
ABC2A: A Straightforward and Fast Method for the Accurate Backmapping of RNA Coarse-Grained Models to All-Atom Structures
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
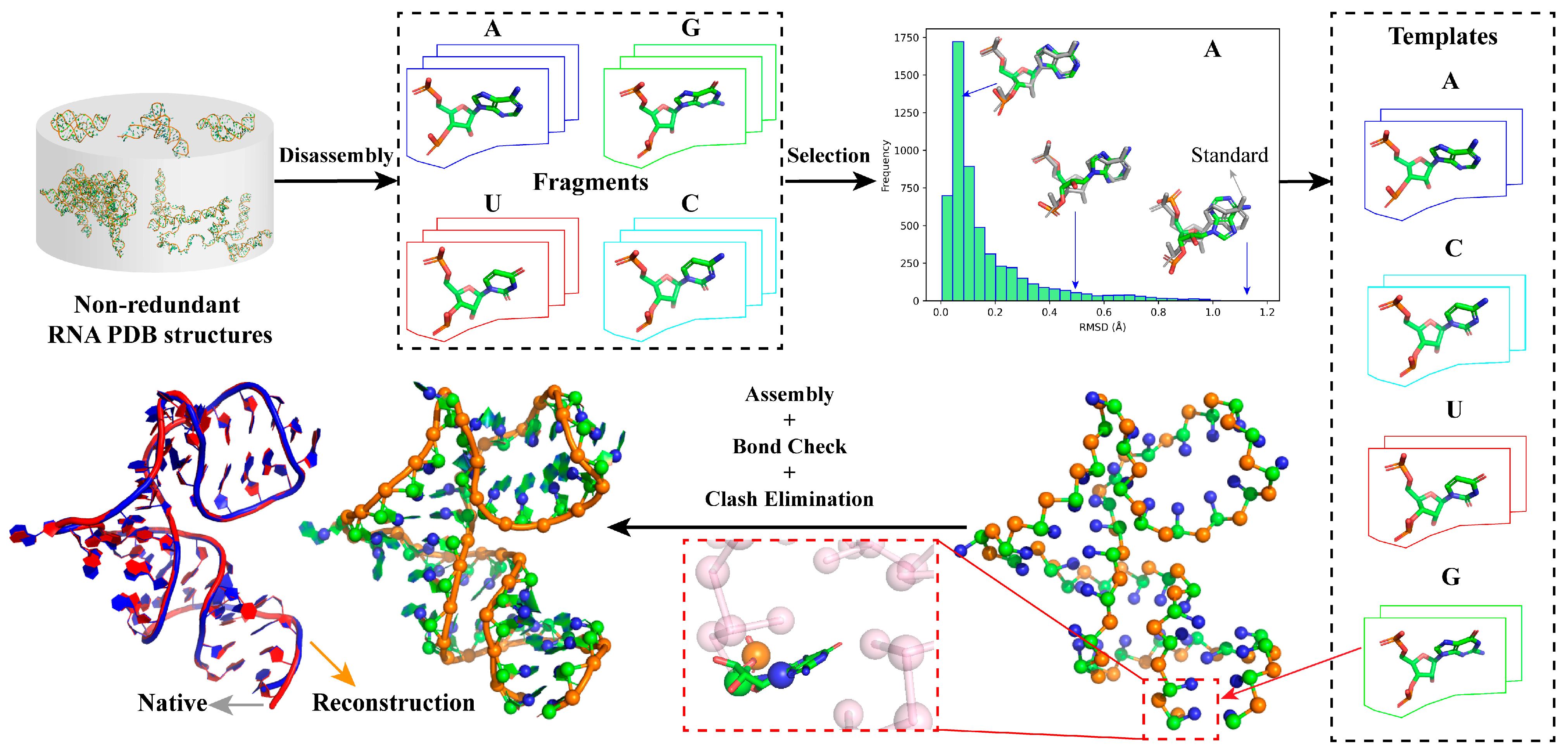
2.1. Overview of ABC2A
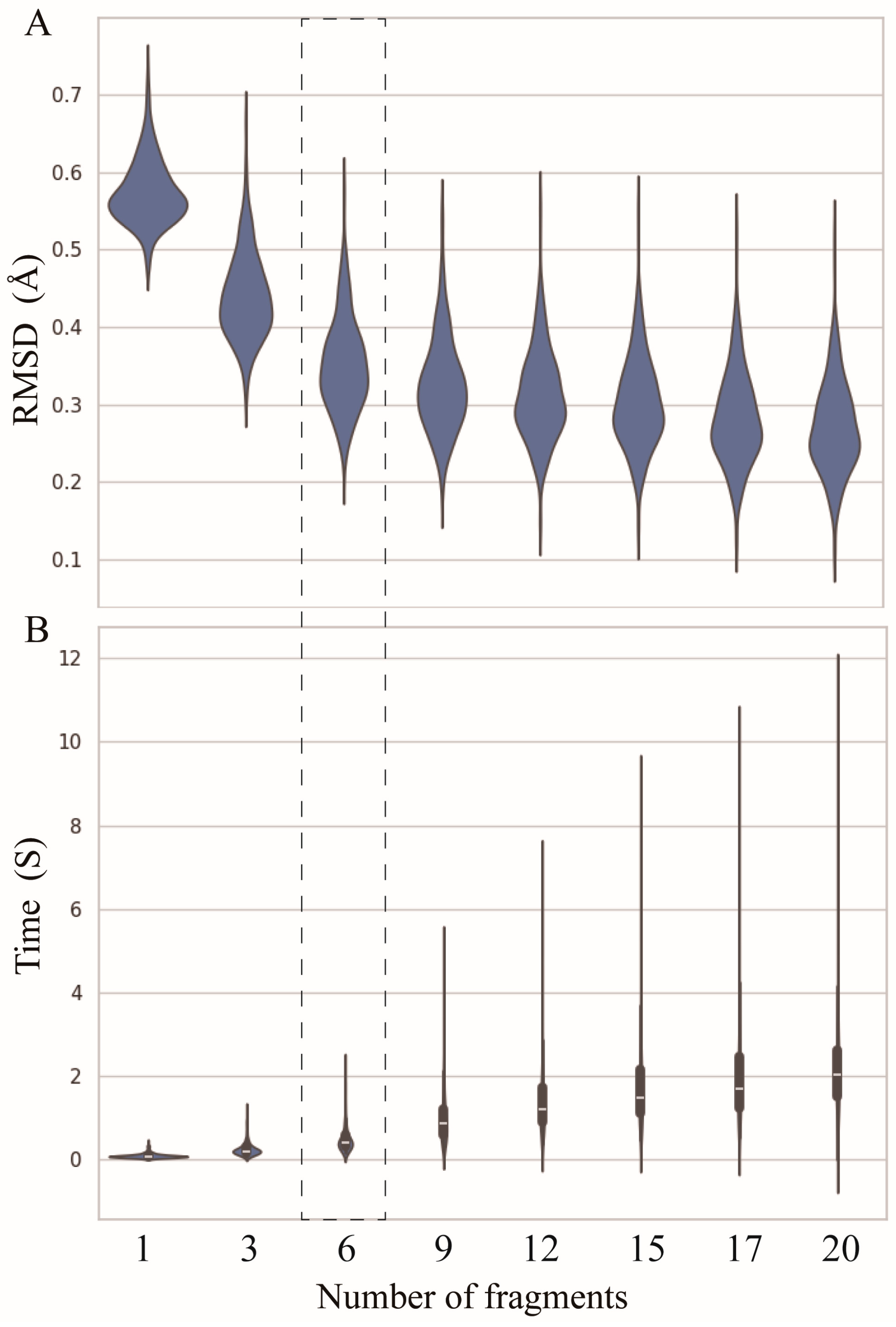
2.2. Number of Fragments
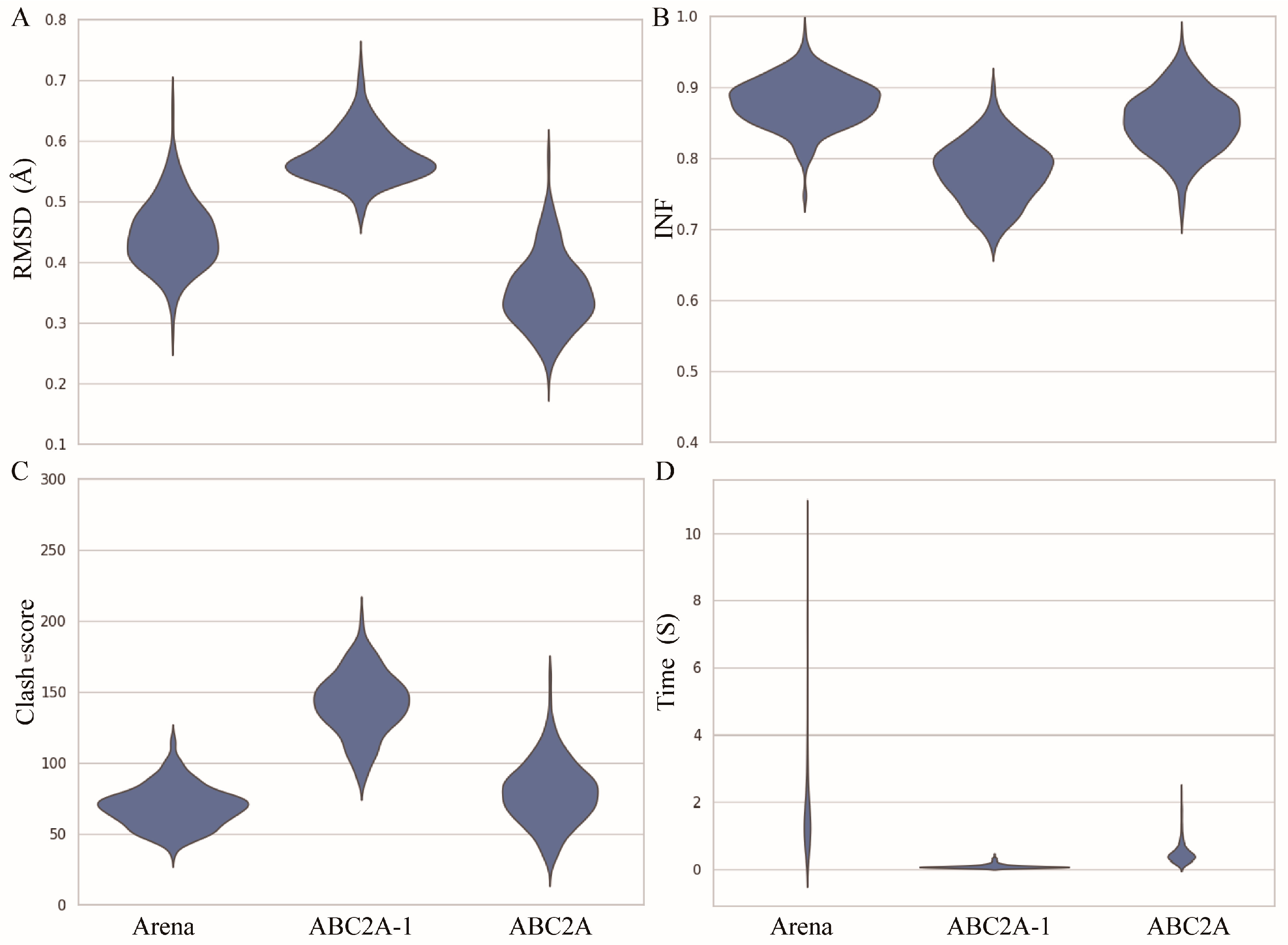
2.3. Performance of ABC2A
3. Discussion
4. Materials and Methods
4.1. The Three-Bead Coarse-Grained Model
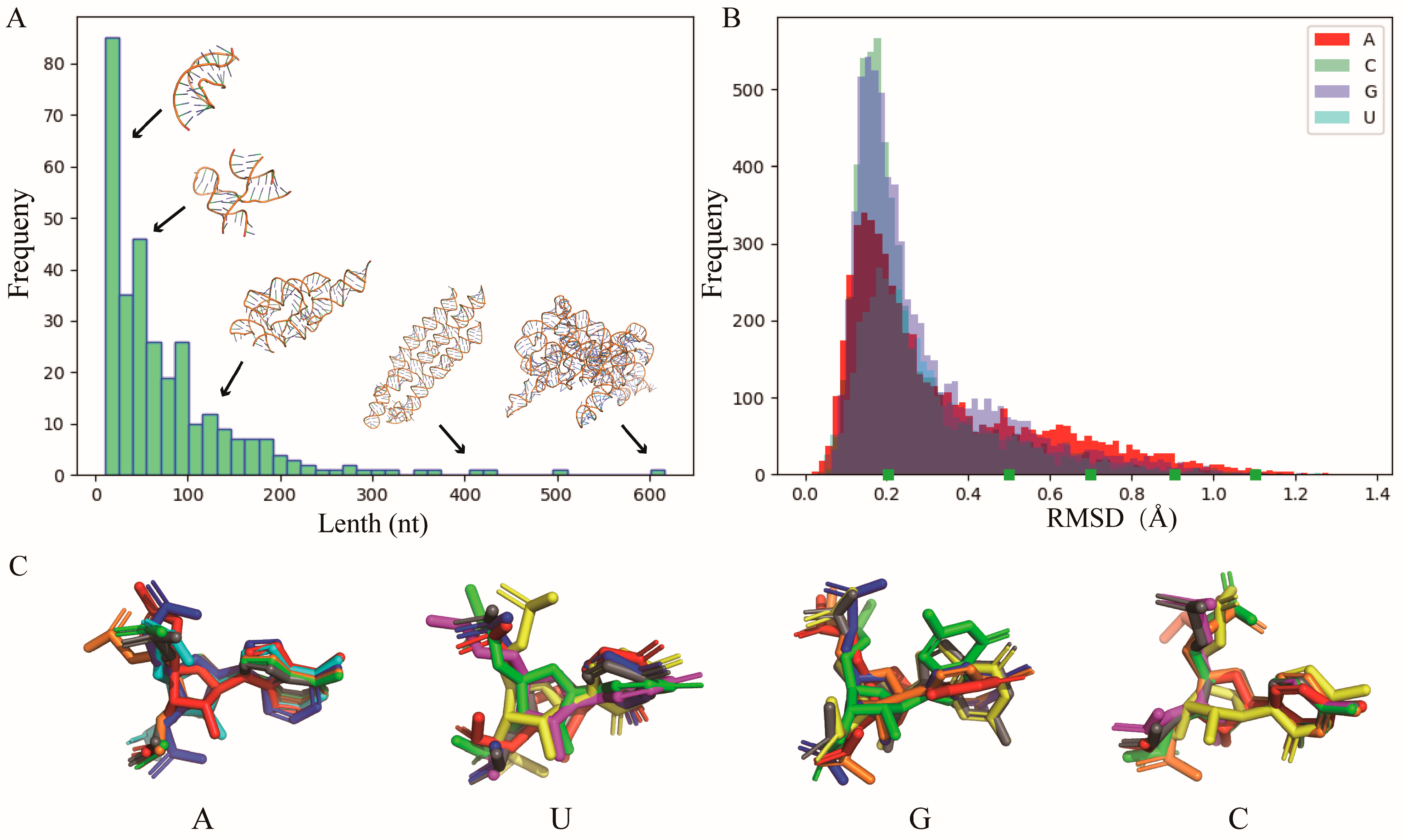
4.2. Construction of Nucleotide Template Library
4.3. Full Atomic Structure Assembly
4.4. Structure Refinement
4.5. Test Sets and Performance Evaluation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar] [CrossRef]
- Zhang, J.; Fei, Y.; Sun, L.; Zhang, Q.C. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat. Methods 2022, 19, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. RNA Structure: Advances and Assessment of 3D Structure Prediction. Annu. Rev. Biophys. 2017, 46, 483–503. [Google Scholar] [CrossRef]
- Ou, X.; Zhang, Y.; Xiong, Y.; Xiao, Y. Advances in RNA 3D Structure Prediction. J. Chem. Inf. Model. 2022, 62, 5862–5874. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, S.; Lou, E.; Tan, Y.L.; Tan, Z.J. RNA 3D Structure Prediction: Progress and Perspective. Molecules 2023, 28, 5532. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gong, Z.; Zhao, Y. Methods and applications in proteins and RNAs. Life 2023, 13, 672. [Google Scholar] [CrossRef]
- Wu, K.E.; Zou, J.Y.; Chang, H. Machine learning modeling of RNA structures: Methods, challenges and future perspectives. Brief. Bioinform. 2023, 24, bbad210. [Google Scholar] [CrossRef]
