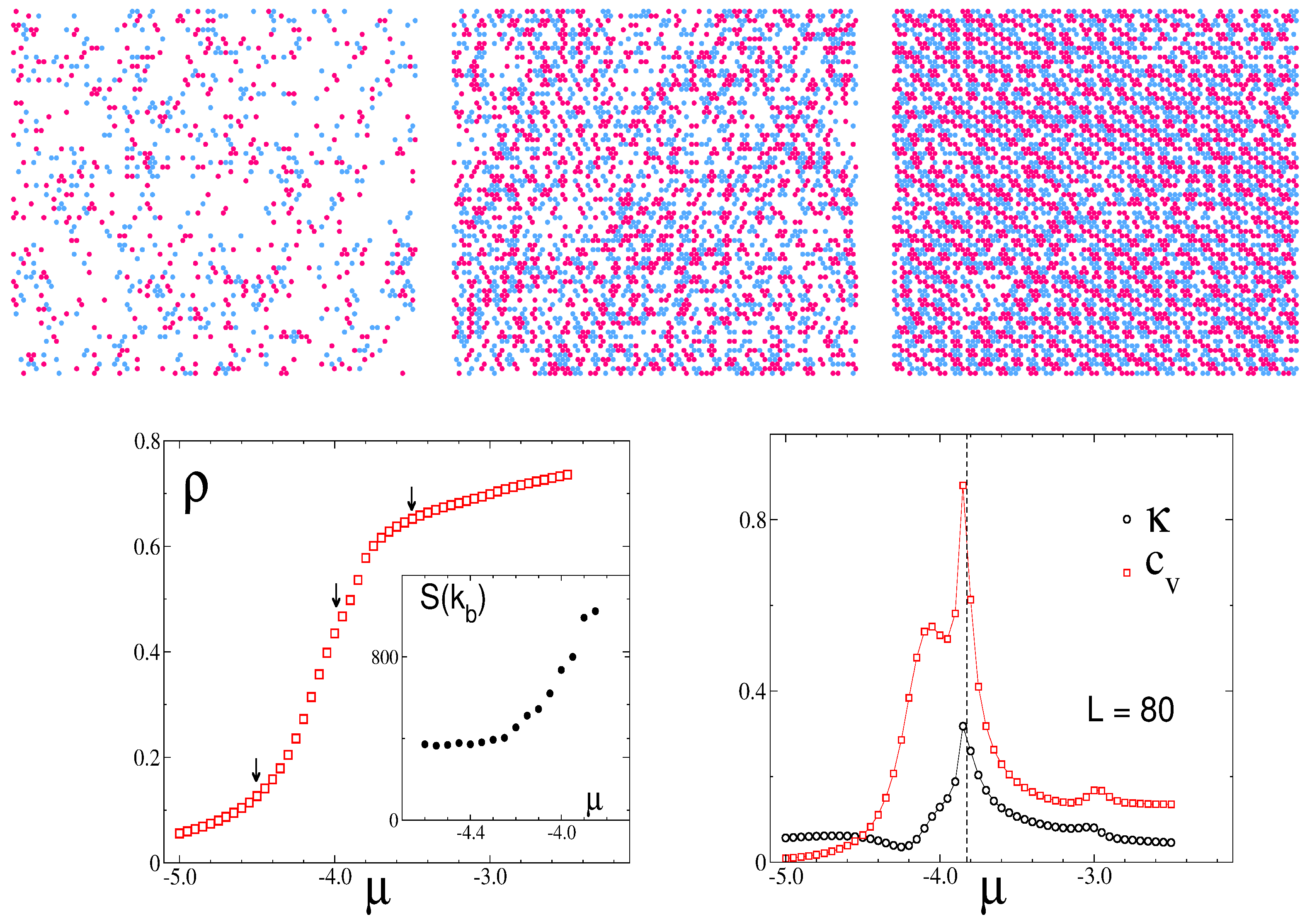
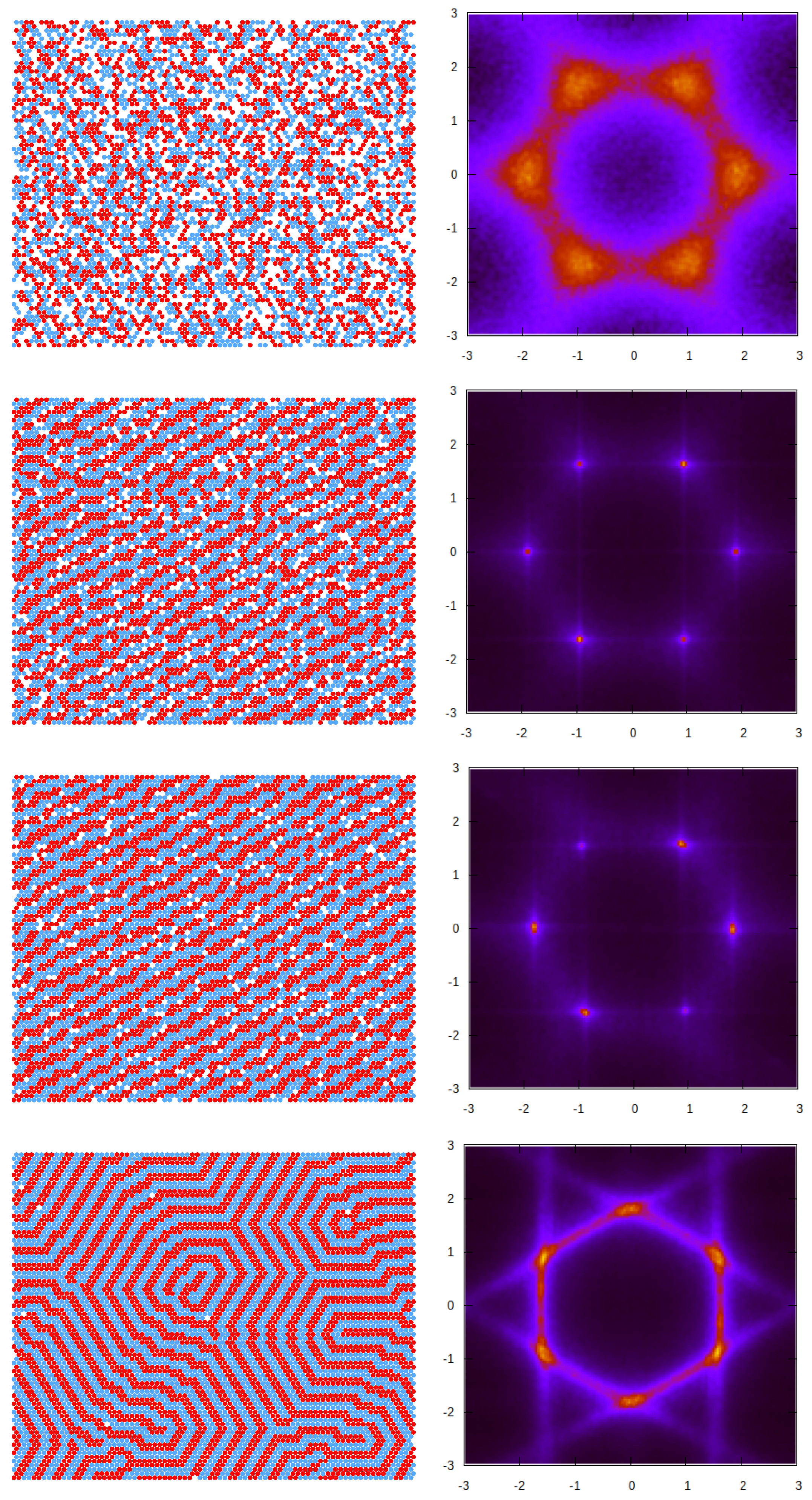
3.2. Simulation Results
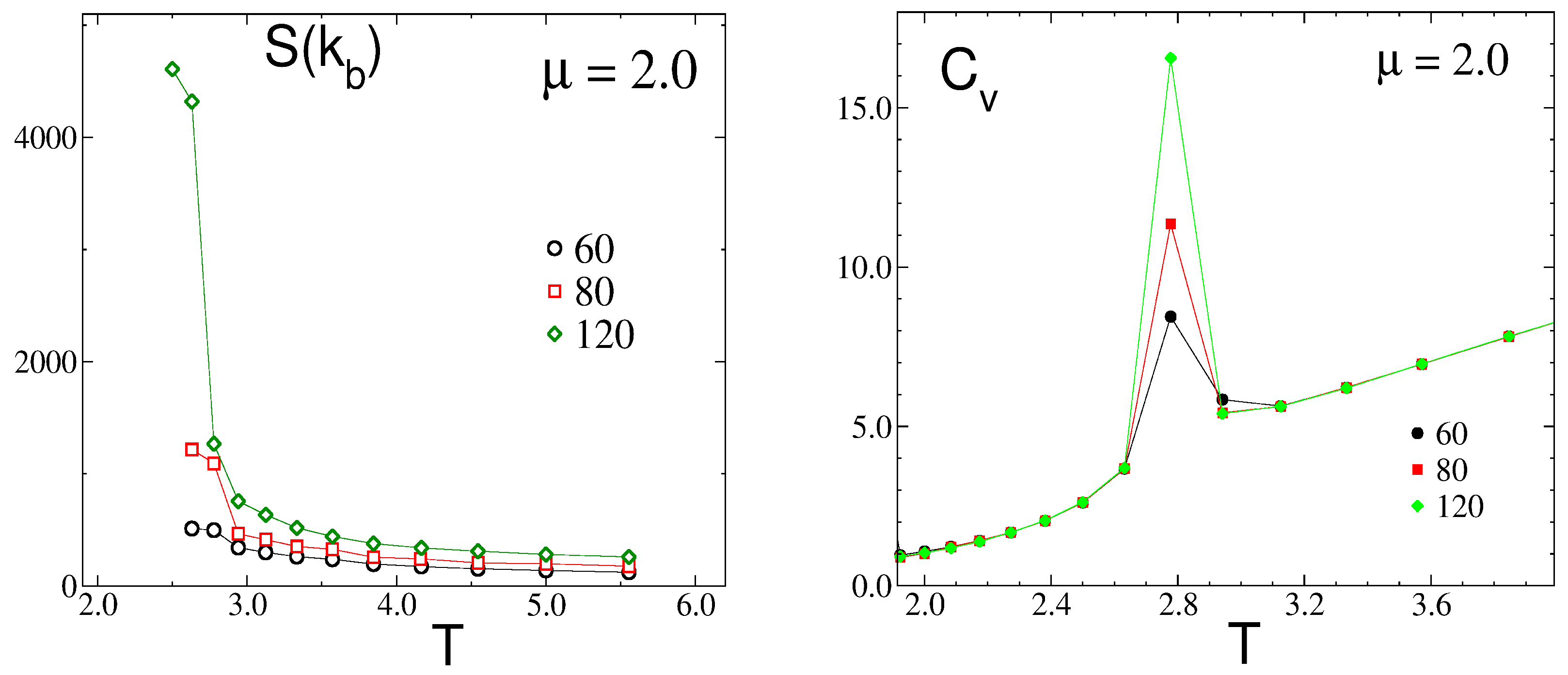
3.2.1. Structure at High Temperatures: The Disordered Fluid
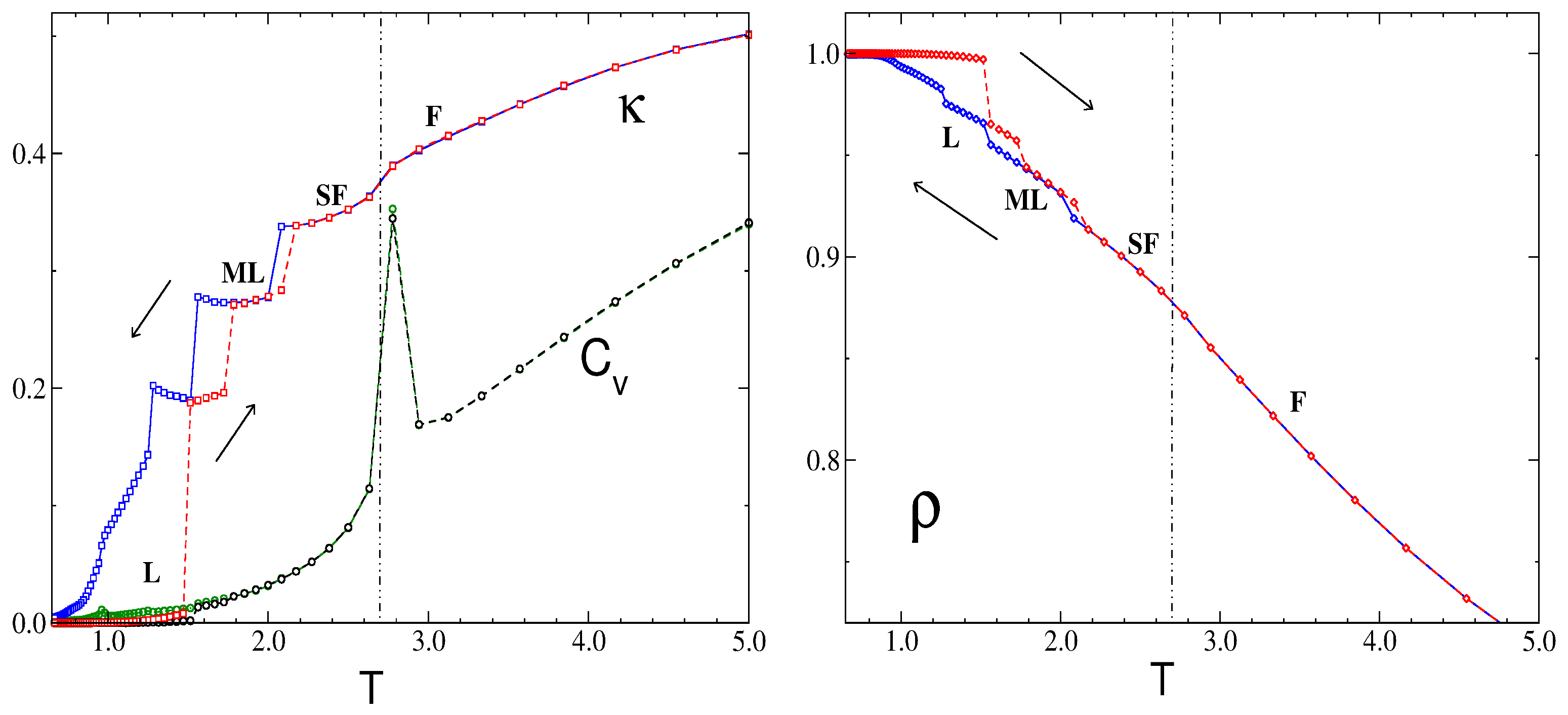
3.2.2. Phase Transitions of Lamellas
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stradner, A.; Sedgwick, H.; Cardinaux, F.; Poon, W.; Egelhaaf, S.; Schurtenberger, P. Equilibrium cluster formation in concentrated protein solutions and colloids. Nature 2004, 432, 492. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.; Campbell, A.I. Three-Dimensional Binary Superlattices of Oppositely Charged Colloids. Phys. Rev. Lett. 2005, 95, 128302. [Google Scholar] [CrossRef] [PubMed]
- Imperio, A.; Reatto, L. Microphase separation in two-dimensional systems with competing interactions. J. Chem. Phys. 2006, 124, 164712. [Google Scholar] [CrossRef] [PubMed]
- Archer, A.J.; Wilding, N.B. Phase behavior of a fluid with competing attractive and repulsive interactions. Phys. Rev. E 2007, 76, 031501. [Google Scholar] [CrossRef] [PubMed]
- Archer, A.J. Two-dimensional fluid with competing interactions exhibiting microphase separation: Theory for bulk and interfacial properties. Phys. Rev. E 2008, 78, 031402. [Google Scholar] [CrossRef] [PubMed]
