

Exploring Deactivation Reasons of Biomass-Based Phosphorus-Doped Carbon as a Metal-Free Catalyst in the Catalytic Dehydroaromatization of n-Heptane

Abstract

1. Introduction
2. Results
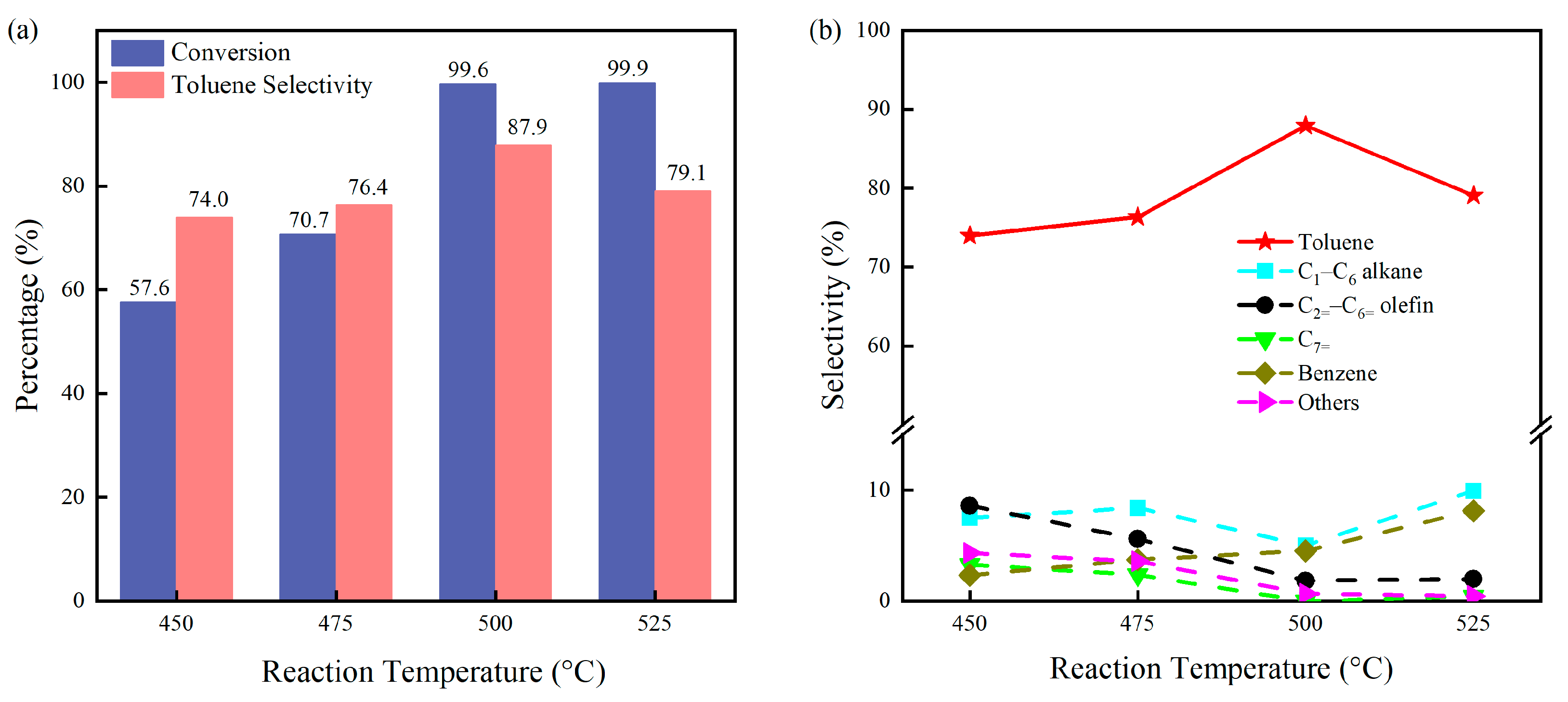
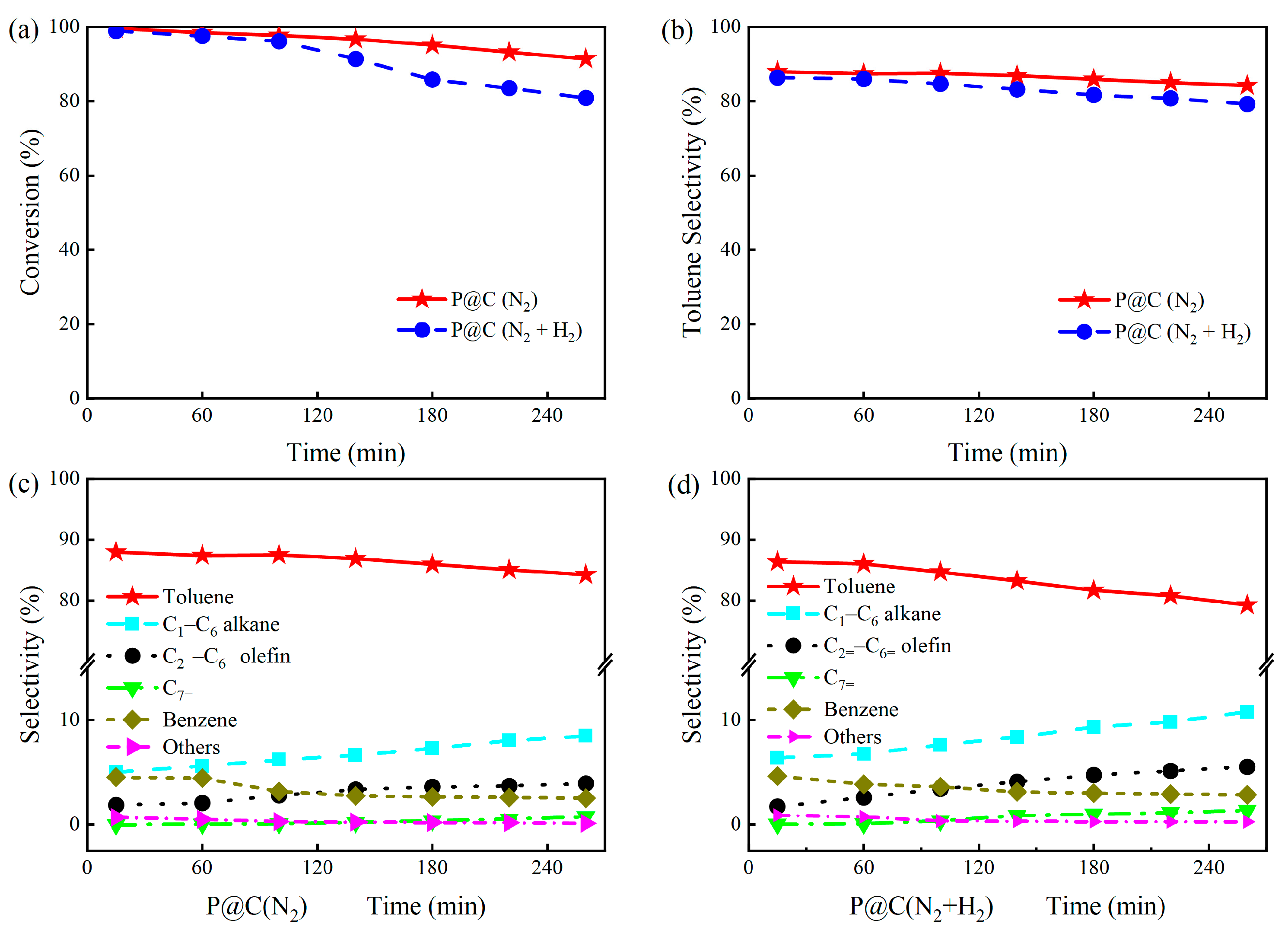
2.1. Catalytic Performance of P@C
2.2. Heat Treatment in H2 Atmosphere
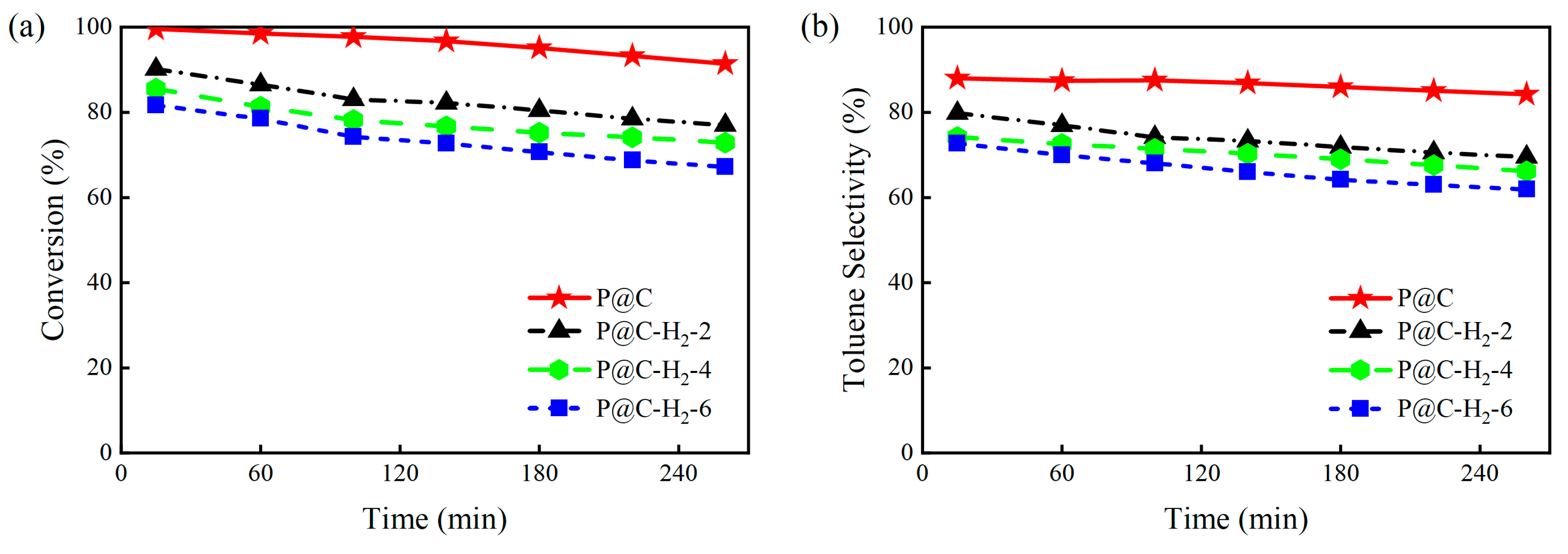
2.2.1. Catalytic Performance of P@C-H2-x
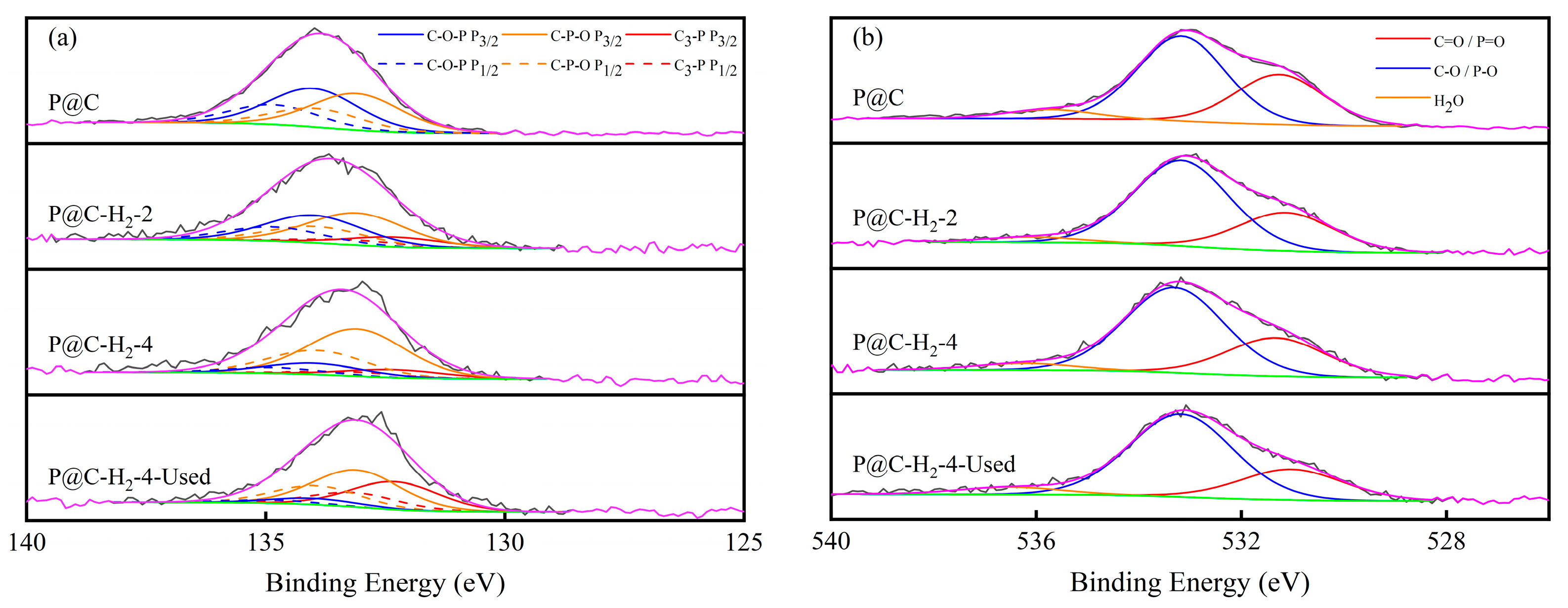
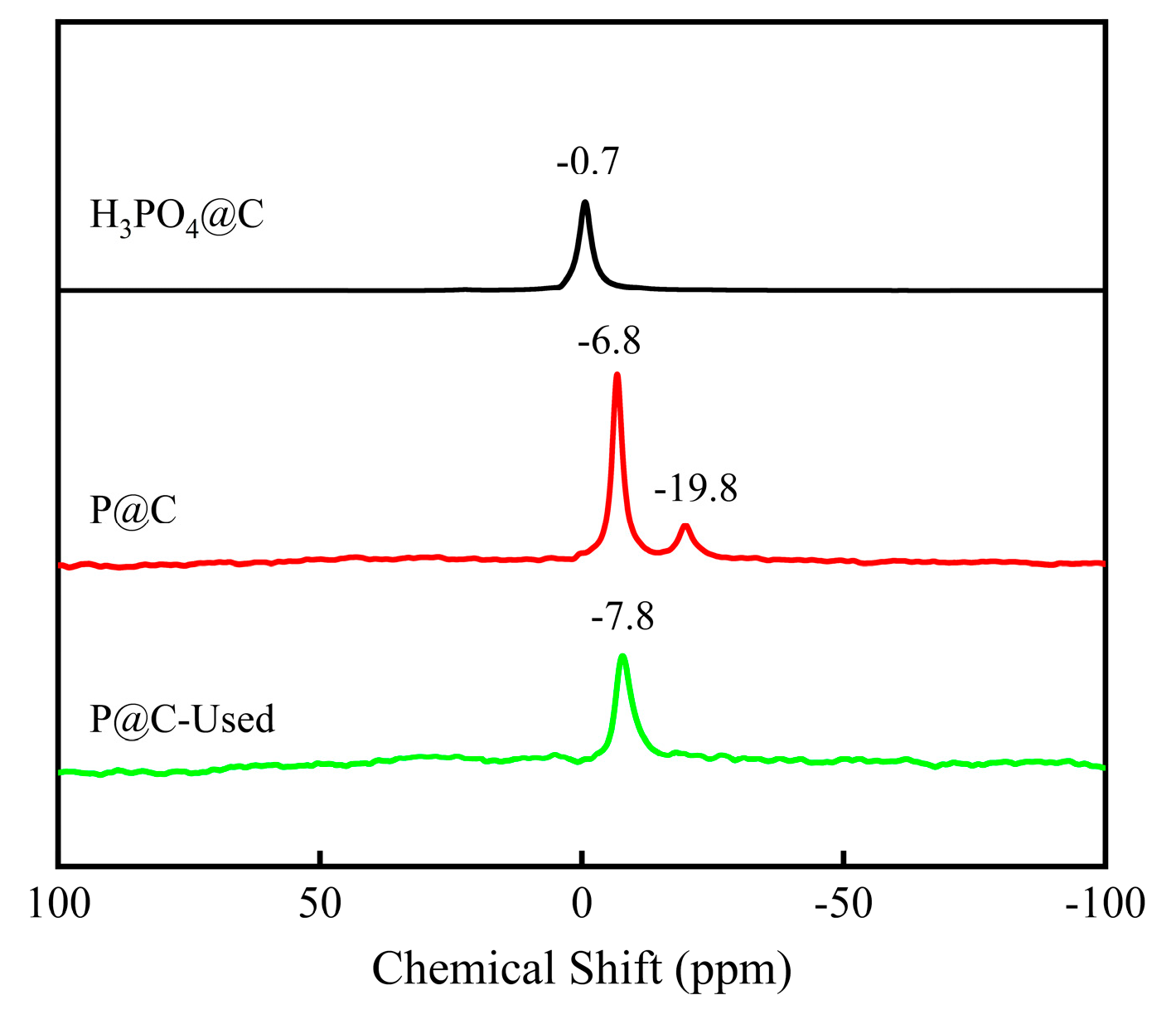
2.2.2. Characterization of P@C-H2-x
2.2.3. Verification in Different H2 Concentrations
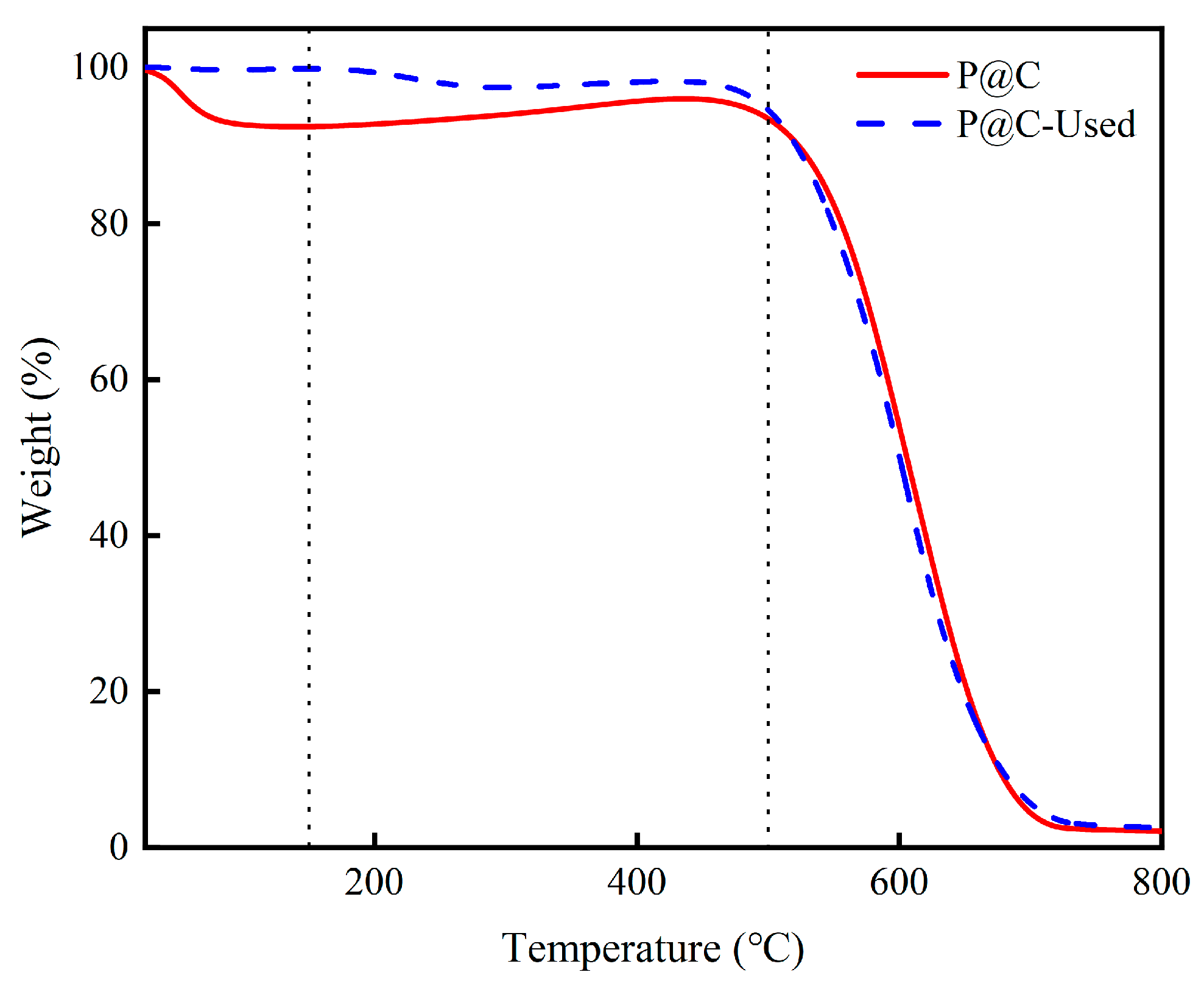
2.3. Coke Analysis
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalyst Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giuseppe, G.; Reinaldo, M.; Roberto, G. Transformation of LPG into aromatic hydrocarbons and hydrogen over zeolite catalysts. Catal. Rev. Sci. Eng. 1994, 36, 271–304. [Google Scholar] [CrossRef]
- Marzocca, A.J. Evaluation of the polymer–solvent interaction parameter χ for the system cured styrene butadiene rubber and toluene. Eur. Polym. J. 2007, 43, 2682–2689. [Google Scholar] [CrossRef]
- Gao, Z.; Ma, B.; Chen, S.; Tian, J.; Zhao, C. Converting waste PET plastics into automobile fuels and antifreeze components. Nat. Commun. 2022, 13, 3343. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cao, L.; Zhang, G.; Zhao, L.; Gao, J.; Xu, C. Catalytic conversion of light alkanes to aromatics by metal-containing HZSM-5 zeolite catalysts—A review. Fuel Process. Technol. 2021, 216, 106770. [Google Scholar] [CrossRef]