- Zhang, J.; Lang, M.; Zhou, Y.; Zhang, Y. Predicting RNA structures and functions by artificial intelligence. Trends Genet. 2024, 40, 94–107. [Google Scholar] [CrossRef]
- Watkins, A.M.; Rangan, R.; Das, R. FARFAR2: Improved de novo rosetta prediction of complex global RNA folds. Structure 2020, 28, 963–976.e6. [Google Scholar] [CrossRef] [PubMed]
- Parisien, M.; Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008, 452, 51–55. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xiao, Y. 3dRNA: 3D Structure Prediction from Linear to Circular RNAs. J. Mol. Biol. 2022, 434, 167452. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, X.; Yu, S.; Tan, Y.L.; Tan, Z.J. FebRNA: An automated fragment-ensemble-based model for building RNA 3D structures. Biophys. J. 2022, 121, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Wu, R.; Zhan, J.; Zhou, Y. Pairing a high-resolution statistical potential with a nucleobase-centric sampling algorithm for improving RNA model refinement. Nat. Commun. 2021, 12, 2777. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, W.; Wang, J.; Li, W.; Gong, S.; Zhang, J.; Wang, W. RNA3DCNN: Local and global quality assessments of RNA 3D structures using 3D deep convolutional neural networks. PLoS Comput. Biol. 2018, 14, e1006514. [Google Scholar] [CrossRef]