- Pȩkalski, J.; Ciach, A.; Almarza, N.G. Periodic ordering of clusters and stripes in a two-dimensional lattice model. I. Ground state, mean-field phase diagram and structure of the disordered phases. J. Chem. Phys. 2014, 140, 114701. [Google Scholar] [CrossRef]
- Almarza, N.G.; Pȩkalski, J.; Ciach, A. Two-dimensional lattice model for periodic ordering of clusters and stripes. II. Monte Carlo simulations. J. Chem. Phys. 2014, 140, 164708. [Google Scholar] [CrossRef] [PubMed]
- Sweatman, M.B.; Fartaria, R.; Lue, L. Cluster formation in fluids with competing short-range and long-range interactions. J. Chem. Phys. 2014, 140, 124508. [Google Scholar] [CrossRef]
- Lindquist, B.A.; Jadrich, R.B.; Truskett, T.M. Assembly of nothing: Equilibrium fluids with designed structrued porosity. Soft Matter 2016, 12, 2663–2667. [Google Scholar] [CrossRef]
- Zhuang, Y.; Zhang, K.; Charbonneau, P. Equilibrium Phase Behavior of a Continuous-Space Microphase Former. Phys. Rev. Lett. 2016, 116, 098301. [Google Scholar] [CrossRef]
- Zhuang, Y.; Charbonneau, P. Equilibrium phase behavior of the square-well linear microphase-forming model. J. Phys. Chem. B 2016, 120, 6178–6188. [Google Scholar] [CrossRef]
- Pini, D.; Parola, A. Pattern formation and self-assembly driven by competing interactions. Soft Matter 2017, 13, 9259. [Google Scholar] [CrossRef] [PubMed]
- Royall, C.P. Hunting mermaids in real space: Known knowns, known unknowns and unknown unknowns. Soft Matter 2018, 14, 4020. [Google Scholar] [CrossRef]
