
Bacterial Chaperone Domain Insertions Convert Human FKBP12 into an Excellent Protein-Folding Catalyst—A Structural and Functional Analysis
 ,
,
Abstract
1. Introduction
2. Results
2.1. The Chimeric Proteins Are Stable and Catalytically Active
2.2. Chimeric Fusion Proteins Catalyze the Protein Refolding Very Well
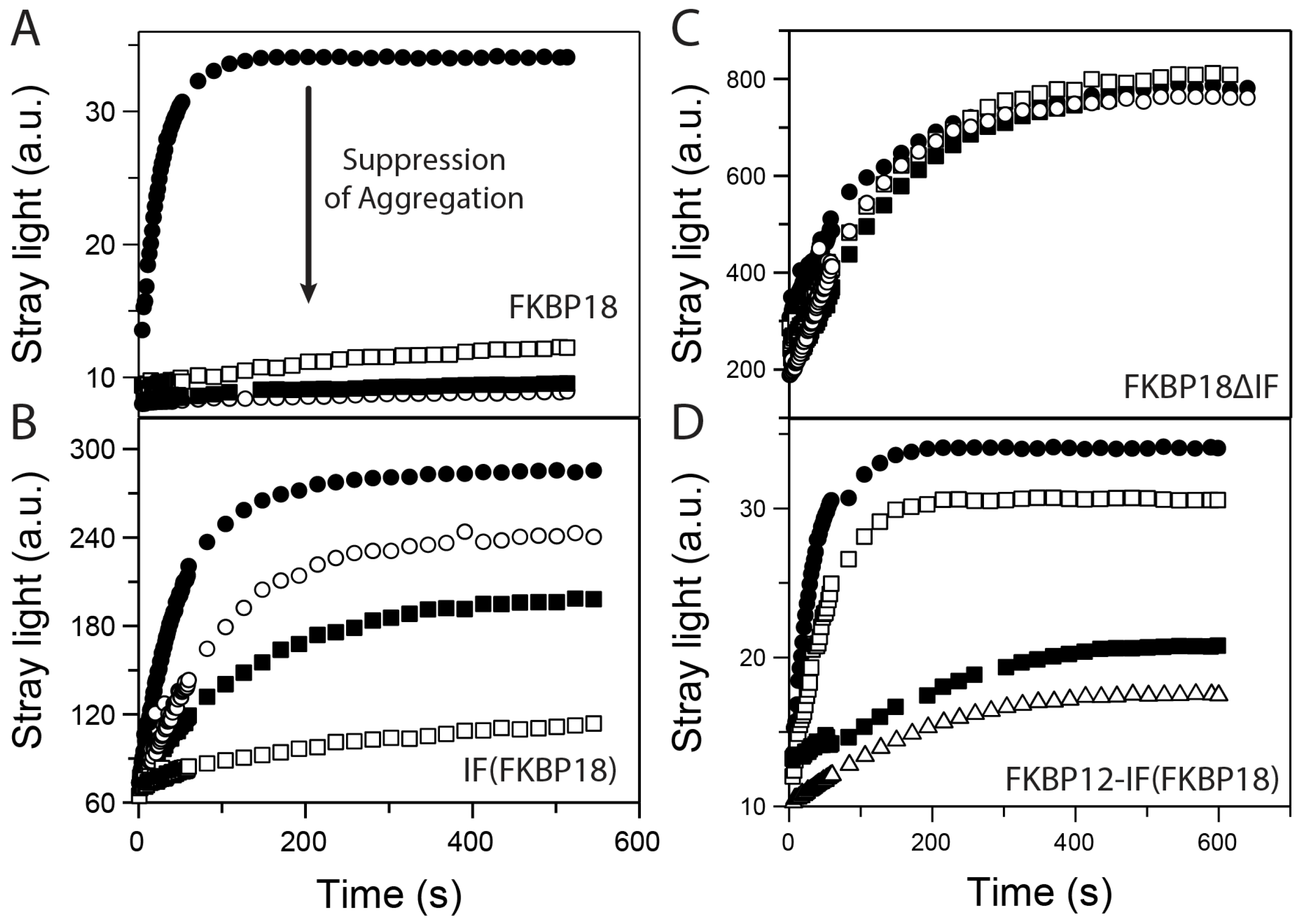
2.3. Chimeric Fusion Proteins Are Good Chaperones
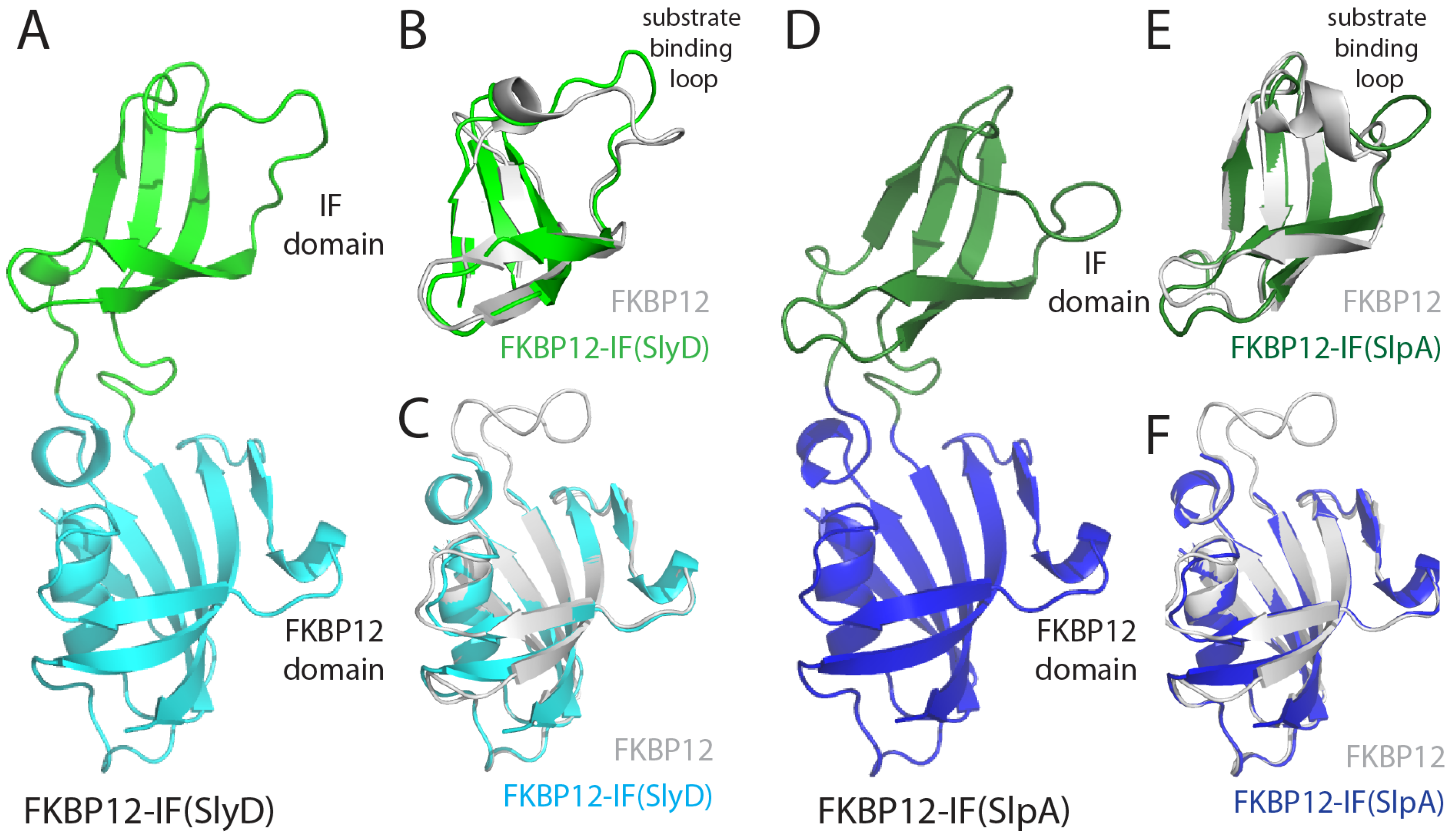
2.4. FKBP12-IF(SlyD) and FKBP12-IF(SlpA) Crystal Structures
3. Discussion
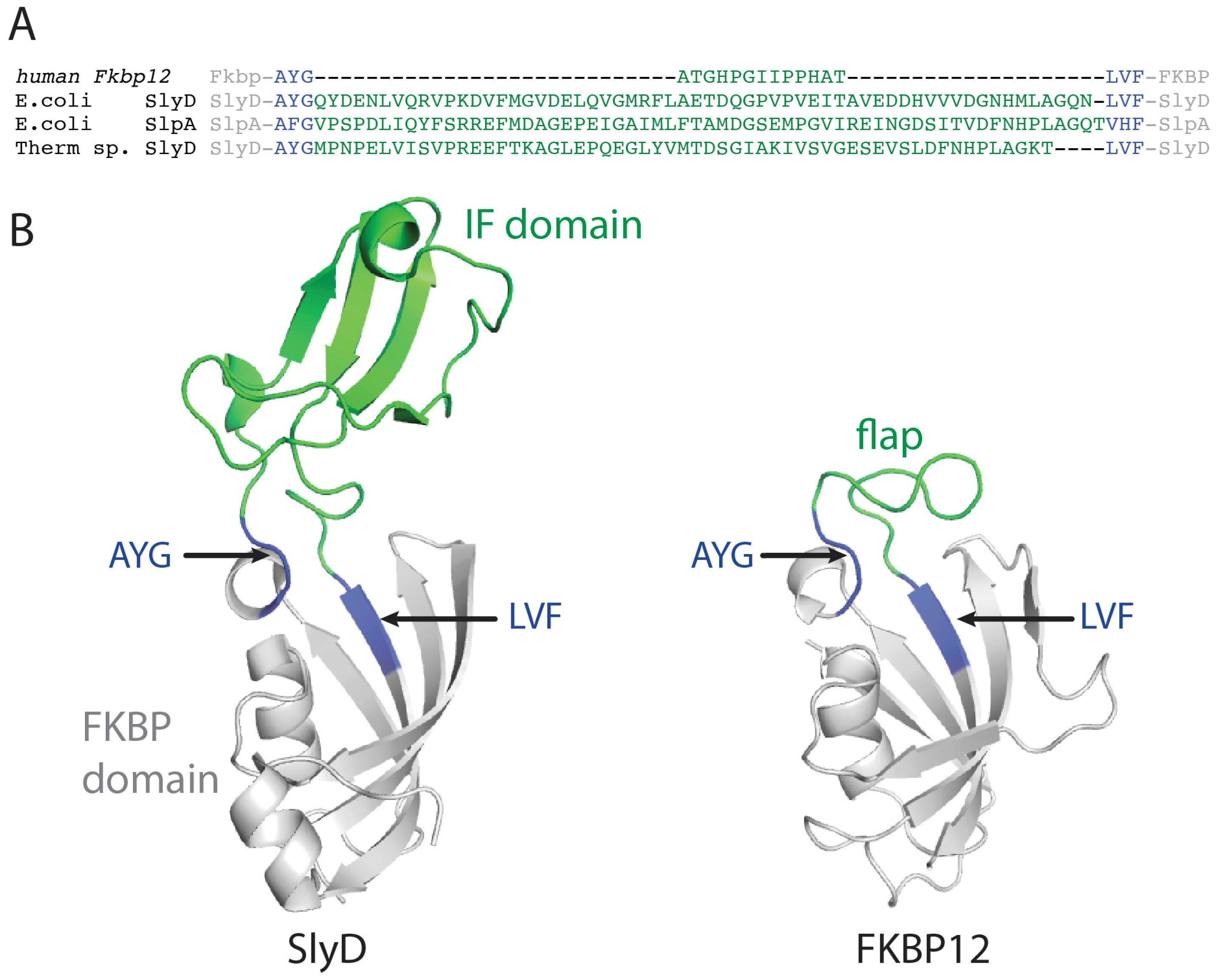
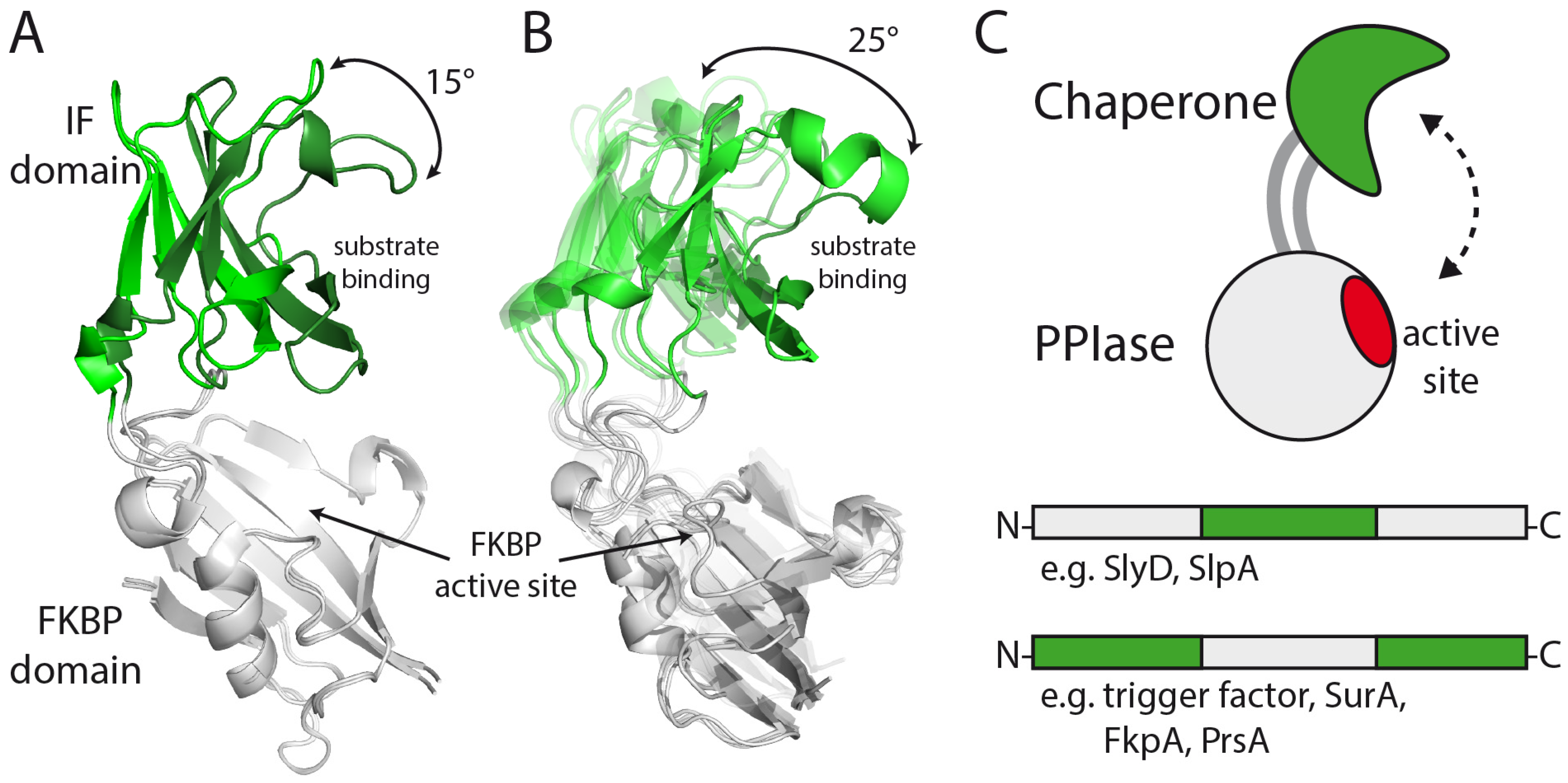
3.1. Domain Exchange between FKBP12 and SlyD-like Proteins
3.2. The Folding Efficiency of FKBP12 Is Improved by Chaperone Domains
3.3. Structure and Large-Scale Dynamics of Chimeric Proteins
4. Materials and Methods
4.1. GdmCl and Urea-Induced Unfolding Transitions
4.2. Prolyl Isomerase and Chaperone Activity Assays
4.3. Protein Crystallization and Structure Determination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schindler, T.; Herrler, M.; Marahiel, M.A.; Schmid, F.X. Extremely rapid folding in the absence of intermediates: The cold-shock protein from Bacillus subtilis. Nat. Struct. Biol. 1995, 2, 663–673. [Google Scholar] [CrossRef]
- Ferguson, N.; Fersht, A.R. Early events in protein folding. Curr. Opin. Struct. Biol. 2003, 13, 75–81. [Google Scholar] [CrossRef]
- Huang, G.S.; Oas, T.G. Submillisecond folding of monomeric lambda repressor. Proc. Natl. Acad. Sci. USA 1995, 92, 6878–6882. [Google Scholar] [CrossRef]
- Mayor, U.; Johnson, C.M.; Daggett, V.; Fersht, A.R. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc. Natl. Acad. Sci. USA 2000, 97, 13518–13522. [Google Scholar] [CrossRef] [PubMed]
- Schmidpeter, P.A.; Schmid, F.X. Prolyl isomerization and its catalysis in protein folding and protein function. J. Mol. Biol. 2015, 427, 1609–1631. [Google Scholar] [CrossRef]