- Zeng, D.; Zhu, G.; Xia, C. Recent advances of aromatization catalysts for C4 hydrocarbons. Fuel Process. Technol. 2022, 226, 107087. [Google Scholar] [CrossRef]
- Mehdad, A.; Lobo, R.F. Ethane and ethylene aromatization on zinc-containing zeolites. Catal. Sci. Technol. 2017, 7, 3562–3572. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Wang, S.; Qin, Z.F.; Dong, M.; Wang, J.G.; Fan, W.B. Research progress on aromatization of C6+ n-alkanes. J. Fuel Chem. Technol. 2023, 51, 1529–1539. [Google Scholar] [CrossRef]
- Sharifi, K.; Halladj, R.; Royaee, S.; Towfighi, F.; Firoozi, S.; Yousefi, H. Effective factors on performance of zeolite based metal catalysts in light hydrocarbon aromatization. Rev. Chem. Eng. 2023, 39, 513–540. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, Q.; Qin, Z.; Wu, Z.; Jiao, W.; Dong, M.; Fan, W.; Wang, J. Promoting effect of alkali metal on the catalytic performance of hierarchical Pt/Beta in the aromatization of n-heptane. Microporous Mesoporous Mater. 2022, 343, 112189. [Google Scholar] [CrossRef]
- Akhtar, M.N.; Aitani, A.M.; Ummer, A.C.; Alasiri, H.S. Review on the Catalytic conversion of naphtha to aromatics: Advances and outlook. Energy Fuels 2023, 37, 2586–2607. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, S.; Wu, Z.; Qin, Z.; Dong, M.; Wang, J.; Fan, W. Aromatization of n-C7–n-C9 alkanes on a Pt/KZSM-5(DeAl) catalyst. Catal. Sci. Technol. 2023, 13, 1009–1020. [Google Scholar] [CrossRef]
- Hughes, T.R.; Buss, W.C.; Tamm, P.W.; Jacobson, R.L. Aromatization of hydrocarbons over platinum alkaline earth zeolites. Stud. Surf. Sci. Catal. 1986, 28, 725–732. [Google Scholar] [CrossRef]