- Marolt, K.; Zimmermann, M.; Roth, R. Microphase separation in a two-dimensional colloidal system with competing attractive critical Casimir and repulsive magnetic dipole interactions. Phys. Rev. E 2019, 100, 052602. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Franco, J.; Zaccarelli, E. On the role of competing interactions in charged colloids with short-range attraction. Annu. Rev. Condens. Matter Phys. 2021, 12, 51. [Google Scholar] [CrossRef]
- Serna, H.; Pozuelo, A.D.; Noya, E.G.; Góźdź, W.T. Formation and internal ordering of periodic microphases in colloidal models with competing interactions. Soft Matter 2021, 17, 4957. [Google Scholar] [CrossRef]
- Kumar, A.; Molinero, V. Self-Assembly of Mesophases from Nanoparticles. J. Phys. Chem. Lett. 2017, 8, 5053. [Google Scholar] [CrossRef]
- Workineh, Z.G.; Pellicane, G.; Tsige, M. Tuning Solvent Quality Induces Morphological Phase Transitions in Miktoarm Star Polymer Films. Macromolecules 2020, 53, 6151. [Google Scholar] [CrossRef]
- McManus, J.J.; Charbonneau, P.; Zaccarelli, E.; Asherie, N. The Physics of Protein Self-Assembly. Curr. Opin. Colloid Interface Sci. 2016, 22, 73. [Google Scholar] [CrossRef]
- Khalil, K.; Sagastegui, A.; Li, Y.; Tahir, M.; Socolar, J.; Wiley, B.; Yellen, B. Binary colloidal structures assembled through Ising interactions. Nat. Commun. 2012, 3, 794. [Google Scholar] [CrossRef]