- Wang, W.; Feng, C.; Han, R.; Wang, Z.; Ye, L.; Du, Z.; Wei, H.; Zhang, F.; Peng, Z.; Yang, J. trRosettaRNA: Automated prediction of RNA 3D structure with transformer network. Nat. Commun. 2023, 14, 7266. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Feng, C.; Pearce, R.; Lydia Freddolino, P.; Zhang, Y. Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction. Nat. Commun. 2023, 14, 5745. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.J. RNA 3D structure prediction using coarse-grained models. Front. Mol. Biosci. 2021, 8, 720937. [Google Scholar] [CrossRef]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016, 44, e63. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ding, F.; Dokholyan, N.V. iFoldRNA: Three-dimensional RNA structure prediction and folding. Bioinformatics 2008, 24, 1951–1952. [Google Scholar] [CrossRef] [PubMed]
- Jonikas, M.A.; Radmer, R.J.; Laederach, A.; Das, R.; Pearlman, S.; Herschlag, D.; Altman, R.B. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 2009, 15, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Chen, S.J. Physics-based de novo prediction of RNA 3D structures. J. Phys. Chem. B 2011, 115, 4216–4226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.J. RNAJP: Enhanced RNA 3D structure predictions with non-canonical interactions and global topology sampling. Nucleic Acids Res. 2023, 51, 3341–3356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Chen, S.J. IsRNA1: De novo prediction and blind screening of RNA 3D structures. J. Chem. Theory Comput. 2021, 17, 1842–1857. [Google Scholar] [CrossRef]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Doye, J.P.; Louis, A.A. A nucleotide-level coarse-grained model of RNA. J. Chem. Phys. 2014, 140, 235102. [Google Scholar] [CrossRef]