- Schiene-Fischer, C.; Aumüller, T.; Fischer, G. Peptide bond cis/trans isomerases: A biocatalysis perspective of conformational dynamics in proteins. Top. Curr. Chem. 2013, 328, 35–67. [Google Scholar] [CrossRef]
- Stewart, D.E.; Sarkar, A.; Wampler, J.E. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 1990, 214, 253–260. [Google Scholar] [CrossRef]
- Schmid, F.X. Prolyl isomerases. Adv. Protein Chem. 2002, 59, 243–282. [Google Scholar]
- Fischer, G. Peptidyl-prolyl cis/trans isomerases and their effectors. Angew. Chem. Int. Ed. Engl. 1994, 33, 1415–1436. [Google Scholar] [CrossRef]
- Schiene-Fischer, C. Multidomain peptidyl prolyl cis/trans Isomerases. Biochim. Biophys. Acta 2015, 1850, 2005–2016. [Google Scholar] [CrossRef]
- Galat, A. Peptidylprolyl cis/trans isomerases (immunophilins): Biological diversity—Targets—functions. Curr. Top. Med. Chem. 2003, 3, 1315–1347. [Google Scholar] [CrossRef] [PubMed]
- Ferbitz, L.; Maier, T.; Patzelt, H.; Bukau, B.; Deuerling, E.; Ban, N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 2004, 431, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Rahfeld, J.U.; Rucknagel, K.P.; Stoller, G.; Horne, S.M.; Schierhorn, A.; Young, K.D.; Fischer, G. Isolation and amino acid sequence of a new 22-kDa FKBP-like peptidyl-prolyl cis/trans-isomerase of Escherichia coli. Similarity to Mip-like proteins of pathogenic bacteria. J. Biol. Chem. 1996, 271, 22130–22138. [Google Scholar] [CrossRef] [PubMed]
- Saul, F.A.; Arie, J.P.; Vulliez-le Normand, B.; Kahn, R.; Betton, J.M.; Bentley, G.A. Structural and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J. Mol. Biol. 2004, 335, 595–608. [Google Scholar] [CrossRef]
- Hottenrott, S.; Schumann, T.; Plückthun, A.; Fischer, G.; Rahfeld, J.U. The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem. 1997, 272, 15697–15701. [Google Scholar] [CrossRef]
