

Enhanced Stability of Dimethyl Ether Carbonylation through Pyrazole Tartrate on Tartaric Acid-Complexed Cobalt–Iron-Modified Hydrogen-Type Mordenite

Abstract
1. Introduction
2. Results and Discussion
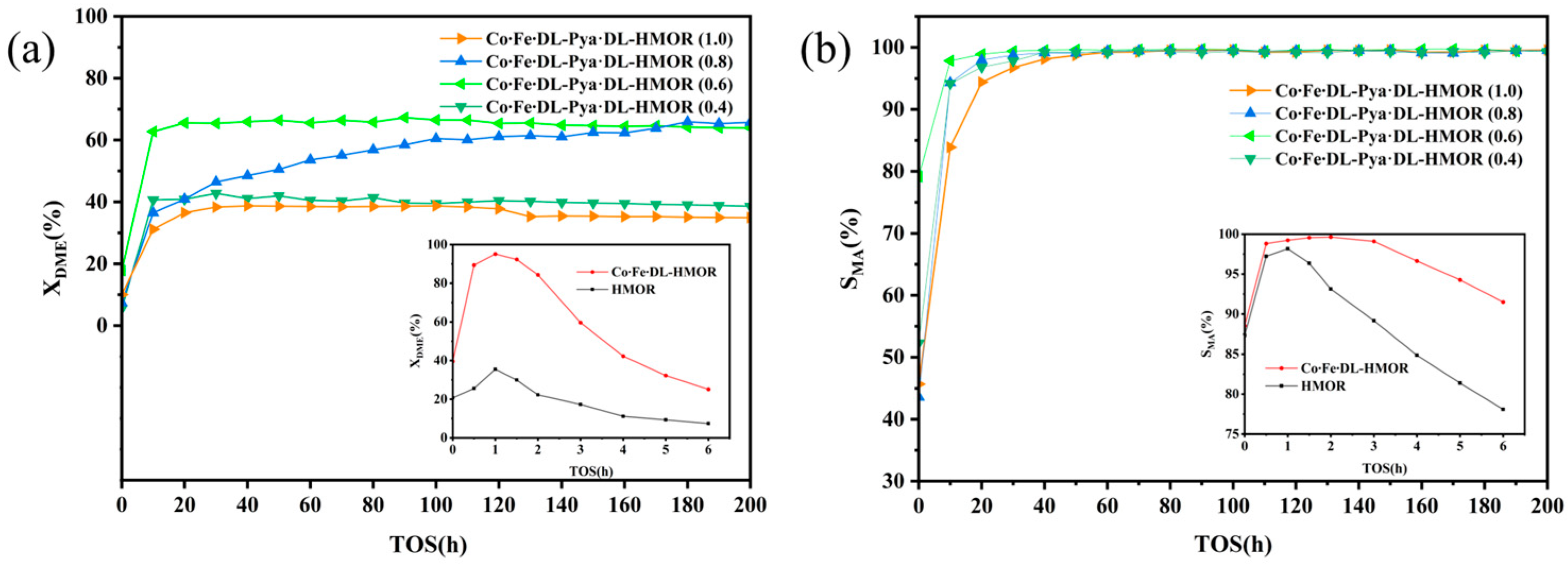
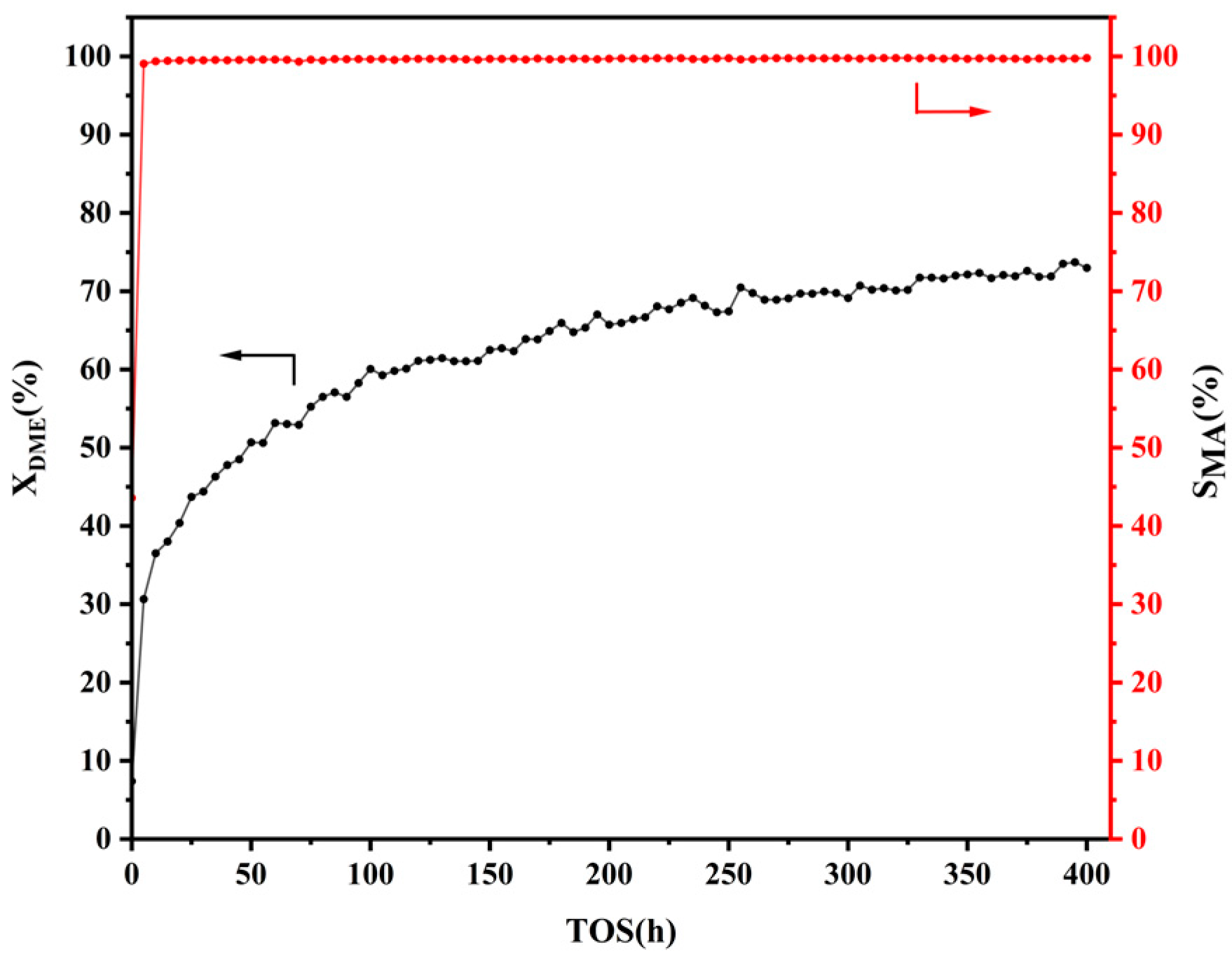
2.1. Catalytic Performance
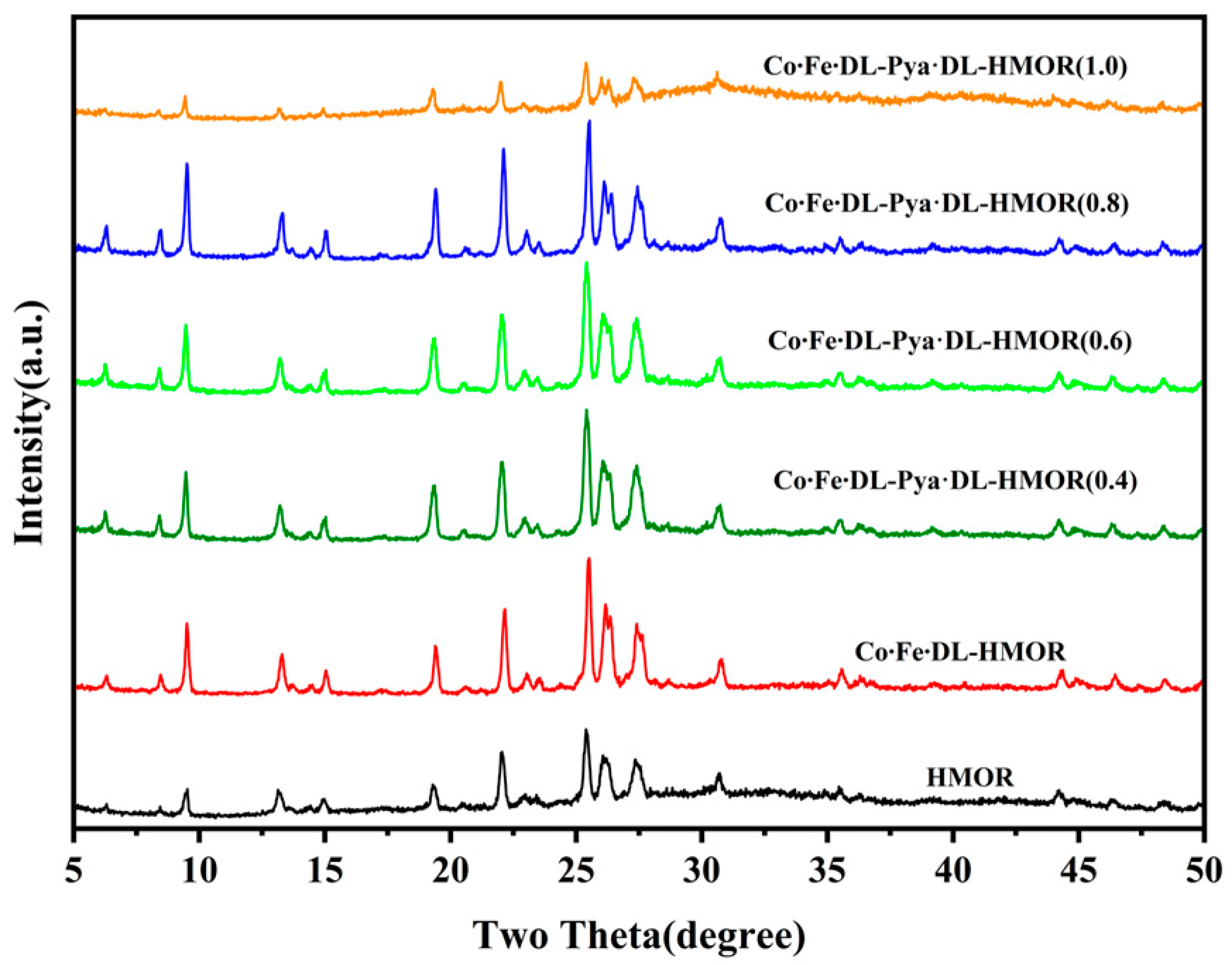
2.2. XRD Analysis
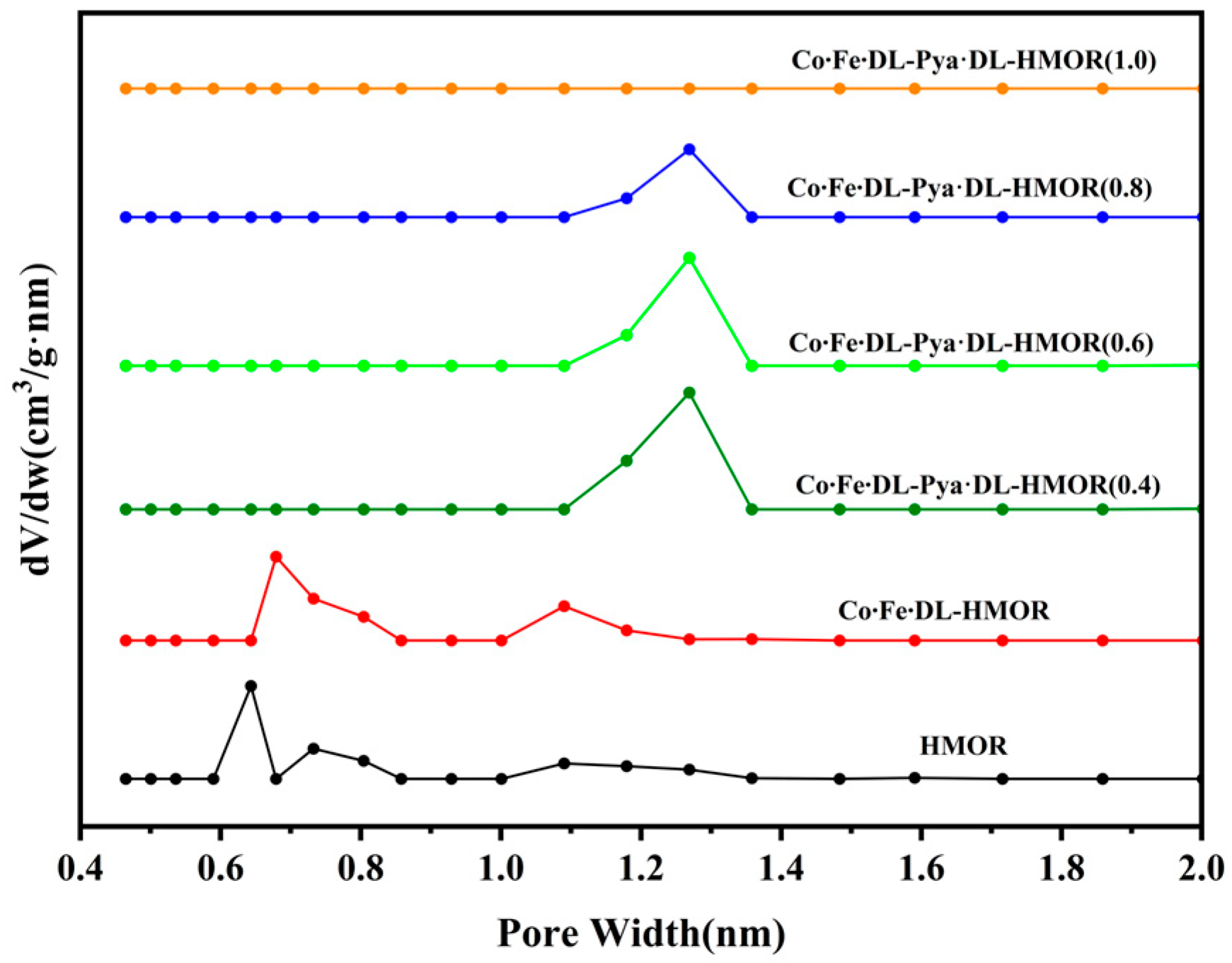
2.3. BET Test
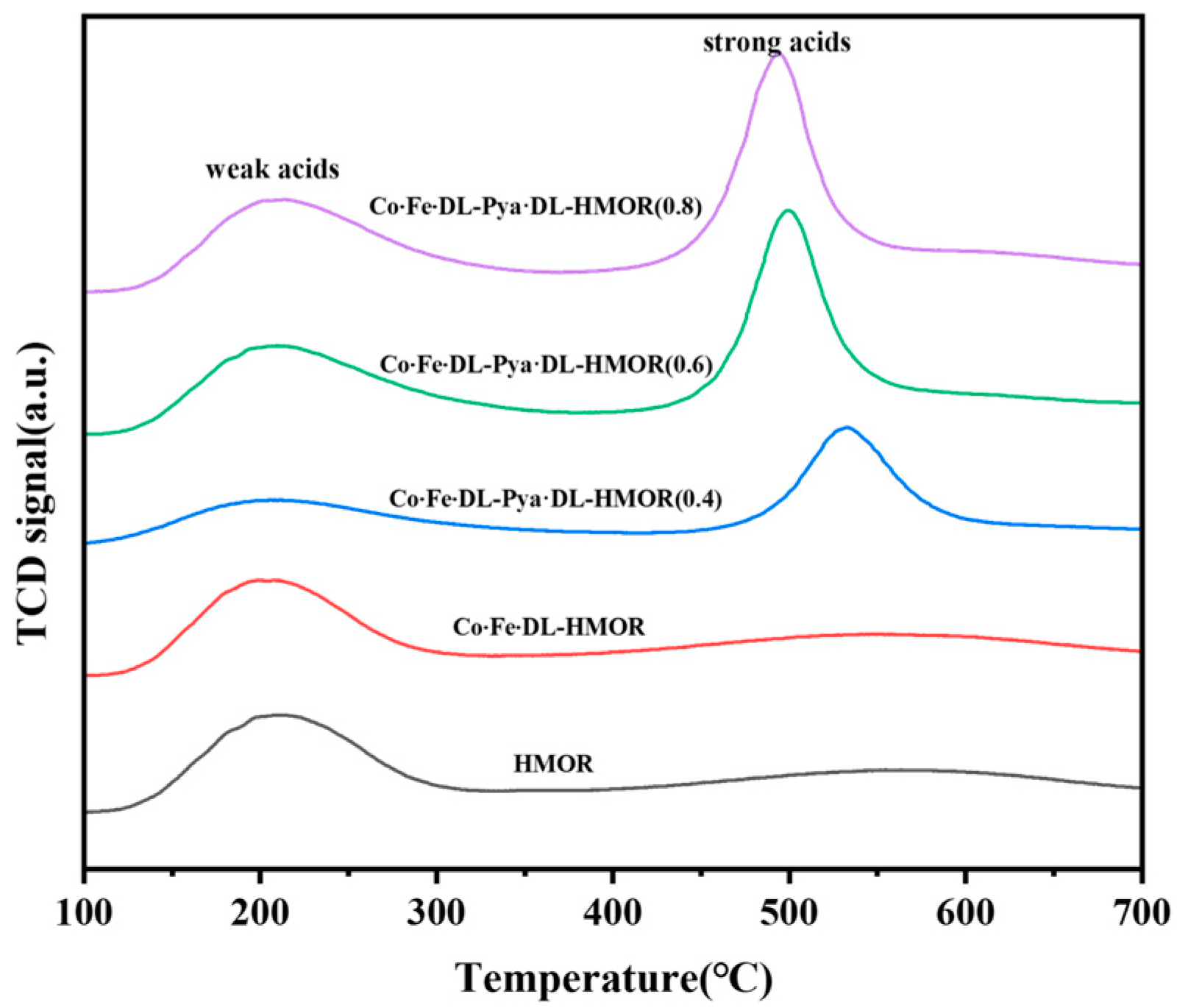
2.4. NH3-TPD Analysis
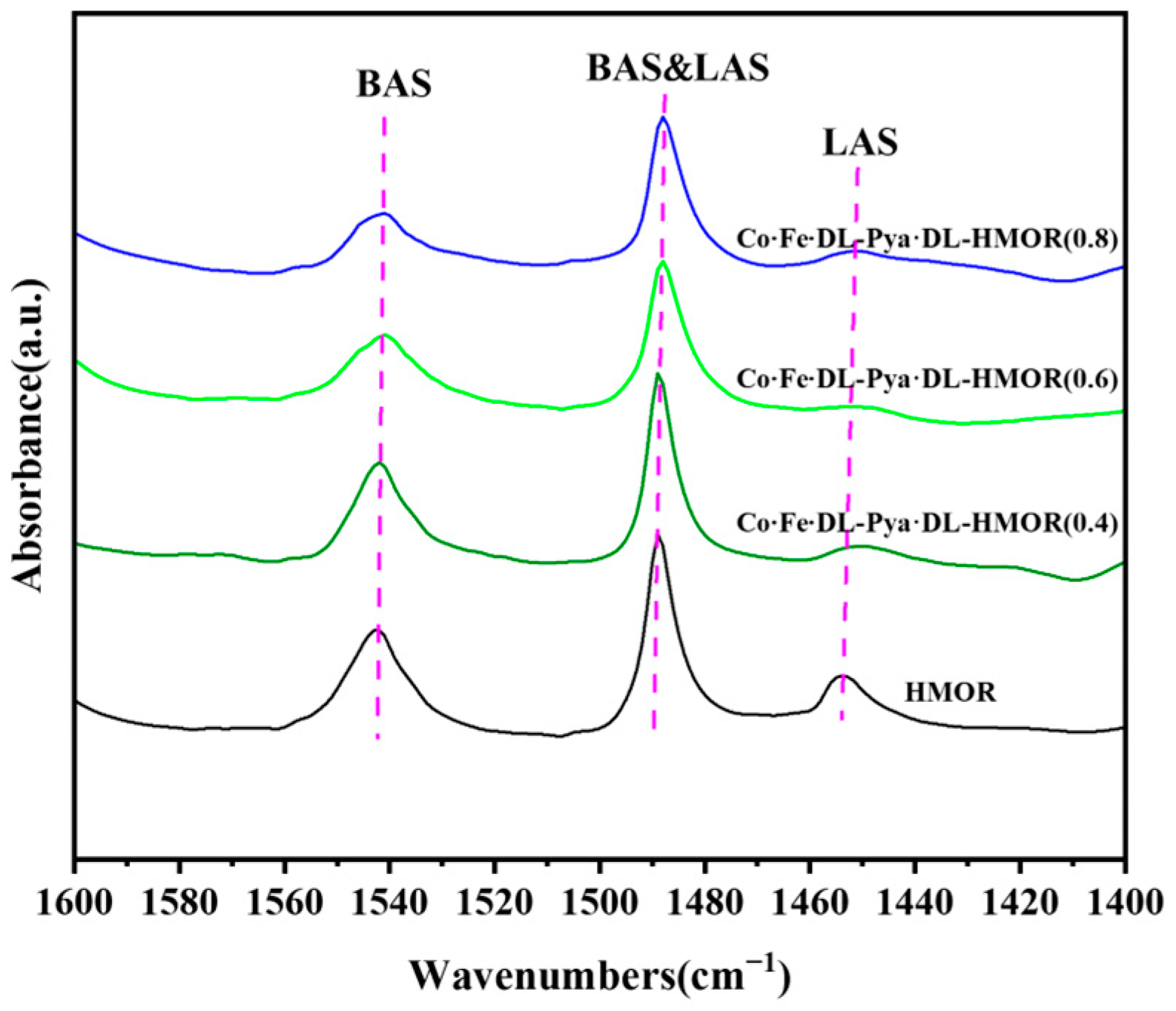
2.5. Py-IR Analysis
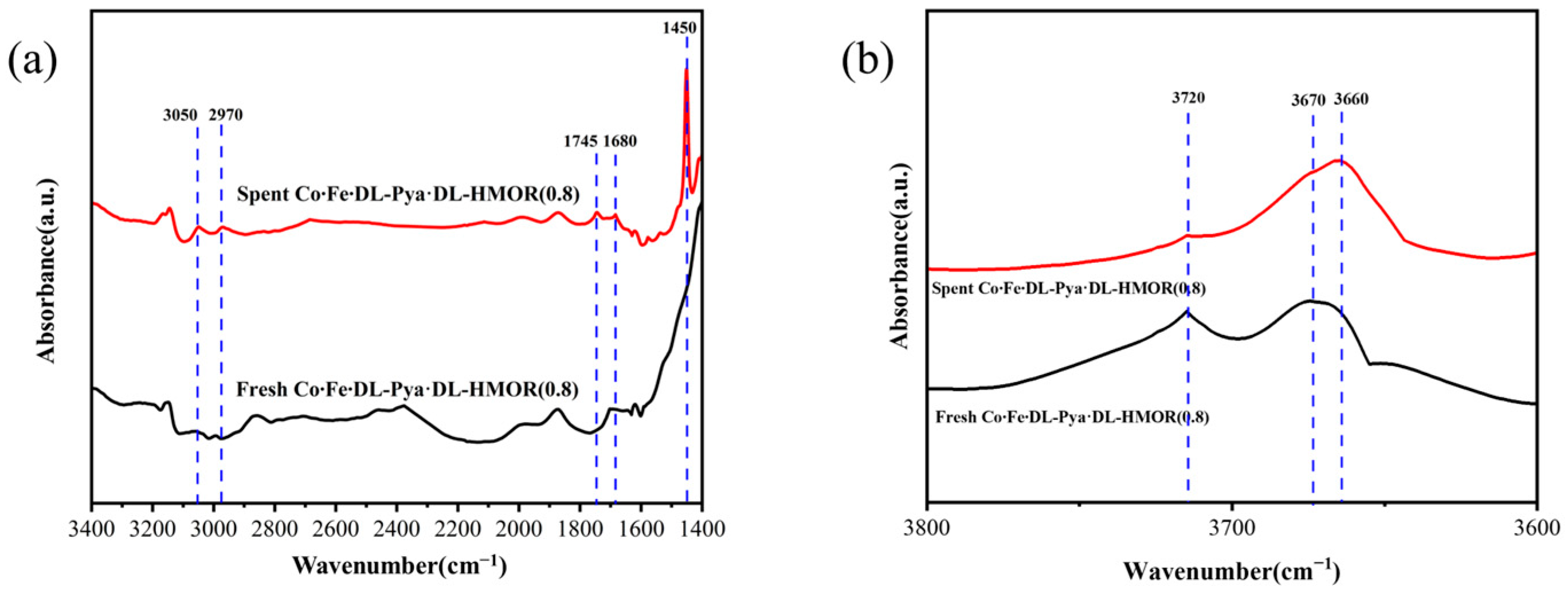
2.6. FT-IR Analysis
2.7. TG Analysis
3. Materials and Methods
3.1. Materials
3.2. Preparation of the Catalyst
3.3. Characterization
3.4. DME Carbonylation Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luk, H.T.; Mondelli, C.; Ferré, D.C.; Stewart, J.A.; Pérez-Ramírez, J. Status and Prospects in Higher Alcohols Synthesis from Syngas. Chem. Soc. Rev. 2017, 46, 1358–1426. [Google Scholar] [CrossRef]
- Cheung, P.; Bhan, A.; Sunley, G.; Law, D.; Iglesia, E. Site Requirements and Elementary Steps in Dimethyl Ether Carbonylation Catalyzed by Acidic Zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Blasco, T.; Boronat, M.; Concepción, P.; Corma, A.; Law, D.; Vidal-Moya, J.A. Carbonylation of Methanol on Metal–Acid Zeolites: Evidence for a Mechanism Involving a Multisite Active Center. Angew. Chem. Int. Ed. 2007, 46, 3938–3941. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Ren, P.; Liu, X.; Xu, S.; Han, X.; Bao, X. Direct Observation of DME Carbonylation in the Different Channels of H-MOR Zeolite by Continuous-Flow Solid-State NMR Spectroscopy. Chem. Commun. 2015, 51, 16868–16870. [Google Scholar] [CrossRef]