- Xu, D.; Wang, S.; Wu, B.; Huo, C.; Qin, Y.; Zhang, B.; Yin, J.; Huang, L.; Wen, X.; Yang, Y.; et al. Tailoring Pt locations in KL zeolite by improved atomic layer deposition for excellent performance in n-heptane aromatization. J. Catal. 2018, 365, 163–173. [Google Scholar] [CrossRef]
- Wang, S.; Gao, Y.; Yi, F.; Yan, M.; Zhu, D.; Xu, D.; Li, Y. Regulation of sub-nanometric platinum on BaKL zeolite for boosting n-heptane aromatization. Fuel 2022, 328, 125281. [Google Scholar] [CrossRef]
- Xu, D.; Wei, L.; Yan, M.; Yi, F.; Zhao, G.; Jia, A.; Zhu, D.; Wang, S.; Li, Y. Zinc-assisted nanometric Pt cluster stabilized on KL zeolite via atomic layer deposition for the n-heptane aromatization. Appl. Catal. A 2023, 663, 119308. [Google Scholar] [CrossRef]
- Xu, D.; Wang, S.; Wu, B.; Zhang, B.; Qin, Y.; Huo, C.; Huang, L.; Wen, X.; Yang, Y.; Li, Y. Highly dispersed single-atom Pt and Pt clusters in the Fe-Modified KL zeolite with enhanced selectivity for n-heptane aromatization. ACS Appl. Mater. Interfaces 2019, 11, 29858–29867. [Google Scholar] [CrossRef]
- Wang, S.; Xu, D.; Zhu, D.; Zhao, B.; Guan, H.; Qin, Y.; Wu, B.; Yang, Y.; Li, Y. Elucidating the restructuring-induced highly active bimetallic Pt–Co/KL catalyst for the aromatization of n-heptane. Chem. Commun. 2020, 56, 892–895. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Chen, S.; Bao, S.; Xing, F.; Jiang, B. Phosphorus-doped activated carbon catalyst for n-hexane dehydroaromatization reaction. Catal. Commun. 2021, 156, 106318. [Google Scholar] [CrossRef]