- Weininger, U.; Haupt, C.; Schweimer, K.; Graubner, W.; Kovermann, M.; Bruser, T.; Scholz, C.; Schaarschmidt, P.; Žoldák, G.; Schmid, F.X.; et al. NMR solution structure of SlyD from Escherichia coli: Spatial separation of prolyl isomerase and chaperone function. J. Mol. Biol. 2009, 387, 295–305. [Google Scholar] [CrossRef]
- Löw, C.; Neumann, P.; Tidow, H.; Weininger, U.; Haupt, C.; Friedrich-Epler, B.; Scholz, C.; Stubbs, M.T.; Balbach, J. Crystal structure determination and functional characterization of the metallochaperone SlyD from Thermus thermophilus. J. Mol. Biol. 2010, 398, 375–390. [Google Scholar] [CrossRef]
- Van Duyne, G.D.; Standaert, R.F.; Karplus, P.A.; Schreiber, S.L.; Clardy, J. Atomic structure of FKBP-FK506, an immunophilin-immunosuppressant complex. Science 1991, 252, 839–842. [Google Scholar] [CrossRef]
- Harrison, R.K.; Stein, R.L. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: Evidence for the existence of a family of distinct enzymes. Biochemistry 1990, 29, 3813–3816. [Google Scholar] [CrossRef]
- Jakob, R.P.; Žoldák, G.; Aumueller, T.; Schmid, F.X. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc. Natl. Acad. Sci. USA 2009, 106, 20282–20287. [Google Scholar] [CrossRef]
- Žoldák, G.; Aumüller, T.; Lücke, C.; Hritz, J.; Oostenbrink, C.; Fischer, G.; Schmid, F.X. A library of fluorescent peptides for exploring the substrate specificities of prolyl isomerases. Biochemistry 2009, 48, 10423–10436. [Google Scholar] [CrossRef] [PubMed]
- Knappe, T.A.; Eckert, B.; Schaarschmidt, P.; Scholz, C.; Schmid, F.X. Insertion of a chaperone domain converts FKBP12 into a powerful catalyst of protein folding. J. Mol. Biol. 2007, 368, 1458–1468. [Google Scholar] [CrossRef]
- Jakob, R.P.; Schmid, F.X. Molecular determinants of a native-state prolyl isomerization. J. Mol. Biol. 2009, 387, 1017–1031. [Google Scholar] [CrossRef]