- Yang, G.; San, X.; Jiang, N.; Tanaka, Y.; Li, X.; Jin, Q.; Tao, K.; Meng, F.; Tsubaki, N. A New Method of Ethanol Synthesis from Dimethyl Ether and Syngas in a Sequential Dual Bed Reactor with the Modified Zeolite and Cu/ZnO Catalysts. Catal. Today 2011, 164, 425–428. [Google Scholar] [CrossRef]
- Li, X.; San, X.; Zhang, Y.; Ichii, T.; Meng, M.; Tan, Y.; Tsubaki, N. Direct Synthesis of Ethanol from Dimethyl Ether and Syngas over Combined H-Mordenite and Cu/ZnO Catalysts. ChemSusChem 2010, 3, 1192–1199. [Google Scholar] [CrossRef]
- Wang, D.; Yang, G.; Ma, Q.; Yoneyama, Y.; Tan, Y.; Han, Y.; Tsubaki, N. Facile Solid-State Synthesis of Cu–Zn–O Catalysts for Novel Ethanol Synthesis from Dimethyl Ether (DME) and Syngas (CO+H2). Fuel 2013, 109, 54–60. [Google Scholar] [CrossRef]
- Lu, P.; Yang, G.; Tanaka, Y.; Tsubaki, N. Ethanol Direct Synthesis from Dimethyl Ether and Syngas on the Combination of Noble Metal Impregnated Zeolite with Cu/ZnO Catalyst. Catal. Today 2014, 232, 22–26. [Google Scholar] [CrossRef]
- San, X.; Zhang, Y.; Shen, W.; Tsubaki, N. New Synthesis Method of Ethanol from Dimethyl Ether with a Synergic Effect between the Zeolite Catalyst and Metallic Catalyst. Energy Fuels 2009, 23, 2843–2844. [Google Scholar] [CrossRef]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective Carbonylation of Dimethyl Ether to Methyl Acetate Catalyzed by Acidic Zeolites. Angew. Chem. Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef]