- Machta, B.B.; Veatch, S.L.; Sethna, J.P. Critical Casimir Forces in Cellular Membranes. Phys. Rev. Lett. 2012, 109, 138101. [Google Scholar] [CrossRef]
- Rauh, A.; Rey, M.; Barbera, L.; Zanini, M.; Karg, M.; Isa, L. Compression of hard core-soft shell nanoparticles at liquid-liquid interfaces: Influence of the shell thickness. Soft Matter 2017, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Ickler, M.; Menath, J.; Holstein, L.; Rey, M.; Buzza, D.M.A.; Vogel, N. Interfacial self-assembly of SiO2–PNIPAM core–shell particles with varied crosslinking density. Soft Matter 2022, 18, 5585. [Google Scholar] [CrossRef]
- Nazli, K.O.; Pester, C.; Konradi, A.; Boker, A.; van Rijn, P. Cross-Linking Density and Temperature Effects on the Self-Assembly of SiO2—PNIPAAm Core–Shell Particles at Interfaces. Chem. Eur. J. 2013, 19, 5586. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Elnathan, R.; Ditcovski, R.; Geisel, K.; Zanini, M.; Fernandez-Rodriguez, M.A.; Naik, V.V.; Frutiger, A.; Richtering, W.; Ellenbogen, T.; et al. Fully tunable silicon nanowire arrays fabricated by soft nanoparticle templating. Nano Lett. 2016, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Vasilyev, O.A.; Kluft, B.B.; Stroink, M.J.B.; Kondrat, S.; Schall, P. Controlled deposition of nanoparticles with critical Casimir forces. Nanoscale Horiz. 2021, 6, 751. [Google Scholar] [CrossRef] [PubMed]