- Quistgaard, E.M.; Nordlund, P.; Löw, C. High-resolution insights into binding of unfolded polypeptides by the PPIase chaperone SlpA. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4003–4013. [Google Scholar] [CrossRef]
- Geitner, A.J.; Weininger, U.; Paulsen, H.; Balbach, J.; Kovermann, M. Structure-Based Insights into the Dynamics and Function of Two-Domain SlpA from Escherichia coli. Biochemistry 2017, 56, 6533–6543. [Google Scholar] [CrossRef]
- Suzuki, R.; Nagata, K.; Yumoto, F.; Kawakami, M.; Nemoto, N.; Furutani, M.; Adachi, K.; Maruyama, T.; Tanokura, M. Three-dimensional solution structure of an archaeal FKBP with a dual function of peptidyl prolyl cis-trans isomerase and chaperone-like activities. J. Mol. Biol. 2003, 328, 1149–1160. [Google Scholar] [CrossRef]
- Martinez-Hackert, E.; Hendrickson, W.A. Structural analysis of protein folding by the long-chain archaeal chaperone FKBP26. J. Mol. Biol. 2011, 407, 450–464. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, W.; Logan, T.M.; Li, M.; Marshall, A.G. Human recombinant [C22A] FK506-binding protein amide hydrogen exchange rates from mass spectrometry match and extend those from NMR. Protein Sci. Publ. Protein Soc. 1997, 6, 2203–2217. [Google Scholar] [CrossRef]
- Egan, D.A.; Logan, T.M.; Liang, H.; Matayoshi, E.; Fesik, S.W.; Holzman, T.F. Equilibrium Denaturation of Recombinant Human FK Binding Protein in Urea. Biochemistry 1993, 32, 1920–1927. [Google Scholar] [CrossRef]
- Janowski, B.; Wöllner, S.; Schutkowski, M.; Fischer, G. A protease-free assay for peptidyl-prolyl cis/trans isomerases using standard peptide substrates. Anal. Biochem. 1997, 252, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Mücke, M.; Schmid, F.X. Folding mechanism of ribonuclease T1 in the absence of the disulfide bonds. Biochemistry 1994, 33, 14608–14619. [Google Scholar] [CrossRef]