- Huo, H.; Peng, L.; Gan, Z.; Grey, C.P. Solid-State MAS NMR Studies of Brønsted Acid Sites in Zeolite H-Mordenite. J. Am. Chem. Soc. 2012, 134, 9708–9720. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, J.; Xu, C.; Zhu, W.; Wu, P. An Insight into Crystal Morphology-Dependent Catalytic Properties of MOR-Type Titanosilicate in Liquid-Phase Selective Oxidation. J. Catal. 2015, 325, 101–110. [Google Scholar] [CrossRef]
- Bhan, A.; Iglesia, E. A Link between Reactivity and Local Structure in Acid Catalysis on Zeolites. Acc. Chem. Res. 2008, 41, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, S.; Cheng, Z.; Wang, S.; Ge, Q.; Ma, X. Synergy between Cu and Brønsted Acid Sites in Carbonylation of Dimethyl Ether over Cu/H-MOR. J. Catal. 2018, 365, 440–449. [Google Scholar] [CrossRef]
- Wang, S.; Guo, W.; Zhu, L.; Wang, H.; Qiu, K.; Cen, K. Methyl Acetate Synthesis from Dimethyl Ether Carbonylation over Mordenite Modified by Cation Exchange. J. Phys. Chem. C 2015, 119, 524–533. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Ma, X.; Liu, Y.; Zhu, W.; Liu, Z. Identifying and Controlling the Acid Site Distributions in Mordenite Zeolite for Dimethyl Ether Carbonylation Reaction by Means of Selective Ion-Exchange. Catal. Sci. Technol. 2020, 10, 4663–4672. [Google Scholar] [CrossRef]
- Liu, S.; Fang, X.; Liu, Y.; Liu, H.; Ma, X.; Zhu, W.; Liu, Z. Dimethyl Ether Carbonylation over Mordenite Zeolite Modified by Alkyimidazolium Ions. Catal. Commun. 2020, 147, 106161. [Google Scholar] [CrossRef]