- Prestipino, S.; Pini, D.; Costa, D.; Malescio, D.; Munao, G. A density functional theory and simulation study of stripe phases in symmetric colloidal mixtures. J. Chem. Phys. 2023, 159, 204902. [Google Scholar] [CrossRef] [PubMed]
- Patsahan, O.; Litniewski, M.; Ciach, A. Self-assembly in mixtures with competing interactions. Soft Matter 2021, 17, 2883. [Google Scholar] [CrossRef] [PubMed]
- Munao, G.; Costa, D.; Malescio, D.; Bomont, J.M.; Prestipino, S. Like aggregation from unlike attraction: Stripes in symmetric mixtures of cross-attracting hard spheres. Phys. Chem. Chem. Phys. 2023, 25, 16227. [Google Scholar] [CrossRef]
- Patsahan, O.; Meyra, A.; Ciach, A. Spontaneous pattern formation in monolayers of binary mixtures with competing interactions. Soft Matter 2024, 20, 1410–1424. [Google Scholar] [CrossRef]
- Hou, J.; Li, M.; Song, Y. Patterned Colloidal Photonic Crystals. Angew. Chem. Int. Ed. 2018, 57, 2544. [Google Scholar] [CrossRef] [PubMed]
- Hensley, A.; Videbaek, T.E.; Seyforth, H.; Jacobs, W.M.; Rogers, W.B. Macroscopic photonic single crystals via seeded growth of DNA-coated colloids. Nat. Commun. 2023, 14, 4237. [Google Scholar] [CrossRef] [PubMed]
- Mamba, S.; Perry, D.S.; Tsige, M.; Pellicane, G. Toward the Rational Design of Organic Solar Photovoltaics: Application of Molecular Structure Methods to Donor Polymers. J. Phys. Chem. A 2021, 125, 10593. [Google Scholar] [CrossRef] [PubMed]