- Li, Y.; Bao, S.; Zhao, H.; Feng, B.; Chen, S.; Gu, T.; Yang, B.; Jiang, B. Unprecedented high selectivity of n-hexane dehydroaromatization to benzene over metal-free phosphorus-doped activated carbon catalysts. Chem. Commun. 2021, 57, 4166–4169. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Chiu, C.C.; Gong, J. Molecular understandings on the activation of light hydrocarbons over heterogeneous catalysts. Chem. Sci. 2015, 6, 4403–4425. [Google Scholar] [CrossRef]
- Yalan, W.; Ping, H.; Jia, Y.; Yi-An, Z.; De, C. C–H bond activation in light alkanes: A theoretical perspective. Chem. Soc. Rev. 2021, 50, 4299–4358. [Google Scholar] [CrossRef]
- Li, K.; Chang, Q.; Yin, J.; Zhao, C.; Huang, L.; Tao, Z.; Yun, Y.; Zhang, C.; Xiang, H.; Yang, Y.; et al. Deactivation of Pt/KL catalyst during n-heptane aromatization reaction. J. Catal. 2018, 361, 193–203. [Google Scholar] [CrossRef]
- Cimino, S.; Lisi, L. Catalyst deactivation, poisoning and regeneration. Catalysts 2019, 9, 668. [Google Scholar] [CrossRef]
- Saito, H.; Sekine, Y. Catalytic conversion of ethane to valuable products through non-oxidative dehydrogenation and dehydroaromatization. RSC Adv. 2020, 10, 21427–21453. [Google Scholar] [CrossRef]
- Treacy, M.M.J. Pt Agglomeration and entombment in single channel zeolites: Pt/LTL. Microporous Mesoporous Mater. 1999, 28, 271–292. [Google Scholar] [CrossRef]
- Fung, S.C. Deactivation and regeneration/redispersion chemistry of Pt/KL-Zeolite. Stud. Surf. Sci. Catal. 2001, 139, 399–406. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Phillips, J.; Xia, B.; Radovic, L.R. On the modification and characterization of chemical surface properties of activated carbon: In the search of carbons with stable basic properties. Langmuir 1996, 12, 4404–4410. [Google Scholar] [CrossRef]