- Cragnolini, T.; Derreumaux, P.; Pasquali, S. Coarse-grained simulations of RNA and DNA duplexes. J. Phys. Chem. B 2013, 117, 8047–8060. [Google Scholar] [CrossRef]
- Shi, Y.Z.; Wang, F.H.; Wu, Y.Y.; Tan, Z.J. A coarse-grained model with implicit salt for RNAs: Predicting 3D structure, stability and salt effect. J. Chem. Phys. 2014, 141, 105102. [Google Scholar] [CrossRef]
- Shi, Y.Z.; Jin, L.; Feng, C.J.; Tan, Y.L.; Tan, Z.J. Predicting 3D structure and stability of RNA pseudoknots in monovalent and divalent ion solutions. PLoS Comput. Biol. 2018, 14, e1006222. [Google Scholar] [CrossRef]
- Shi, Y.Z.; Jin, L.; Wang, F.H.; Zhu, X.L.; Tan, Z.J. Predicting 3D structure, flexibility, and stability of RNA hairpins in monovalent and divalent ion solutions. Biophys. J. 2015, 109, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tan, Y.L.; Wu, Y.; Wang, X.; Shi, Y.Z.; Tan, Z.J. Structure folding of RNA kissing complexes in salt solutions: Predicting 3D structure, stability, and folding pathway. RNA 2019, 25, 1532–1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, Y.L.; Yu, S.; Shi, Y.Z.; Tan, Z.J. Predicting 3D structures and stabilities for complex RNA pseudoknots in ion solutions. Biophys. J. 2023, 122, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.G.; Qiu, H.H.; Jiang, J.; Liu, J.; Shi, Y.Z. 3D structure stability of the HIV-1 TAR RNA in ion solutions: A coarse-grained model study. J. Chem. Phys. 2019, 151, 165101. [Google Scholar] [CrossRef] [PubMed]