- Buchner, J.; Schmidt, M.; Fuchs, M.; Jaenicke, R.; Rudolph, R.; Schmid, F.X.; Kiefhaber, T. GroE Facilitates Refolding of Citrate Synthase by Suppressing Aggregation. Biochemistry 1991, 30, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Eckert, B.; Hagn, F.; Schaarschmidt, P.; Balbach, J.; Schmid, F.X. SlyD proteins from different species exhibit high prolyl isomerase and chaperone activities. Biochemistry 2006, 45, 20–33. [Google Scholar] [CrossRef]
- Martino, L.; He, Y.; Hands-Taylor, K.L.; Valentine, E.R.; Kelly, G.; Giancola, C.; Conte, M.R. The interaction of the Escherichia coli protein SlyD with nickel ions illuminates the mechanism of regulation of its peptidyl-prolyl isomerase activity. FEBS J. 2009, 276, 4529–4544. [Google Scholar] [CrossRef] [PubMed]
- Skjaerven, L.; Hollup, S.M.; Reuter, N. Normal mode analysis for proteins. J. Mol. Struct. THEOCHEM 2009, 898, 42–48. [Google Scholar] [CrossRef]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Collinet, B.; Herve, M.; Pecorari, F.; Minard, P.; Eder, O.; Desmadril, M. Functionally accepted insertions of proteins within protein domains. J. Biol. Chem. 2000, 275, 17428–17433. [Google Scholar] [CrossRef]
- Ay, J.; Gotz, F.; Borriss, R.; Heinemann, U. Structure and function of the Bacillus hybrid enzyme GluXyn-1: Native-like jellyroll fold preserved after insertion of autonomous globular domain. Proc. Natl. Acad. Sci. USA 1998, 95, 6613–6618. [Google Scholar] [CrossRef]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef]
- Baumlova, A.; Chalupska, D.; Róźycki, B.; Jovic, M.; Wisniewski, E.; Klima, M.; Dubankova, A.; Kloer, D.P.; Nencka, R.; Balla, T.; et al. The crystal structure of the phosphatidylinositol 4-kinase II α. EMBO Rep. 2014, 15, 1085–1092. [Google Scholar] [CrossRef]
- Žoldák, G.; Schmid, F.X. Cooperation of the prolyl isomerase and chaperone activities of the protein folding catalyst SlyD. J. Mol. Biol. 2011, 406, 176–194. [Google Scholar] [CrossRef]