- Kundu, S.; Wang, Y.; Xia, W.; Muhler, M. Thermal stability and reducibility of oxygen-containing functional groups on multiwalled carbon nanotube surfaces: A quantitative high-resolution XPS and TPD/TPR study. J. Phys. Chem. C 2008, 112, 16869–16878. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, Y.; Fan, S.; Wang, S.; Qin, Z.; Dong, M.; Wang, J.; Fan, W. Development and catalytic mechanism of a highly efficient Pt/Kβ catalyst for n-heptane aromatization. Fuel 2023, 337, 126874. [Google Scholar] [CrossRef]
- Li, K.; Yan, M.; Wang, H.; Cai, L.; Wang, P.; Chen, H. Enhanced stability of Pt@S-1 with the aid of potassium ions for n-hexane and n-heptane aromatization. Fuel Process. Technol. 2023, 252, 107982. [Google Scholar] [CrossRef]
- Lin, C.; Yang, Z.; Pan, H.; Cui, J.; Lv, Z.; Liu, X.; Tian, P.; Xiao, Z.; Li, P.; Xu, J.; et al. Ce-introduced effects on modification of acidity and Pt electronic states on Pt-Sn/γ-Al2O3 catalysts for catalytic reforming. Appl. Catal. A 2021, 617, 118116. [Google Scholar] [CrossRef]
- Wang, F.; Cheong, J.Y.; He, Q.; Duan, G.; He, S.; Zhang, L.; Zhao, Y.; Kim, I.D.; Jiang, S. Phosphorus-doped thick carbon electrode for high-energy density and long-life supercapacitors. Chem. Eng. J. 2021, 414, 128767. [Google Scholar] [CrossRef]
- Zhang, G.; Qian, Z.; Mao, Y.; Sun, B.; Zuo, P.; Ma, Y.; Du, C.; Yin, G.; Xie, J. Phosphorus-doped carbon as cathode material for high energy nonaqueous Li-O2 batteries. Appl. Surf. Sci. 2021, 543, 148864. [Google Scholar] [CrossRef]
- Rey-Raap, N.; Granja, M.A.C.; Pereira, M.F.R.; Figueiredo, J.L. Phosphorus-doped carbon/carbon nanotube hybrids as high-performance electrodes for supercapacitors. Electrochim. Acta 2020, 354, 136713. [Google Scholar] [CrossRef]