- Ciach, A.; Pȩkalski, J.; Góźdź, W.T. Origin of similarity of phase diagrams in amphiphilic and colloidal systems with competing interactions. Soft Matter 2013, 9, 6301. [Google Scholar] [CrossRef]
- Ciach, A. Mesoscopic theory for inhomogeneous mixtures. Mol. Phys. 2011, 109, 1101–1119. [Google Scholar] [CrossRef]
- Veatch, S.L.; Soubias, O.; Keller, S.L.; Gawrisch, K. Critical fluctuations in domain-forming lipid mixtures. Proc. Nat. Acad. Sci. USA 2007, 104, 17650–17655. [Google Scholar] [CrossRef]
- Grishina, V.S.; Vikhrenko, V.S.; Ciach, A. Structural and thermodynamic peculiarities of core-shell particles at fluid interfaces from triangular lattice models. Entropy 2020, 32, 1215. [Google Scholar] [CrossRef]
- Ciach, A.; DeVirgiliis, A.; Meyra, A.; Litniewski, M. Pattern Formation in Two-Component Monolayers of Particles with Competing Interactions. Molecules 2023, 28, 1366. [Google Scholar] [CrossRef] [PubMed]
- Ciach, A. Combined density functional and Brazovskii theories for systems with spontaneous inhomogeneities. Soft Matter 2018, 14, 5497. [Google Scholar] [CrossRef]
- Ciach, A.; Patsahan, O. Structure of ionic liquids and concentrated electrolytes from a mesoscopic theory. J. Mol. Liq. 2023, 377, 121453. [Google Scholar] [CrossRef]
- Brazovskii, S.A. Phase transition of an isotropic system to a nonuniform state. Sov. Phys. JETP 1975, 41, 85. [Google Scholar]