- Kovermann, M.; Schmid, F.X.; Balbach, J. Molecular function of the prolyl cis/trans isomerase and metallochaperone SlyD. Biol. Chem. 2013, 394, 965–975. [Google Scholar] [CrossRef]
- Kovermann, M.; Zierold, R.; Haupt, C.; Löw, C.; Balbach, J. NMR relaxation unravels interdomain crosstalk of the two domain prolyl isomerase and chaperone SlyD. Biochim. Biophys. Acta 2011, 1814, 873–881. [Google Scholar] [CrossRef]
- Kahra, D.; Kovermann, M.; Löw, C.; Hirschfeld, V.; Haupt, C.; Balbach, J.; Hübner, C.G. Conformational plasticity and dynamics in the generic protein folding catalyst SlyD unraveled by single-molecule FRET. J. Mol. Biol. 2011, 411, 781–790. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef]
- Aroul-Selvam, R.; Hubbard, T.; Sasidharan, R. Domain insertions in protein structures. J. Mol. Biol. 2004, 338, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Ekman, D.; Bjorklund, A.K.; Frey-Skott, J.; Elofsson, A. Multi-domain proteins in the three kingdoms of life: Orphan domains and other unassigned regions. J. Mol. Biol. 2005, 348, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Herbst, D.A.; Jakob, R.P.; Zahringer, F.; Maier, T. Mycocerosic acid synthase exemplifies the architecture of reducing polyketide synthases. Nature 2016, 531, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Leibundgut, M.; Ban, N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Wriggers, W.; Chakravarty, S.; Jennings, P.A. Control of protein functional dynamics by peptide linkers. Biopolymers 2005, 80, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl a- chymotrypsin using different denaturants. Biochemistry 1988, 27, 8063–8068. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; Yamashita, M.M.; Sintchak, M.D.; Rotstein, S.H.; Murcko, M.A.; Boger, J.; Thomson, J.A.; Fitzgibbon, M.J.; Black, J.R.; Navia, M.A. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr. Sect. D Biol. Crystallogr. 1995, 51, 511–521. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prolyl Isomerase | [D]M (M) | m (kJ mol−1 M−1) | ΔG (kJ mol−1) | Reference |