- Schwartz, V.; Xie, H.; Meyer, H.M.; Overbury, S.H.; Liang, C. Oxidative dehydrogenation of isobutane on phosphorous-modified graphitic mesoporous carbon. Carbon 2011, 49, 659–668. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, S.; Yang, J.; Yoon, S.H. Evolution of phosphorus-containing groups on activated carbons during heat treatment. Langmuir 2017, 33, 3112–3122. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR studies of phosphoric acid activated carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Strelko, V.; Streat, M.; Kozynchenko, O. Preparation, characterisation and sorptive properties of polymer based phosphorus-containing carbon. React. Funct. Polym. 1999, 41, 245–253. [Google Scholar] [CrossRef]
- Albero, J.; Vidal, A.; Migani, A.; Concepción, P.; Blancafort, L.; García, H. Phosphorus-doped graphene as a metal-free material for thermochemical water reforming at unusually mild conditions. ACS Sustain. Chem. Eng. 2019, 7, 838–846. [Google Scholar] [CrossRef]
- Wang, B.; Yu, L.; Zhang, J.; Pu, Y.; Zhang, H.; Li, W. Phosphorus-doped carbon supports enhance gold-based catalysts for acetylene hydrochlorination. RSC Adv. 2014, 4, 15877–15885. [Google Scholar] [CrossRef]
- Yaxin, L.; Xian, Z.; Ruiguang, Y.; Guiying, L.; Changwei, H. The role of H3PO4 in the preparation of activated carbon from NaOH-treated rice husk residue. RSC Adv. 2015, 5, 32626–32636. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Reaction Condition 1 | Con 2 (%) | SArom 3 (%) | Lifetime (h) | Ref. |
|---|---|---|---|---|---|
| Pt/KZSM-5(deAL) | 550 °C, ambient pressure, WHSV = 2 h−1, H2/n-heptane = 6 | 96.1 | B + T: 75.4 | 234 | [11] |