- Ma, M.; Zhan, E.; Huang, X.; Ta, N.; Xiong, Z.; Bai, L.; Shen, W. Carbonylation of Dimethyl Ether over Co-HMOR. Catal. Sci. Technol. 2018, 8, 2124–2130. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, W.; Shi, L.; Liu, H.; Liu, S.; Xu, S.; Ni, Y.; Liu, Y.; Li, L.; Liu, Z. Promotion Effect of Fe in Mordenite Zeolite on Carbonylation of Dimethyl Ether to Methyl Acetate. Catal. Sci. Technol. 2015, 5, 1961–1968. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, Y.; Geng, J.; Dong, X. Enhancing the Dimethyl Ether Carbonylation Performance over Hydrogen-Type Mordenites Modified by Pyrazole Hydrochloride. RSC Adv. 2022, 12, 123–128. [Google Scholar] [CrossRef]
- Reule, A.A.C.; Prasad, V.; Semagina, N. Effect of Cu and Zn Ion-Exchange Locations on Mordenite Performance in Dimethyl Ether Carbonylation. Microporous Mesoporous Mater. 2018, 263, 220–230. [Google Scholar] [CrossRef]
- Zhang, L.-Y.; Feng, X.-B.; He, Z.-M.; Chen, F.; Su, C.; Zhao, X.-Y.; Cao, J.-P.; He, Y.-R. Enhancing the Stability of Dimethyl Ether Carbonylation over Fe-Doped MOR Zeolites with Tunable 8-MR Acidity. Chem. Eng. Sci. 2022, 256, 117671. [Google Scholar] [CrossRef]
- Liu, R.; Zeng, S.; Sun, T.; Xu, S.; Yu, Z.; Wei, Y.; Liu, Z. Selective Removal of Acid Sites in Mordenite Zeolite by Trimethylchlorosilane Silylation to Improve Dimethyl Ether Carbonylation Stability. ACS Catal. 2022, 12, 4491–4500. [Google Scholar] [CrossRef]
- Reule, A.A.C.; Semagina, N. Zinc Hinders Deactivation of Copper-Mordenite: Dimethyl Ether Carbonylation. ACS Catal. 2016, 6, 4972–4975. [Google Scholar] [CrossRef]
- Ban, S.; van Laak, A.N.C.; Landers, J.; Neimark, A.V.; de Jongh, P.E.; de Jong, K.P.; Vlugt, T.J.H. Insight into the Effect of Dealumination on Mordenite Using Experimentally Validated Simulations. J. Phys. Chem. C 2010, 114, 2056–2065. [Google Scholar] [CrossRef]