|---|---|---|---|---|
| FKBP12 | 2.6 | 7.8 | 20 | urea |
| SlyD | 2.6 | 6.3 | 16.2 | urea a |
| SlyD ΔIF | 3.2 | 4.8 | 15.2 | urea a |
| IF(SlyD) | - | - | unfolded | urea a |
| FKBP12-IF(SlyD) | 1.9 | 8.1 | 15.2 | urea a |
| SlpA | 1.7 | 11.6 | 19.9 | GdmCl |
| SlpaΔIF | 1.4 | 9.8 | 14.1 | GdmCl |
| IF(SlpA) | 1.2 | 8.2 | 10.1 | GdmCl |
| FKBP12-IF(SlpA) | 1.3 | 20.0 | 25.0 | GdmCl |
| FKBP18 | 3.6 | 10.0 | 36.0 | GdmCl |
| FKBP18ΔIF | 3.8 | 7.4 | 28.3 | GdmCl |
| IF(FKBP18) | 0.6 | 10.8 | 6.3 | GdmCl |
| FKBP12-IF(FKBP18) | 5.0 | 5.6 | 27.8 | urea |
| Protein | Tetrapeptide a kcat/KM (M−1·s−1) | RCM-T1 b kcat/KM (M−1·s−1) | Chaperone Activity d |
|---|---|---|---|
| FKBP12 c | 0.7 × 106 | 1.50 × 104 | − |
| SlyD c | 0.25 × 106 | 0.82 × 106 | +++ |
| SlyD ΔIF c | 0.23 × 106 | no activity | − |
| IF(SlyD) c | no activity | no activity | − |
| FKBP12-IF(SlyD) c | 0.71 × 106 | 2.9 × 106 | ++ |
| SlpA | 0.01 × 106 | 2.48 × 103 | ++ |
| SlpaΔIF | 0.01 × 106 | 2.50 × 103 | − |
| IF(SlpA) | no activity | no activity | + |
| FKBP12-IF(SlpA) | 0.48 × 106 | 0.36 × 106 | ++ |
| FKBP18 | 0.21 × 106 | 0.68 × 106 | +++ |
| FKBP18ΔIF | 0.30 × 106 | 2.0 × 103 | − |
| IF(FKBP18) | no activity | no activity | ++ |
| FKBP12-IF(FKBP18) | 0.42 × 106 | 0.66 × 106 | ++ |
| Data Set | FKBP12-IF(SlyD) | FKBP12-IF(SlpA) |
|---|---|---|
| PDB ID | 5I7P | 5I7Q |
| Wavelength (Å) | 1.5418 | 1.5418 |
| Space group | P 41212 | P 212121 |
| Unit cell (Å, °) | 60.2 60.2 120.5 90 90 90 | 54.8 63.6 46.7 90 90 90 |
| Resolution (Å) | 30–2.0 (2.1–2.0) * | 35.0–1.9 (2.0–1.9) * |
| Total reflections | 92,295 | 45,572 |
| Unique reflections | 15,492 | 12,984 |
| Multiplicity | 5.9 (5.8) | 3.5 (3.6) |
| Completeness (%) | 99.1 (99.2) | 97.2 (99.7) |
| Mean I/sigma(I) | 22.0 (2.6) | 14.3 (3.0) |