| Pt/KBeta | 550 °C, 0.1 MPa, WHSV = 2 h−1, H2/n-heptane = 6 | 80–100 | 70–80.7 | 160 | [29] |
| Pt/Beta-Rb | 550 °C, 0.1 MPa, WHSV = 2 h−1, H2/n-heptane = 6 | 78.5 | T: 94.6 | 15.7 | [9] |
| KPt@S-1 | 500 °C, 0.1 MPa, WHSV = 3 h−1, H2/n-heptane = 2 | 98 | T: 62 | 180 | [30] |
| 6Pt/BaKL | 420 °C, 0.1 MPa, WHSV = 0.68 h−1, H2/n-heptane = 6 | 97 | T: 92 | 20 | [14] |
| PtZn3/KL | 500 °C, 0.1 MPa, WHSV = 0.68 h−1, H2/n-heptane = 6 | 92 | T: 86 | 25 | [15] |
| Pt-Ce/γ-Al2O3 | 500 °C, 0.6 MPa, WHSV = 3 h−1, H2/n-heptane = 10 | 94.2 | T: 29.2 | / | [31] |
| Pt-5/KL | 420 °C, ambient pressure, WHSV = 0.68 h−1, H2/n-heptane = 6 | 90 | T: 89 | 27 | [17] |
| PtFe-1/KL | 420 °C, 0.1 MPa, WHSV = 0.68 h−1, H2/n-heptane = 6 | 90 | T: 90 | 30 | [16] |
| 10Pt/KL | 420 °C, 0.1 MPa, WHSV = 0.68 h−1, H2/n-heptane = 6 | 78 (2 h) | T: 82 | 20 | [13] |
| P@C | 500 °C, ambient pressure, WHSV = 0.68 h−1 | 99.6 | T: 87.9 | / | This Work |
| Sample | C (at %) * | O (at %) | P (at %) | ||||
|---|---|---|---|---|---|---|---|
| C=O/P=O | C–O/P–O | H2O | C–O–P | C–P–O | C3–P | ||
| P@C | 87.52 | 3.53 | 5.98 | 0.66 | 1.13 | 1.18 | 0.00 |
| 34.7% | 58.8% | 6.5% | 48.7% | 51.3% | 0.0% | ||
| P@C-H2-2 | 93.03 | 1.74 | 3.87 | 0.23 | 0.45 | 0.52 | 0.16 |
| 29.8% | 66.2% | 4.0% | 39.8% | 46.3% | 13.9% | ||
| P@C-H2-4 | 93.78 | 1.59 | 3.53 | 0.28 | 0.10 | 0.57 | 0.15 |
| 29.4% | 65.4% | 5.2% | 12.2% | 69.5% | 18.3% | ||
| P@C-H2-4-Used | 94.26 | 1.25 | 3.37 | 0.30 | 0.07 | 0.43 | 0.32 |
| 25.3% | 68.5% | 6.2% | 8.6% | 52.4% | 39.0% | ||
| Sample | P (at %) 1 | Total Acidity 2 (μmol g−1) | SBET (m2 g−1) | Vt 3 (cm3 g−1) | D 4 (Å) | Carbon Deposition 5 (molCarbon/gCat./h) |