- Fredrickson, G.H.; Helfand, E. Fluctuation effects in the theory of microphase separation in block copolymers. J. Chem. Phys. 1987, 87, 697–705. [Google Scholar] [CrossRef]
- Ciach, A.; Stell, G. Mesoscopic field theory of ionic systems. Int. J. Mod. Phys. B 2005, 19, 3309. [Google Scholar] [CrossRef]
- Ciach, A. Effect of charge-density fluctuations on the order-disorder transition in the lattice restricted primitive models of electrolytes. Phys. Rev. E 2004, 70, 046103. [Google Scholar] [CrossRef] [PubMed]



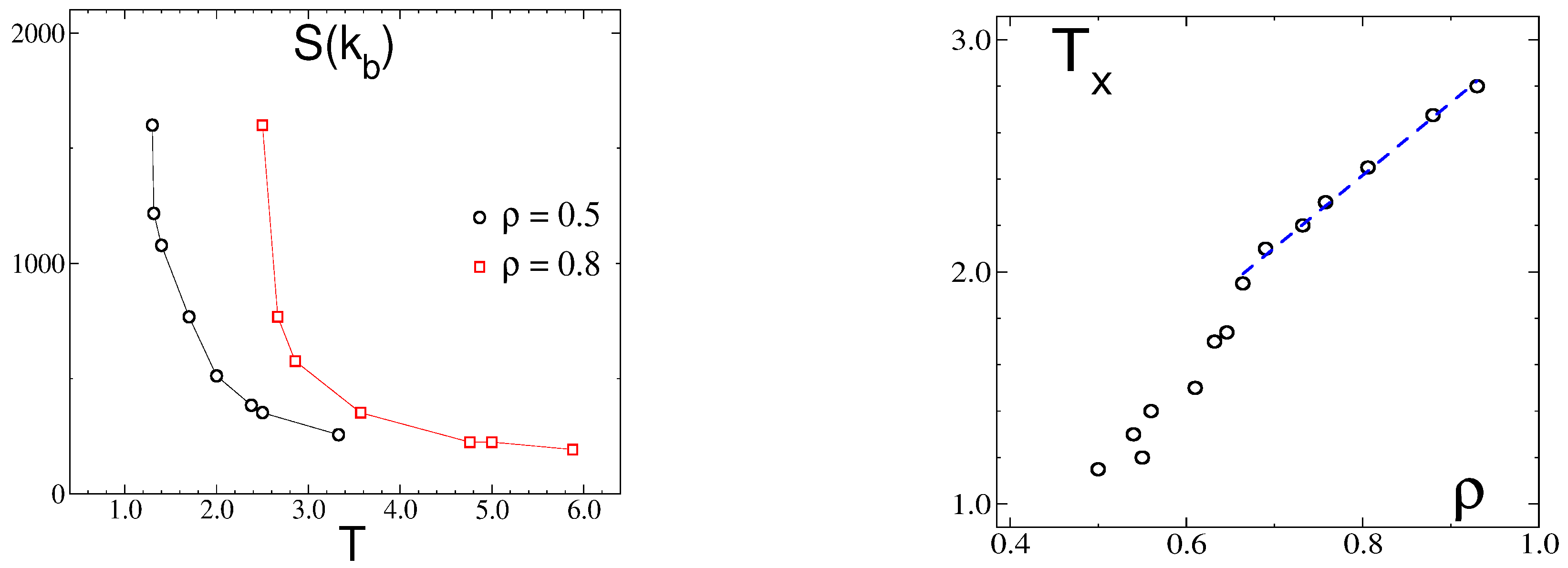

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Virgiliis, A.; Meyra, A.; Ciach, A. Lattice Model Results for Pattern Formation in a Mixture with Competing Interactions. Molecules 2024, 29, 1512. https://doi.org/10.3390/molecules29071512
De Virgiliis A, Meyra A, Ciach A. Lattice Model Results for Pattern Formation in a Mixture with Competing Interactions. Molecules. 2024; 29(7):1512. https://doi.org/10.3390/molecules29071512
Chicago/Turabian StyleDe Virgiliis, Andres, Ariel Meyra, and Alina Ciach. 2024. "Lattice Model Results for Pattern Formation in a Mixture with Competing Interactions" Molecules 29, no. 7: 1512. https://doi.org/10.3390/molecules29071512
APA StyleDe Virgiliis, A., Meyra, A., & Ciach, A. (2024). Lattice Model Results for Pattern Formation in a Mixture with Competing Interactions. Molecules, 29(7), 1512. https://doi.org/10.3390/molecules29071512