- Reule, A.A.C.; Sawada, J.A.; Semagina, N. Effect of Selective 4-Membered Ring Dealumination on Mordenite-Catalyzed Dimethyl Ether Carbonylation. J. Catal. 2017, 349, 98–109. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, J.; Gu, Y.; Zhang, W.; Cao, K.; Cui, W.; Xu, S.; Fan, D.; Tian, P.; Liu, Z. Designed Synthesis of MOR Zeolites Using Gemini-Type Bis(Methylpyrrolidinium) Dications as Structure Directing Agents and Their DME Carbonylation Performance. J. Mater. Chem. A 2022, 10, 8334–8343. [Google Scholar] [CrossRef]
- Wang, M.; Huang, S.; Lü, J.; Cheng, Z.; Li, Y.; Wang, S.; Ma, X. Modifying the Acidity of H-MOR and Its Catalytic Carbonylation of Dimethyl Ether. Chin. J. Catal. 2016, 37, 1530–1537. [Google Scholar] [CrossRef]
- Zhao, P.; Qian, W.; Ma, H.; Sheng, H.; Zhang, H.; Ying, W. Effect of Zr Incorporation on Mordenite Catalyzed Dimethyl Ether Carbonylation. Catal. Lett. 2021, 151, 940–954. [Google Scholar] [CrossRef]
- Zhao, N.; Cheng, Q.; Lyu, S.; Guo, L.; Tian, Y.; Ding, T.; Xu, J.; Ma, X.; Li, X. Promoting Dimethyl Ether Carbonylation over Hot-Water Pretreated H-Mordenite. Catal. Today 2020, 339, 86–92. [Google Scholar] [CrossRef]
- Liu, J.; Xue, H.; Huang, X.; Wu, P.-H.; Huang, S.-J.; Liu, S.-B.; Shen, W. Stability Enhancement of H-Mordenite in Dimethyl Ether Carbonylation to Methyl Acetate by Pre-Adsorption of Pyridine. Chin. J. Catal. 2010, 31, 729–738. [Google Scholar] [CrossRef]
- Li, Y.; Huang, S.; Cheng, Z.; Cai, K.; Li, L.; Milan, E.; Lv, J.; Wang, Y.; Sun, Q.; Ma, X. Promoting the Activity of Ce-Incorporated MOR in Dimethyl Ether Carbonylation through Tailoring the Distribution of Brønsted Acids. Appl. Catal. B Environ. 2019, 256, 117777. [Google Scholar] [CrossRef]