- Stasiewicz, J.; Mukherjee, S.; Nithin, C.; Bujnicki, J.M. QRNAS: Software tool for refinement of nucleic acid structures. BMC Struct. Biol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. Computational reconstruction of atomistic protein structures from coarse-grained models. Comput. Struct. Biotechnol. J. 2019, 18, 162–176. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, C.; Ma, R.; Zhang, Z. Backmapping from Multiresolution Coarse-Grained Models to Atomic Structures of Large Biomolecules by Restrained Molecular Dynamics Simulations Using Bayesian Inference. J. Chem. Theory Comput. 2019, 15, 3344–3353. [Google Scholar] [CrossRef]
- Shimizu, M.; Takada, S. Reconstruction of atomistic structures from coarse-grained models for protein-DNA complexes. J. Chem. Theory Comput. 2018, 14, 1682–1694. [Google Scholar] [CrossRef]
- Keating, K.S.; Pyle, A.M. Semiautomated model building for RNA crystallography using a directed rotameric approach. Proc. Natl. Acad. Sci. USA 2010, 107, 8177–8182. [Google Scholar] [CrossRef]
- Jonikas, M.A.; Radmer, R.J.; Altman, R.B. Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models. Bioinformatics 2009, 25, 3259–3266. [Google Scholar] [CrossRef]
- Golon, L.; Sieradzan, K. NARall: A novel tool for reconstruction of the all-atom structure of nucleic acids from heavily coarse-grained model. Chem. Pap. 2023, 77, 2437–2445. [Google Scholar] [CrossRef]
- Perry, Z.R.; Pyle, A.M.; Zhang, C. Arena: Rapid and accurate reconstruction of full atomic RNA structures from coarse-grained models. J. Mol. Biol. 2023, 435, 168210. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Golon, Ł.; Liwo, A. Prediction of DNA and RNA structure with the NARES-2P force field and conformational space annealing. Phys. Chem. Chem. Phys. 2018, 20, 19656–19663. [Google Scholar] [CrossRef]