|---|---|---|---|---|---|---|
| P@C | 2.31 | 137.60 | 1733.7 | 0.7876 | 5.50 | 1.20 × 10−3 |
| P@C-H2-2 | 1.13 | 59.48 | 1699.8 | 0.7872 | 5.52 | 1.07 × 10−3 |
| P@C-H2-4 | 0.82 | 25.83 | 1710.2 | 0.8014 | 5.54 | 0.60 × 10−3 |
| P@C-H2-6 | 0.72 | 19.20 | 1723.8 | 0.8112 | 5.55 | 0.30 × 10−3 |
| Sample | Total Acidity 1 (μmol g−1) | SBET (m2 g−1) | Vt 2 (cm3 g−1) | D 3 (Å) | Carbon Deposition 4 (molCarbon/gCat./h) |
|---|---|---|---|---|---|
| P@C(N2)-Used | 29.56 | 946.3 | 0.4302 | 5.49 | 1.20 × 10−3 |
| P@C(N2 + H2)-Used | 28.54 | 1263.9 | 0.5759 | 5.40 | 0.81 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.; Liu, S.; Liu, B. Exploring Deactivation Reasons of Biomass-Based Phosphorus-Doped Carbon as a Metal-Free Catalyst in the Catalytic Dehydroaromatization of n-Heptane. Molecules 2024, 29, 1288. https://doi.org/10.3390/molecules29061288
Yu F, Liu S, Liu B. Exploring Deactivation Reasons of Biomass-Based Phosphorus-Doped Carbon as a Metal-Free Catalyst in the Catalytic Dehydroaromatization of n-Heptane. Molecules. 2024; 29(6):1288. https://doi.org/10.3390/molecules29061288
Chicago/Turabian StyleYu, Fei, Siyuan Liu, and Bo Liu. 2024. "Exploring Deactivation Reasons of Biomass-Based Phosphorus-Doped Carbon as a Metal-Free Catalyst in the Catalytic Dehydroaromatization of n-Heptane" Molecules 29, no. 6: 1288. https://doi.org/10.3390/molecules29061288
APA StyleYu, F., Liu, S., & Liu, B. (2024). Exploring Deactivation Reasons of Biomass-Based Phosphorus-Doped Carbon as a Metal-Free Catalyst in the Catalytic Dehydroaromatization of n-Heptane. Molecules, 29(6), 1288. https://doi.org/10.3390/molecules29061288