- Xia, Y.; Li, Z.; Li, Y.; Cai, K.; Liu, Y.; Lv, J.; Huang, S.; Ma, X. Promotion Effect and Mechanism of Ga Modification on Dimethyl Ether Carbonylation Catalyzed by Mordenite. Catal. Today 2022, 405–406, 152–158. [Google Scholar] [CrossRef]
- Chen, J.G.; Basu, P.; Ballinger, T.H.; Yates, J.T. A Transmission Infrared Spectroscopic Investigation of the Reaction of Dimethyl Ether with Alumina Surfaces. Langmuir 1989, 5, 352–356. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, D.; Liu, Z.; Su, B. Highly Efficient Catalytic Conversion of Chloromethane to Light Olefins over HSAPO-34 as Studied by Catalytic Testing and In Situ FTIR. J. Catal. 2006, 238, 46–57. [Google Scholar] [CrossRef]
- Bhan, A.; Allian, A.D.; Sunley, G.J.; Law, D.J.; Iglesia, E. Specificity of Sites within Eight-Membered Ring Zeolite Channels for Carbonylation of Methyls to Acetyls. J. Am. Chem. Soc. 2007, 129, 4919–4924. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Du, Y.; Ye, C.; Wang, J.; Ding, T.; Li, H. Acid Sites and Hydration Behavior of Dealuminated Zeolite HZSM-5: A High-Resolution Solid State NMR Study. J. Phys. Chem. 1995, 99, 15208–15214. [Google Scholar] [CrossRef]
- Hayashi, S.; Kojima, N. Acid Properties of H-Type Mordenite Studied by Solid-State NMR. Microporous Mesoporous Mater. 2011, 141, 49–55. [Google Scholar] [CrossRef]
- Boronat, M.; Martínez-Sánchez, C.; Law, D.; Corma, A. Enzyme-like Specificity in Zeolites: A Unique Site Position in Mordenite for Selective Carbonylation of Methanol and Dimethyl Ether with CO. J. Am. Chem. Soc. 2008, 130, 16316–16323. [Google Scholar] [CrossRef]