| Wilson B-factor (Å2) | 29.4 | 23.5 |
| R-merge | 0.061 (0.560) | 0.144 (0.527) |
| CC1/2 | 99.9 (92.0) | 99.9 (82.2) |
| R-work | 0.206 (0.237) | 0.203 (0.216) |
| R-free | 0.250 (0.295) | 0.238 (0.258) |
| Number of atoms | 2566 | 2716 |
| Macromolecules | 1205 | 1238 |
| Water | 175 | 240 |
| Protein residues | 153 | 156 |
| RMS(bonds) (Å) | 0.005 | 0.006 |
| RMS(angles) (°) | 0.95 | 1.00 |
| Ramachandran favored (%) | 99 | 98 |
| Ramachandran outliers (%) | 0 | 0 |
| Clashscore | 2.1 | 2.0 |
| Average B-factor (Å2) | 36.8 | 18.6 |
| Macromolecules | 36.1 | 17.1 |
| Solvent | 41.8 | 26.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žoldák, G.; Knappe, T.A.; Geitner, A.-J.; Scholz, C.; Dobbek, H.; Schmid, F.X.; Jakob, R.P. Bacterial Chaperone Domain Insertions Convert Human FKBP12 into an Excellent Protein-Folding Catalyst—A Structural and Functional Analysis. Molecules 2024, 29, 1440. https://doi.org/10.3390/molecules29071440
Žoldák G, Knappe TA, Geitner A-J, Scholz C, Dobbek H, Schmid FX, Jakob RP. Bacterial Chaperone Domain Insertions Convert Human FKBP12 into an Excellent Protein-Folding Catalyst—A Structural and Functional Analysis. Molecules. 2024; 29(7):1440. https://doi.org/10.3390/molecules29071440
Chicago/Turabian StyleŽoldák, Gabriel, Thomas A. Knappe, Anne-Juliane Geitner, Christian Scholz, Holger Dobbek, Franz X. Schmid, and Roman P. Jakob. 2024. "Bacterial Chaperone Domain Insertions Convert Human FKBP12 into an Excellent Protein-Folding Catalyst—A Structural and Functional Analysis" Molecules 29, no. 7: 1440. https://doi.org/10.3390/molecules29071440
APA StyleŽoldák, G., Knappe, T. A., Geitner, A.-J., Scholz, C., Dobbek, H., Schmid, F. X., & Jakob, R. P. (2024). Bacterial Chaperone Domain Insertions Convert Human FKBP12 into an Excellent Protein-Folding Catalyst—A Structural and Functional Analysis. Molecules, 29(7), 1440. https://doi.org/10.3390/molecules29071440