- Zhang, C.; Pyle, A.M. CSSR: Assignment of secondary structure to coarse-grained RNA tertiary structures. Acta Crystallogr. D Struct. Biol. 2022, 78, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Parisien, M.; Cruz, J.A.; Westhof, E.; Major, F. New metrics for comparing and assessing discrepancies between RNA 3D structures and models. RNA 2009, 15, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Chou, F.C.; Das, R. Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol. 2015, 553, 35–64. [Google Scholar] [PubMed]
- Lan, P.; Tan, M.; Zhang, Y.; Niu, S.; Chen, J.; Shi, S.; Qiu, S.; Wang, X.; Peng, X.; Cai, G.; et al. Structural insight into precursor tRNA processing by yeast ribonuclease P. Science 2018, 362, eaat6678. [Google Scholar] [CrossRef] [PubMed]
- Coureux, P.D.; Lazennec-Schurdevin, C.; Bourcier, S.; Mechulam, Y.; Schmitt, E. Cryo-EM study of an archaeal 30S initiation complex gives insights into evolution of translation initiation. Commun. Biol. 2020, 3, 58. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Olson, W.K.; Lu, X.J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 2019, 47, W26–W34. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Das, R.; Kretsch, R.C.; Simpkin, A.J.; Mulvaney, T.; Pham, P.; Rangan, R.; Bu, F.; Keegan, R.M.; Topf, M.; Rigden, D.J.; et al. Assessment of three-dimensional RNA structure prediction in CASP15. Proteins 2023, 91, 1747–1770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Gong, S.; Zhang, C.; Zhang, Y. RNA-align: Quick accurate alignment of RNA 3D structures based on size-independent, TM-scoreRNA. Bioinformatics 2019, 35, 4459–4461. [Google Scholar] [CrossRef]