- He, T.; Liu, X.; Xu, S.; Han, X.; Pan, X.; Hou, G.; Bao, X. Role of 12-Ring Channels of Mordenite in DME Carbonylation Investigated by Solid-State NMR. J. Phys. Chem. C 2016, 120, 22526–22531. [Google Scholar] [CrossRef]
- Liu, Z.; Yi, X.; Wang, G.; Tang, X.; Li, G.; Huang, L.; Zheng, A. Roles of 8-Ring and 12-Ring Channels in Mordenite for Carbonylation Reaction: From the Perspective of Molecular Adsorption and Diffusion. J. Catal. 2019, 369, 335–344. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Q.; Huang, S.; Cheng, Z.; Cai, K.; Lv, J.; Ma, X. Dimethyl Ether Carbonylation over Pyridine-Modified MOR: Enhanced Stability Influenced by Acidity. Catal. Today 2018, 311, 81–88. [Google Scholar] [CrossRef]
- Cao, K.; Fan, D.; Li, L.; Fan, B.; Wang, L.; Zhu, D.; Wang, Q.; Tian, P.; Liu, Z. Insights into the Pyridine-Modified MOR Zeolite Catalysts for DME Carbonylation. ACS Catal. 2020, 10, 3372–3380. [Google Scholar] [CrossRef]
- Zhao, N.; Tian, Y.; Zhang, L.; Cheng, Q.; Lyu, S.; Ding, T.; Hu, Z.; Ma, X.; Li, X. Spacial Hindrance Induced Recovery of Over-Poisoned Active Acid Sites in Pyridine-Modified H-Mordenite for Dimethyl Ether Carbonylation. Chin. J. Catal. 2019, 40, 895–904. [Google Scholar] [CrossRef]
- Xue, H.; Huang, X.; Zhan, E.; Ma, M.; Shen, W. Selective Dealumination of Mordenite for Enhancing Its Stability in Dimethyl Ether Carbonylation. Catal. Commun. 2013, 37, 75–79. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET/(m2/g) | Vtotal/(cm3/g) |
|---|---|---|
| HMOR | 352 | 0.257 |