- Magnus, M.; Antczak, M.; Zok, T.; Wiedemann, J.; Lukasiak, P.; Cao, Y.; Bujnicki, J.M.; Westhof, E.; Szachniuk, M.; Miao, Z. RNA-Puzzles toolkit: A computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools. Nucleic Acids Res. 2020, 48, 576–588. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.-Z.; Wu, H.; Li, S.-S.; Li, H.-Z.; Zhang, B.-G.; Tan, Y.-L. ABC2A: A Straightforward and Fast Method for the Accurate Backmapping of RNA Coarse-Grained Models to All-Atom Structures. Molecules 2024, 29, 1244. https://doi.org/10.3390/molecules29061244
Shi Y-Z, Wu H, Li S-S, Li H-Z, Zhang B-G, Tan Y-L. ABC2A: A Straightforward and Fast Method for the Accurate Backmapping of RNA Coarse-Grained Models to All-Atom Structures. Molecules. 2024; 29(6):1244. https://doi.org/10.3390/molecules29061244
Chicago/Turabian StyleShi, Ya-Zhou, Hao Wu, Sha-Sha Li, Hui-Zhen Li, Ben-Gong Zhang, and Ya-Lan Tan. 2024. "ABC2A: A Straightforward and Fast Method for the Accurate Backmapping of RNA Coarse-Grained Models to All-Atom Structures" Molecules 29, no. 6: 1244. https://doi.org/10.3390/molecules29061244
APA StyleShi, Y.-Z., Wu, H., Li, S.-S., Li, H.-Z., Zhang, B.-G., & Tan, Y.-L. (2024). ABC2A: A Straightforward and Fast Method for the Accurate Backmapping of RNA Coarse-Grained Models to All-Atom Structures. Molecules, 29(6), 1244. https://doi.org/10.3390/molecules29061244