| Co∙Fe∙DL-HMOR | 335 | 0.211 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.4) | 238 | 0.171 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.6) | 142 | 0.118 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.8) | 53.1 | 0.087 |
| Co∙Fe∙DL-Pya·DL-HMOR (1.0) | 6.07 | 0.043 |
| Sample | BAS in the 12 MR/(µmol/g) | LAS in the 12 MR/(µmol/g) | BAS and LAS in the 12 MR/(µmol/g) |
|---|---|---|---|
| HMOR | 237 | 57 | 294 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.4) | 172 | 28 | 200 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.6) | 113 | 18 | 131 |
| Co∙Fe∙DL-Pya·DL-HMOR (0.8) | 58 | 16 | 74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, G.; Dong, X. Enhanced Stability of Dimethyl Ether Carbonylation through Pyrazole Tartrate on Tartaric Acid-Complexed Cobalt–Iron-Modified Hydrogen-Type Mordenite. Molecules 2024, 29, 1510. https://doi.org/10.3390/molecules29071510
Fu G, Dong X. Enhanced Stability of Dimethyl Ether Carbonylation through Pyrazole Tartrate on Tartaric Acid-Complexed Cobalt–Iron-Modified Hydrogen-Type Mordenite. Molecules. 2024; 29(7):1510. https://doi.org/10.3390/molecules29071510
Chicago/Turabian StyleFu, Guangtao, and Xinfa Dong. 2024. "Enhanced Stability of Dimethyl Ether Carbonylation through Pyrazole Tartrate on Tartaric Acid-Complexed Cobalt–Iron-Modified Hydrogen-Type Mordenite" Molecules 29, no. 7: 1510. https://doi.org/10.3390/molecules29071510
APA StyleFu, G., & Dong, X. (2024). Enhanced Stability of Dimethyl Ether Carbonylation through Pyrazole Tartrate on Tartaric Acid-Complexed Cobalt–Iron-Modified Hydrogen-Type Mordenite. Molecules, 29(7), 1510. https://doi.org/10.3390/molecules29071510